Introduction
Cellular metabolic alterations represent a
well-known aspect of cancer, which have been linked to controlling
the invasion and spread of cancer cells (1,2).
Glioblastoma (GBM), which is characterized by high invasiveness and
metabolic flexibility, demonstrates this connection. Known as the
most aggressive type of primary brain tumor in adults, GBM rapidly
progresses, infiltrates nearby healthy brain tissue and is
associated with a poor prognosis (3).
The recurrence of GBM, driven by its invasive
behavior, typically occurs in close proximity to the initial tumor
location, ultimately resulting in the death of almost all patients,
which is globally reflected in the fact that >200,000 patients
die from this condition each year (3). The invasiveness of GBM poses a
significant challenge to its treatment, as the tumor cells
infiltrating healthy brain tissue evade surgical removal and show
limited responsiveness to existing therapies (4,5).
Enhancing the understanding of how invasion and metabolism
function, along with their connections, in the progression,
maintenance and dissemination of tumors, may result in the
identification of innovative therapies that can ultimately prolong
patient survival.
Previous research has highlighted that mitochondria,
fundamental organelles present in the majority of eukaryotic cells,
are integral to cellular metabolism, energy generation and
communication pathways, with implications extending to disease
progression, including cancer (6,7).
Various alterations in mitochondrial function have been detected in
GBM, encompassing structural and functional modifications that
influence mitogenic, hemodynamic, bioenergetic and apoptotic
signaling, suggesting that the oxidative phosphorylation (OXPHOS)
system and energy coupling in glioma cells may operate with
diminished efficiency (8).
Alterations in mitochondrial DNA (mtDNA) copy number
(CN) have an important role in tumorigenesis, including in GBM, as
they influence the quantity of mitochondrial genome copies within
cells, potentially impacting cellular metabolism, energy production
and overall mitochondrial function (9,10).
Enhanced mitochondrial biogenesis and metabolic flexibility are
associated with an increased mtDNA-CN, supporting the proliferation
and survival of tumor cells (11).
Conversely, a reduced mtDNA-CN leads to mitochondrial dysfunction,
altered bioenergetics and increased resistance to apoptosis
(7,12). These mtDNA-CN alterations are
associated with clinical outcomes, including tumor aggressiveness,
therapy response and patient prognosis (13,14).
Understanding these changes may provide insights into mitochondrial
dynamics in GBM and could suggest potential therapeutic strategies
that target mitochondrial function. Therefore, comprehending the
significance of mtDNA-CN alterations in the development of GBM
carcinogenesis is crucial. To address this, the present article
reviewed these occurrences, with a particular focus on variations
in mtDNA-CN and their association with GBM, based on articles
published between 1996 and 2024 from databases such as Scopus,
PubMed and Google Scholar.
GBM
Derived from glial cells in the brain and spinal
cord, gliomas are the most prevalent type of primary tumors in the
central nervous system, varying in aggressiveness and posing
significant global public health risks due to their high mortality
rate, impact on patient quality of life and association with
reduced life expectancy (15).
Gliomas, which are classified based on their histological origin
into types such as ependymoma, oligodendroglioma, astrocytoma, GBM
or mixed glioma, have been reported to account for 26.3% of all
tumors, which refers to a global figure, and have been graded by
the World Health Organization (WHO) from grade 1, the least
aggressive, to grade 4, the most aggressive, reflecting their
degree of cellular alteration and malignancy (16).
Astrocytomas, originating from astrocytic cells, are
the most common type of glioma in adults. The WHO classifies
astrocytomas into three grades: i) Diffuse astrocytoma (WHO grade
2) is characterized by diffuse infiltration, high cellularity and
atypia, but lacks significant endothelial proliferation, mitoses or
necrosis; ii) anaplastic astrocytoma (WHO grade 3) exhibits
increased cellularity, more pronounced nuclear atypia,
hyperchromasia and mitotic activity, but shows no notable
endothelial proliferation or necrosis; iii) GBM (WHO grade 4) is
highly aggressive with dense cellularity, pleomorphism, mitotic
activity and microvascular proliferation or necrosis, often
invading adjacent brain areas but rarely spreading beyond the brain
(17,18).
GBM is not only one of the most severe malignancies
but also the most prevalent malignant primary tumor of the brain
and central nervous system, constituting 14.5% of all central
nervous system tumors and 48.6% of malignant central nervous system
tumors, with a notably short median overall survival time of 15
months (19). Global research
conducted by Ostrom et al (20) revealed that GBM has an incidence
rate of <10 cases per 100,000 people worldwide, with a rising
trend over the last decade. According to projections by the
American Cancer Society, 25,400 individuals in the United States
are expected to be diagnosed with malignant brain and spinal cord
tumors in 2024, leading to 18,760 deaths from this condition
(21).
Distinguished by several defining characteristics,
this aggressive tumor features unique histological traits, rapid
cellular proliferation and a notable capacity to invade surrounding
healthy brain tissue, while also demonstrating pronounced
angiogenesis that enhances blood supply and supports tumor growth
(22). The high likelihood of
recurrence, combined with an inherent resistance to apoptosis,
complicates treatment efforts and verifies the status of GBM as one
of the most treatment-resistant types of cancer, ultimately leading
to a particularly unfavorable prognosis for affected patients, with
typical survival rates of <2 years (5,23).
At present, the primary treatment modalities for GBM
include surgery, chemotherapy and radiotherapy; however, this tumor
type is considered particularly challenging to treat, as it
exhibits resistance to conventional chemotherapy, is frequently
identified at advanced stages and predominantly displays invasive
growth, which results in partial tumor removal through surgery
(24). The inadequate
effectiveness of existing glioma treatments is associated with
several critical factors, including modifications in intricate
signaling pathways that are essential for cellular communication,
the significance of the blood-brain barrier, which restricts the
delivery of therapeutic agents, and the persistence of stem-like
cells within GBM that contribute to tumor resilience and recurrence
(25).
Historically, GBM had been classified into primary
and secondary types based on its clinical presentation (26). Primary GBM has been established to
account for ~90% of GBM cases, emerging de novo in patients
without a preceding history of brain tumors and being more
prevalent among older adults, characterized by specific molecular
alterations, such as amplification of the epidermal growth factor
receptor (EGFR) gene and loss of the tumor suppressor gene
phosphatase and tensin homolog (PTEN) (26). By contrast, secondary GBM
constitutes ~10% of cases, tends to occur in younger patients and
develops from lower-grade tumors, exhibiting a more favorable
prognosis and often harboring mutations in the isocitrate
dehydrogenase (IDH)1 and tumor protein 53 (TP53) genes (26). In the 2016 update of the WHO
classification system for CNS tumors, experts formally classified
GBM based on the presence or absence of IDH gene mutations (either
IDH1 or IDH2), acknowledging that IDH-mutant GBM, closely aligned
with secondary GBM, and IDH-wild-type GBM, defined as primary or
de novo, differ fundamentally in tumorigenesis, biological
progression and therapeutic outcomes (27). The classification of GBM has been
further updated in the fifth edition of the WHO classification of
CNS tumors, released in 2021 to include more molecular features,
such as EGFR gene amplification, telomerase reverse transcriptase
(TERT) promoter mutations, and the simultaneous gain of chromosome
7 along with the loss of chromosome 10 (+7/-10), all of which meet
the criteria for diagnosing IDH-wild-type GBM (28). Additionally, the 2016
classification of secondary GBM was revised to astrocytoma,
IDH-mutant, central nervous system WHO grade 4, marked by mutations
in IDH1 or IDH2, along with ATRX and TP53 gene alterations, and
CDKN2A/B homozygous deletions (28).
A hallmark trait of GBM is its marked molecular and
cellular heterogeneity, which includes a variety of genetic
alterations, epigenetic modifications and cellular phenotypes
(29). This heterogeneity is
widely recognized as a significant factor contributing to the high
malignancy and likelihood of recurrence of GBM, creating
considerable challenges in the development of effective treatment
strategies. Traditionally, studies on tumor initiation and
progression have primarily focused on nuclear genetic changes, with
GBM serving as a key example where these alterations have been
thoroughly documented (8,9,29).
The genetic alterations in GBM are typically defined
by three fundamental biological mechanisms associated with
triggering tumor growth, circumventing cellular aging and
facilitating sustained proliferation, and flaws in each mechanism
appear vital for glioma tumorigenesis through essential signaling
pathways. Three core signaling pathways, the activation of the
receptor tyrosine kinase/Ras/PI3K pathway, the inhibition of the
p53 pathway, and the disruption of the retinoblastoma protein
pathway, are commonly dysregulated in GBM (30). The primary genetic alterations
recognized in GBM thus far encompass TP53 mutations, IDH1
mutations, alterations in the promoter region of the TERT gene,
ATRX mutations, deletions of the PTEN tumor suppressor gene,
O6-methylguanine DNA methyltransferase promoter methylation, and
both the amplification and overexpression of EGFR, all of which
have demonstrated promise in forecasting survival outcomes and
treatment responses in patients with GBM (31).
A previous study revealed that the TP53 mutation
spectrum in IDH-mutant astrocytoma, as opposed to IDH-wild-type
astrocytoma, is primarily driven by a single enriched hotspot
mutation, R273C, which is associated with poor outcomes, especially
in male patients, in spite of histologic and transcriptomic
findings indicating lower proliferation (32). The unique IDH1 R132H mutation has
been identified as a robust prognostic and predictive biomarker
linked to improved clinical outcomes for patients with glioma, as
those diagnosed with secondary GBM harboring this mutation exhibit
a more favorable prognosis compared with patients with primary GBM
possessing the wild-type IDH1 gene (33). Additional significant genetic
alterations noted in GBM comprise CDKN2A/B, H3F3A, NF1, PDGFRA and
PIK3CA (34).
Despite significant efforts to clarify the etiology
of GBM and advancements in molecular assessment, considerable
uncertainty persists regarding the specific mechanisms underlying
its tumorigenesis, while the identification of effective therapies
remains challenging, yielding limited improvements in survival
rates. The intricate genomic changes observed in GBM cells have led
researchers to redirect their attention toward another genome;
beyond the nuclear genome, the mitochondrial genome also warrants
investigation. Exploring alterations in the mitochondrial genome
has created new possibilities for targeted therapies in GBM.
Mitochondrial genome: Heredity, organization
and preservation
Mitochondria are essential organelles within
mammalian cells, responsible for generating energy via adenosine
triphosphate (ATP) through the Krebs cycle and OXPHOS (35). They also regulate key physiological
processes, such as apoptosis, β-oxidation of fatty acids,
iron-sulfur cluster biogenesis, and maintenance of calcium and
redox homeostasis (36,37). Mitochondria-derived reactive oxygen
species (ROS), byproducts of OXPHOS, are implicated in several
conditions, including neurodegenerative disorders, diabetes, cancer
and the aging process (38).
Mitochondria house their own genetic material,
mtDNA, a circular double-stranded DNA (dsDNA) structure of 16,569
base pairs that lacks introns (39). Unlike nuclear DNA, which exists in
two copies per cell, mtDNA is present in numerous copies per cell,
varying based on tissue-specific energy requirements. MtDNA is
encapsulated in nucleoids, which are organized nucleoprotein
complexes, rather than existing as a naked molecule such as
bacterial chromosomes (40).
Super-resolution microscopy has shown that most nucleoids contain
only a single mtDNA molecule, indicating that mitochondria operate
autonomously (41,42).
Various proteins interacting with mtDNA nucleoids
have been identified using advanced techniques, such as
immunoprecipitation and biotinylation (43,44).
One key protein, mitochondrial transcription factor A (TFAM),
serves an essential role in both packaging and transcribing mtDNA
(45). TFAM completely coats mtDNA
and induces bends at specific locations, facilitating the
regulation of mitochondrial transcription and replication (46–48).
Throughout evolution, most mitochondrial genes were
either lost or transferred to nuclear DNA; however, mtDNA has
retained 37 genes. These include genes encoding two ribosomal RNAs,
22 transfer RNAs and 13 proteins (ND1-ND6, ND4L, cytochrome
b, COI-COIII, ATPase-6 and ATPase-8), which are integral
components of the electron transport chain (ETC) in the OXPHOS
complexes (49). The majority of
mitochondrial proteins are encoded by nuclear DNA, translated in
the cytoplasm, and imported into the mitochondria, serving crucial
roles in mitochondrial biogenesis and function (50).
Human mtDNA consists of light (L) and heavy (H)
strands with two non-coding regions (NCRs). The displacement loop
(D-loop), spanning 16,024 to 576, contains elements essential for
replication and transcription (10). Another NCRs, including the L-strand
origin of replication (OriL), consists of 30 nucleotides. Within
the NCRs, the L-strand promoter (LSP) and H-strand promoter (HSP)
are located 150 bp apart (51).
HSP drives transcription of 12 mRNAs, 2 rRNAs and 22 tRNAs, whereas
LSP regulates ND6 and 8 tRNAs in the opposite direction (52). These details are illustrated in
Fig. 1.
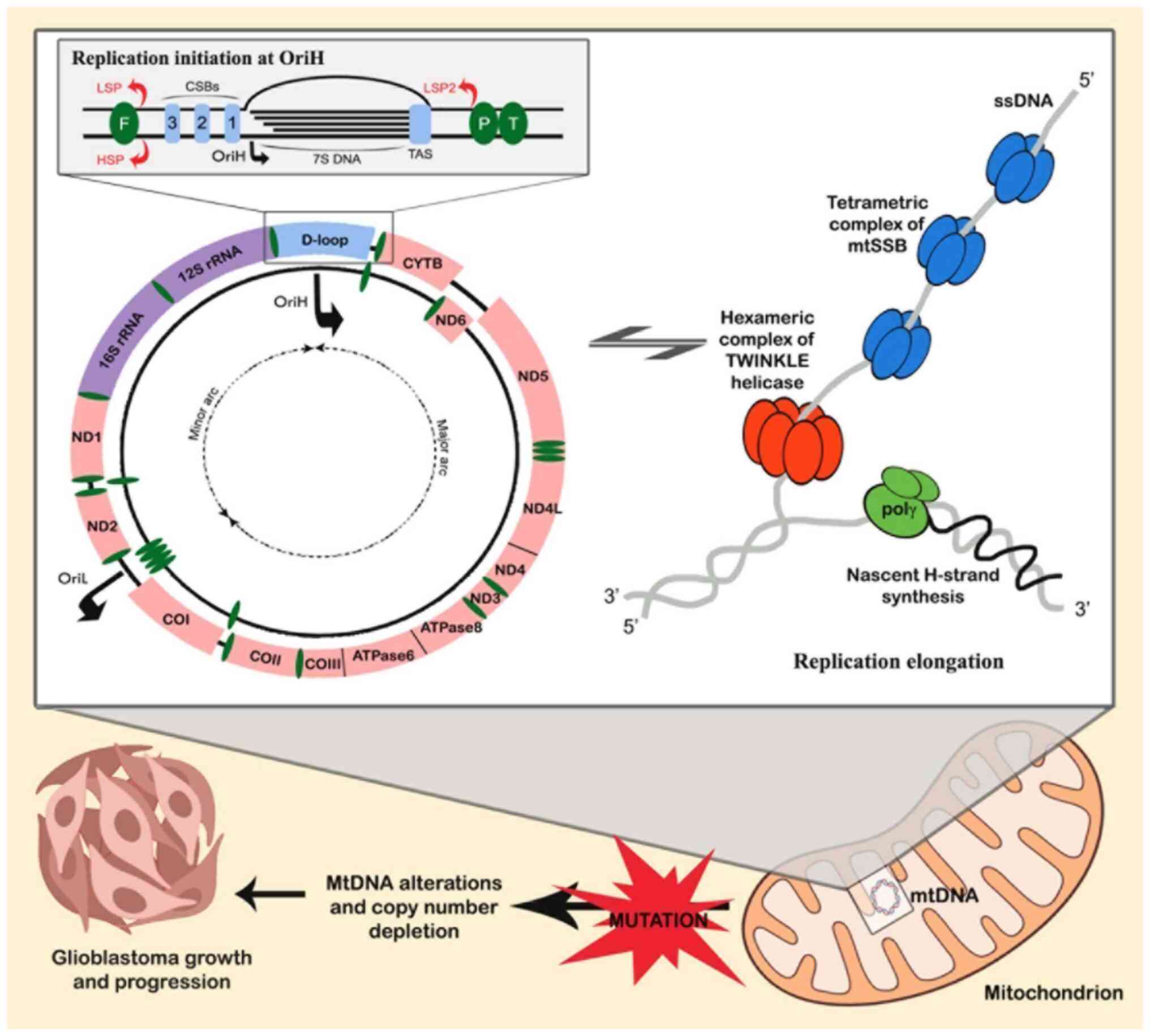 | Figure 1.mtDNA replication mechanisms and copy
number variations in GBM. The mtDNA-encoded genes are depicted as
separate strands on the H-strand and L-strand, with the replication
origins (OriH and OriL) marked accordingly. A close-up view of the
D-loop shows replication initiation at OriH, highlighting all
essential components: The HSP, LSP and LSP2; CSBs 1, 2 and 3; 7S
DNA; and TAS. The 5′ end aligns with the OriH region, while the 3′
end is located at the TAS. It is postulated that mtDNA mutations
from endogenous damage may impair the regulation of mitochondrial
replication elongation, potentially leading to further mtDNA
alterations and depletion of mtDNA-copy number. This disruption
could impact overall mtDNA biogenesis, potentially promoting GBM
growth and progression. GBM, glioblastoma; OriH, heavy-strand
origin of replication; OriL, light-strand origin of replication;
HSP, heavy-strand promoter; LSP, light-strand promoter; CSBs,
conserved sequence blocks; TAS, termination-associated sequence;
mtDNA, mitochondrial DNA; mtSSB, mitochondrial single-stranded
binding protein; polγ, polymerase γ. |
The existence of a second HSP (HSP2) downstream of
HSP was suggested based on early guanylyltransferase capping
studies (53); however, its
functionality has been questioned due to discrepancies in its
transcription start site between in vivo and in vitro
studies, as well as the lack of TFAM requirement during initiation
(54,55). Recent cappable-seq data have
provided no evidence for transcription initiation at the HSP2 site
(56,57). Additionally, a new LSP, LSP2, has
been identified opposite LSP in the NCRs, suggesting a potential
role in transcribing mtDNA genes (57). Further studies are required to
confirm the involvement of LSP2 in mtDNA replication and
transcription.
Transcription in mitochondria follows the
polycistronic model, with long primary transcripts produced from
the LSP and HSP. These transcripts are processed by mitochondrial
RNase P and RNase Z, leading to the release of individual mRNAs,
tRNAs and rRNAs necessary for protein synthesis (52,58).
The mitochondrial RNA polymerase (POLRMT), along with TFAM and
TFB2M, is critical for initiating transcription. TFAM binds to the
promoter regions, facilitating POLRMT recruitment, while TFB2M aids
in unwinding the promoter DNA to initiate transcription (59). The transcription elongation factor
TEFM supports POLRMT during transcription, enhancing the production
of long RNA transcripts.
Human mtDNA replication proceeds continuously and
independently of the cell cycle, which is crucial for maintaining a
high mtDNA-CN per cell (60).
While the replication process has been extensively studied
(55,61,62)
it remains incompletely understood and relies on two distinct
canonical replication origins, the H-strand origin of replication
(OriH) and the OriL, which are oriented oppositely. OriH resides in
the NCR, downstream of the LSP, whereas OriL is positioned amidst a
cluster of five tRNA genes, situated around two-thirds of the
genome's distance (~10 kb downstream of OriH) (63).
The minimal essential proteins necessary for mtDNA
synthesis include the heterotrimeric DNA polymerase γ (POLG),
comprising POLGA and POLGB subunits, the hexameric helicase,
TWINKLE, and the mitochondrial single-stranded DNA (ssDNA)-binding
protein, mtSSB (64,65). During replication of the mtDNA
leading strand, dsDNA unwinding occurs in a 5′ to 3′ direction by
TWINKLE. mtSSB binds to protect the ssDNA, thereby enhancing
TWINKLE-induced dsDNA unwinding and POLG-mediated DNA synthesis, as
shown in Fig. 1. POLRMT is
responsible for producing the RNA primers needed to initiate
synthesis on both DNA strands (64). Replication starts at OriH, where
POLRMT initiates transcription from the LSP, ending at conserved
sequence blocks that form R-loops essential for initiation
(59). RNase H1 processes the 3′
ends of the R-loop, providing primer sites for POLG to begin mtDNA
synthesis (66). At OriL, POLRMT
generates an RNA primer from a poly(T) stretch, forming a stem-loop
structure that enables lagging strand replication (67,68).
This process ensures proper mtDNA replication, with replication
from OriH displacing the OriL region to begin lagging-strand
synthesis (65,66).
Regulation of mtDNA integrity and CN
In humans, the number of mtDNA copies per cell
varies from 100 to 1,000, depending on the cell type, aligning with
tissue-specific metabolic demands. Tissues with higher energy
requirements typically exhibit a higher mtDNA-CN (69). For example, the heart contains
2,000-5,000 copies per nucleus, skeletal muscles have 1,000-3,000
copies, the liver ranges from 500 to 1,000 copies, and blood
leukocytes typically have between 150 and 600 copies (69). By contrast, mammalian erythrocytes
lack mtDNA, sperm have ~5 copies per cell and oocytes may contain
>500,000 copies (70).
Researchers have proposed that the mtDNA-CN per cell serves as an
indicator of mitochondrial health.
Notably, the normal mtDNA-CN in a particular tissue
is not constant and can vary considerably, with numerous studies
(71–73) indicating that, in apparently
healthy individuals, mtDNA-CN can fluctuate between 2 to 10 times
the standard value, and mtDNA content ranging from 40 to 150% of
the average is considered within normal clinical parameters
(74).
By exposing cells to intercalating agents, such as
ethidium bromide or the POLG inhibitor dideoxycytidine, mtDNA
replication is inhibited, allowing researchers to achieve decreased
mtDNA-CN in laboratory experiments; however, certain cells, such as
cancer cells, may inherently exhibit resistance to these treatments
or acquire resistance during therapy, resulting in mtDNA-CN
recovery (75). Traditionally,
scientists quantify mtDNA levels in samples by using quantitative
PCR (qPCR) to count nuclear DNA and mtDNA gene copies, with droplet
digital PCR recognized as an advanced method for determining mtDNA
content, each method having resolution thresholds of 50–60% and
30%, respectively (70,76).
The role of specific proteins in regulating mtDNA-CN
has been debated, with TFAM being particularly scrutinized. TFAM
not only serves a critical role in packaging mtDNA into nucleoids
but also acts as a significant controller of mtDNA-CN, supported by
direct associations observed in diverse genetic models, both in
vitro and in vivo (77–79).
Therefore, researchers have hypothesized that the balanced levels
of TFAM and mtDNA observed in certain studies (78–80)
may result from their mutual stabilization within mitochondrial
nucleoids, contributing to a model that explains the role of TFAM
in mitochondrial biogenesis. Moreover, the regulation of mtDNA
biogenesis is believed to be indirectly affected by Lon protease,
as it targets TFAM for degradation (81). Findings from a previous study
indicated that a moderate rise in TFAM levels can result in an
increased mtDNA-CN, with no impact observed on mitochondrial gene
expression in a BAC-TFAM transgenic mouse model (80). By contrast, another study observed
that knockdown of TFAM in 293 cells could result in a significant
decrease in mtDNA-CN and aggregation of mtDNA nucleoids (82). Despite the evidence supporting the
strong positive association between TFAM expression and mtDNA-CN,
there is considerable conflicting data. Previously, Matsuda et
al (83) illustrated that an
overabundance of TFAM reduced lifespan in Drosophila,
although it had no impact on the mtDNA-CN or transcriptional
activity. In another experiment using human cultured cells,
researchers consistently indicated that transiently boosting TFAM
expression may be adequate to enhance mtDNA transcription but does
not result in an increase in mtDNA-CN (84). Furthermore, in a patient with
myoclonic epilepsy with ragged red fibers, a study revealed that
mtDNA-CNs were increased by 3–7 fold in the predominantly affected
brain areas (hippocampus, cortex and putamen) as well as in
skeletal muscle, whereas TFAM levels remained low (85). Kozhukhar and Alexeyev (86) found conflicting evidence regarding
TFAM expression compared with mtDNA-CN, mitochondrial transcription
and mitochondrial mass. They found that TFAM expression does not
consistently correlate with mtDNA-CN or the expression of
mtDNA-encoded proteins across various experimental systems.
Therefore, it is recommended to cautiously validate the use of TFAM
as a marker of mitochondrial biogenesis (86). Research has indicated that in
certain mtDNA-deficient cells, TFAM expression is lower than in
parental cells with mtDNA, and TFAM release from mtDNA complexes is
facilitated by Lon-mediated degradation (87). These findings imply a necessary
proportional balance between TFAM and mtDNA. Nevertheless, findings
from another study indicated that in a tissue-specific knockout of
POLRMT, TFAM expression remains consistent despite a significant
reduction in mtDNA-CN (88). TFAM
in this scenario remains detached from mtDNA and is unaffected by
Lon-mediated degradation, indicating that the stoichiometric
balance between TFAM and mtDNA may not be universally
applicable.
Findings regarding other proteins have also yielded
inconclusive results. Studies have documented that mutations in
mitofusin 2 are linked to varying levels of mtDNA content, both
higher (89) and lower levels
(90), in the skeletal muscle of
affected patients.
In a recent meta-analysis, researchers identified
two nuclear gene regions near HBS1L/MYB and within the GSDMA gene
that were associated with mtDNA-CN across all participants, as well
as two loci in sex-specific analyses (72). The study also detected one rare
mitochondrial variant [MT:9548_A: corresponds to the cytochrome c
oxidase subunit III (MT-CO3)] linked to mtDNA-CN. These findings
imply that genetic variants in the mitochondrial genome likely have
limited influence on regulating mtDNA-CN, given the rarity of the
associated mitochondrial variant (72).
Researchers have extensively documented alterations
in mtDNA content in the tissues of elderly individuals (91,92),
yet the direction of these changes remains contentious and cannot
be directly linked to any specific protein. The Leiden Longevity
Study, with 2,734 participants, discovered that mtDNA content
declines with age, and low mtDNA content was shown to be associated
with familial longevity, indicating that long-lived families
prioritize preserving mitochondrial function over boosting
mitochondrial biogenesis (91).
Moreover, research has indicated that decreased mtDNA-CN in older
individuals is linked to a gradual decline in both cognitive and
physical functions, along with an elevated risk of mortality
(92). In a recent study, lower
levels of mtDNA-CN were revealed to significantly reduce cumulative
survival rates, revealing a particularly higher mortality risk in
middle-aged individuals with low mtDNA-CN levels, underscoring the
association between leukocyte mtDNA-CN and future mortality risk
(93).
Role of mitochondria in governing GBM
tumorigenesis
Comprehensive research has focused on the importance
of mitochondria in GBM, particularly concerning their genomic
contributions to the advancement of cancer, with increasing
evidence indicating that these organelles experience genetic
modifications, functional disturbances and metabolic reprogramming
within the context of this aggressive malignancy, highlighting
their essential role in tumor development and progression (94). A previous study on the
mitochondrial cancer genome demonstrated that hypermutation,
structural variations, CN alterations and the somatic relocation of
mtDNA into the nuclear genome elevate the risk of cancer
progression, growth and metastasis (95).
Horizontal mitochondrial transfer from astrocytes
has been recognized as a mechanism that promotes tumorigenesis in
GBM, which relies on intercellular connections formed between GBM
cells and astrocytes, mediated by growth-associated protein 43,
which serves a role in neuronal axon regeneration and astrocyte
reactivity, eventually leading to increased mitochondrial
respiration and enhanced tumorigenic potential (96).
The relevance of mtDNA alterations in cancer
progression and sustainability has been extensively studied,
revealing that most mtDNA mutations in brain tumors, particularly
in GBM, are missense transitions irregularly allocated between HSP
and LPS strands, potentially playing a pivotal role in modulating
GBM tumorigenesis (Fig. 1)
(8,9). Most somatic point mutations in mtDNA
occur within the D-loop region, disrupting replication and
transcription processes, which cause fluctuations in mtDNA content
and elevated ROS levels, and may subsequently lead to mitochondrial
impairment; notably, researchers have identified mutations linked
to GBM throughout nearly the entire mitochondrial genome (8,97).
The D-loop region acts as the primary hotspot for somatic mtDNA
mutations, particularly within the polycytosine tract (D310), a
reiterated sequence of cytosines spanning nucleotides 303 to 315,
potentially aiding in the clinical monitoring of GBM (98,99).
The T16189C polymorphism represents another hotspot in the D-loop
region that, despite its common occurrence, fails to display an
association with the pathogenesis of GBM.
Apart from the D-loop, mutations have also been
observed in the coding regions of mtDNA, particularly in those that
participate in the ETC and OXPHOS. It has been documented that
genes encoding the subunits of complex I of the ETC, notably NADH
dehydrogenase 4 (ND4) and NADH dehydrogenase 6 (ND6), are highly
prone to mutations in GBM, indicating that mtDNA variants in
complex I may function as triggers for GBM and confer benefits that
facilitate tumorigenesis (100,101). The A10398G alteration in the NADH
dehydrogenase 3 gene has also been identified in GBM, although its
precise involvement remains uncertain, with hypotheses suggesting
that this defect, in combination with other genetic and
environmental factors, may enhance electron leakage and oxidative
stress, leading to increased ROS levels that promote carcinogenesis
(102).
Genes encoding complexes III and IV have commonly
been identified as being susceptible to mutations in GBM, with
several anticipated to be functional and affecting mitochondrial
respiratory chain performance (103). A previous study revealed that the
germ-line mtDNA mutation T14798C, identified in GBM, may influence
the activity and drug sensitivity of complex III in the
mitochondrial ETC by causing an amino acid substitution (F18L) in
the cytochrome b subunit, thereby potentially altering ROS
production, cellular behavior, and patient outcomes (104).
Large-scale 4,977-bp mtDNA deletions, designated as
the ‘common deletion’, have also been reported in patients with
GBM, but a significant association with the etiology of GBM has not
been established (105).
The significant involvement of mitochondria-related
genes (MRGs) in the onset and development of GBM has been
underscored by multiple studies (8,106,107); however, the precise roles of
proteins encoded by these genes in GBM pathology remain
inadequately defined. Prognostic MRGs have been discovered, and a
novel prognostic model for GBM was previously verified using 12
differentially expressed MRGs in a study by Su et al
(108), which demonstrated that
the risk score was associated with inflammatory responses,
extracellular matrix interactions, and pro-cancer and
immune-related pathways, as well as being strongly linked to gene
mutations and immune cell infiltration. Additionally,
single-stranded DNA-binding protein 1 was found to be significantly
upregulated in GBM, and its knockdown caused mitochondrial
dysfunction and elevated ROS levels, subsequently enhancing the
responsiveness of GBM cells to temozolomide (TMZ) by promoting
ferroptosis. In another study, Peng et al (109) identified nine prognostic MRGs and
created an MRG-based prognostic model validated as an independent
risk predictor for patients with GBM, potentially serving as a
dependable diagnostic tool. Moreover, this previous study revealed
that p66Shc, the longest isoform of SHC1, was upregulated in GBM
tissue, and its silencing impeded the proliferation and migration
of GBM cells by altering mitochondrial ROS synthesis and morphology
(109).
The oxidant-antioxidant balance within a cell is
regulated by mitochondria, and oxidative damage, which is
associated with tumorigenesis, is typically caused by mitochondrial
impairment. It is essential to highlight that a marked tendency to
generate ROS is exhibited by numerous tumors with mutations in ETC
components, emphasizing the vital function of this machinery in
influencing the cancer cell phenotype (110). For example, increased superoxide
production may arise from mutations in complex I components, such
as the ND4 subunit, which can support ROS-dependent oncogenic
pathways and cause damage to mtDNA, thereby promoting tumorigenesis
and metastasis in GBM (111).
The mitochondrial enzyme glutamate dehydrogenase 2
(GLUD2), which is predominantly expressed in the brain, functions
as an essential component in catalyzing the reversible
interconversion of glutamate to α-ketoglutarate and ammonia,
significantly contributing to the tricarboxylic acid cycle, energy
production and ammonia homeostasis, while also being considered to
modulate GBM progression (112).
Alterations in GLUD2 expression levels are known to influence
changes in mitochondrial functions and metabolic phenotypes in
human GBM cells. A study by Franceschi et al (112) demonstrated that overexpression of
GLUD2 was significantly associated with improved overall survival
and lower glioma grades. Furthermore, in vitro functional
studies conducted with human GBM cell lines revealed that GLUD2
overexpression was linked to elevated oxygen consumption and
enhanced ROS production. The rise in ROS generation resulting from
GLUD2 overexpression may account for subsequent cell cycle blockage
in G0/G1, caused by the reduced expression of
cyclin D1/E (113,114).
The increased glucose uptake and ATP production
through glycolysis, which are subsequently accompanied by lactic
acid fermentation even in the presence of oxygen, are identified as
significant characteristics of cancer cells, with this metabolic
transition from mitochondrial OXPHOS to glycolysis commonly
referred to as the Warburg effect. This phenomenon demonstrates an
enhanced rate of glycolysis coupled with suppressed mitochondrial
metabolism, a pattern that is prominently evident in a range of
malignancies, particularly GBM (115). Recent research combining
bioinformatics techniques and laboratory assays has established a
link between the Warburg effect and the prognosis and immune
microenvironment of GBM, indicating that targeting genes associated
with this metabolic shift could offer new therapeutic options
(116).
Mitochondria have been identified as key regulators
in maintaining the quiescent state of GBM stem cells (GSCs), which
are major contributors to the resistance of GBM to therapy,
positioning them as a potential target for overcoming resistance.
OXPHOS serves as a critical metabolic pathway for GSCs, enabling
them to sustain their elevated rates of proliferation, resilience
to therapies and retention of stem-like properties (117). Conversely, when mitochondrial
function is compromised, it can significantly contribute to
tumorigenesis through several interconnected mechanisms, including
disrupted cell cycle regulation, impaired calcium homeostasis,
promotion of the shift of GSCs into a quiescent phase and
suppression of apoptosis. Under conditions of stress, such as
irradiation and hypoxia, GSCs activate mitochondrial stress
pathways as a cytoprotective mechanism to endure the hostile
environment, while proliferating GBM cells display heightened
cytoplasmic glycolysis, in contrast to quiescent GSCs and fully
differentiated GBM cells, which increasingly depend on OXPHOS
(118). A recent study has
revealed that GSCs can uptake exogenous mitochondria from
mesenchymal stem cells, a process that not only provides
significant resistance to TMZ chemotherapy but also triggers a
crucial metabolic transition from metabolizing glucose to
glutamine, ultimately resulting in enhanced orotate synthesis
(119).
Implications of mtDNA-CN alterations in GBM
tumorigenesis
Changes in mtDNA-CN are considered key factors in
the development of GBM, a highly aggressive and frequently
treatment-resistant brain tumor. These alterations can disrupt
cellular metabolism, energy generation and oxidative stress,
thereby impacting tumor growth and survival. Exploring the
consequences of mtDNA-CN variations could provide valuable insights
into the progression and clinical severity of GBM, potentially
paving the way for more precise and targeted treatment
strategies.
Research has demonstrated that dysfunctional mtDNA
POLG can induce changes in mtDNA-CN, disrupting OXPHOS and reducing
ATP production (120). This
decline in mitochondrial respiration and ATP synthesis, coupled
with an increase in glycolysis, commonly occurs in both
mitochondrial diseases and cancer (120,121). Dysregulated mtDNA-CN in GBM may
impair OXPHOS efficiency, driving a metabolic shift toward aerobic
glycolysis that supports rapid tumor growth, survival and
malignancy (122,123).
Tumorigenesis is promoted by mtDNA alterations
through the modification of ROS generation, which subsequently
influences the expression of apoptosis-related proteins, such as
cytochrome c and BCL-2 family members (124,125). In GBM, reduced mtDNA-CN may
impair the initiation of apoptosis, enabling cells to escape
programmed cell death and maintain malignancy. In addition, changes
in mtDNA-CN influence the expression of mitochondrial-encoded genes
and the signaling pathways between the nucleus and mitochondria
(126). This disruption may
affect genes regulating cell proliferation, migration and invasion,
fostering tumorigenesis and enhancing GBM malignancy. It has been
reported in previous studies that altered mtDNA-CN can result in
defective ETC activity, leading to elevated ROS production
(127–129). Consequently, excessive ROS levels
may cause DNA damage, genomic instability, and mutations in both
nuclear DNA and mtDNA, potentially driving GBM progression. A
reduction in POLG and yopoisomerase levels, combined with increased
TFAM, has been shown to alter mtDNA-CN and gene expression
(130). Declines in mtDNA-CN are
believed to trigger epigenetic modifications of nuclear DNA through
retrograde signaling, leading to a reduction in OXPHOS capacity and
the subsequent shift of stem cells toward glycolytic metabolism and
aggressive GBM behavior (130).
Variation in mtDNA-CN in GBM
Mitochondrial mechanisms experience alterations in
numerous prevalent pathologies, including cardiovascular diseases,
neurodegeneration, metabolic syndrome and cancer. These ailments
exhibit specific changes in mtDNA, including variations in CN,
rearrangements, deletions and point mutations, which researchers
have scrutinized as potential risk factors or early diagnostic
markers (8–10,35).
Nevertheless, the precise impact of these alterations on disease
progression remains unclear.
Oxidative stress more readily damages mtDNA than
nuclear DNA, potentially due to its close proximity to OXPHOS,
where excessive ROS are regularly produced (131), a phenomenon frequently observed
in cancer cells. Efficient base excision repair mechanisms
predominantly restore damaged mtDNA molecules, while mtDNA
suffering from double-strand breaks undergoes rapid degradation
rather than repair, resulting in a substantial reduction in
mtDNA-CN compared with nuclear DNA (132). Under various physiological and
environmental conditions, mtDNA-CN serves as a relative indicator,
reflecting variations in mitochondrial health, and can fluctuate
according to energy demands (133). Despite this, mtDNA mutations can
accumulate due to faulty replication and repair, resulting in
mitochondrial impairment and transmission of signals to the nucleus
(134).
mtDNA-CN, acting as a surrogate marker for
mitochondrial function, demonstrates extensive connections with
various diseases, and documented alterations in its levels have
been linked to a higher cancer risk (10,127). Nevertheless, the fluctuating
nature of the association of mtDNA-CN with cancer occurrence,
whether positive or negative, is heavily influenced by diverse
factors such as the source of the sample and the type of cancer.
Although mtDNA exists in multiple copies per cell due to
mitochondrial dynamics, such as fusion and fission, cells regulate
its content within a stable range to sustain energy levels and
ensure proper cell function (10,135). Reports have indicated that
variations in mtDNA content occur early in carcinogenesis,
indicating crucial mutations for neoplastic transformation, which
then cause disruptions in OXPHOS and ATP generation (10,136).
mtDNA-CN alterations have been reported across
multiple types of cancer, including breast cancer, colorectal
cancer, gastric cancer, lung cancer, esophageal cancer and brain
tumors (10). However, the present
review specifically focuses on providing a comprehensive summary of
the existing data and previous findings related to mtDNA level
changes reported in brain tumors, with particular emphasis on
GBM.
Research has provided limited evidence on how mtDNA
content influences GBM tumorigenesis, and its association with
clinicopathological factors and patient outcomes. A comprehensive
literature search was performed using PubMed, Google Scholar,
Scopus and Web of Science platforms to compile information
regarding the involvement of mtDNA-CN alterations in GBM (Table I).
 | Table I.Summary of mtDNA copy number changes
across brain tumor types, emphasizing alterations in GBM. |
Table I.
Summary of mtDNA copy number changes
across brain tumor types, emphasizing alterations in GBM.
| First author(s),
year | Country | Brain tumor sample
type | Technique | mthNA gene | Nuclear DNA
gene | mthNA levels
(relative ratio-fold changes) | Additional
observation | (Refs.) |
|---|
| Liang, 1996 | USA | 15 low grade glioma
tissues | Southern blot
hybridization | MT-RNR2 | ACTB | Increased | All tumors
exhibited mitochondrial sequence localization, which was linked to
elevated mtDNA content. | (137) |
| Liang and Hays,
1996 | USA | 45 glioma
tissues | Southern blot
hybridization | MT-CO1 | ACTB | Increased | 5/11 (46%) glioma
tumors showed a recurrent deletion of a 1.2-kb EcoRI
fragment. | (138) |
| Correia et
al, 2011 | Brazil | 120 astrocytoma
tissues (WHO grade II, III and IV) | qPCR | MT-ND1 | HBB | Decreased | mtDNA reduction was
predominantly observed in GBM samples. | (139) |
|
|
|
|
|
|
|
| POLG expression was
associated with low mtDNA content. |
|
| Soltész et
al, 2022 | Hungary | 44 GBM tissues and
plasma-derived exosomes | qPCR | MT-ND1 &
MT-ND5 | SERPINA1 &
SLCO2B1 | Increased | High mtDNA content
was apparent in brain tissue and exosome samples from the control
group. | (140) |
| Marucci et
al, 2013 | Italy | 10 GBM tissues | qPCR | MT-ND2 | FALSG | Increased | Longer median
survival was observed in oncocytic GBM, at 16 months. | (141) |
| Zhang et al,
2014 | China | 414 blood
lymphocytes of glioma | qPCR | MT-ND1 | HBB | Increased | Elevated mtDNA
content was significantly associated with a higher risk of
glioma. | (142) |
| Shen et al,
2016 | USA | 390 whole blood
samples from glioma patients | qPCR | MT-ND1 | HBB | Increased | High mtDNA levels
were associated with a higher risk of glioma. | (143) |
| Zhang et al,
2015 | China | 124 glioma
tissues | qPCR | MT-ND1 | ACTB | Increased &
decreased | High mtDNA content
was significantly linked to seizures. | (144) |
|
|
|
|
|
|
|
| Low mtDNA content
was found in recurrent cases. |
|
| Dardaud et
al, 2019 | France | 67 GBM tissues | qPCR | N/A | N/A | Increased | High mtDNA content
was associated with extended O in the young adult group. | (136) |
| Sourty et
al, 2022 | France | 232 GBM
tissues | qPCR | MT-CO1 &
MT-ND4 | B2M &
GAPDH | Increased &
decreased | High mtDNA level
was associated with improved OS in younger patients. | (145) |
|
|
|
|
|
|
|
| Low mtDNA levels
were associated with better OS in older patients. |
|
| Ab Radzak et
al, 2024 | Malaysia | 41 brain tumor
tissues | qPCR | MT-ND1 | ACTB | Increased | High mtDNA content
was associated with longer OS, particularly in high-grade
tumors. | (146) |
| Hua et al,
2020 | China | 87 meningioma III
tissues | qPCR | MT-ND1 | HBB | Increased | High mtDNA levels
were associated with improved OS and PFS. | (147) |
| Sravya et
al, 2020 | ndia | 20 GBM tissues | qPCR | MT-ND1 | RNase P | Increased | Patients with low
mtDNA Ilevels in blood revealed high mtDNA content in tumor tissue
samples. | (148) |
| Sravya et
al, 2020 | India | 162 GBM
tissues | qPCR | MT-ND1 | RNase P | Decreased | Low mtDNA levels
were associated with IDH wild-type. | (149) |
| Chen et al,
2015 | China | 336 blood from
patients with glioma | qPCR | MT-ND1 | HBB | Increased | High mtDNA content
was associated with poor OS and PFS | (150) |
| Dickinson et
al, 2013 | Australia | HSR-GBM-1, GBM-L1,
GBM-L2, hNSCs | qPCR | MT-RNR2 | HBB | Increased | High mtDNA content
in hNSCs during differentiation was observed. | (122) |
|
|
|
|
|
|
|
| -Prolonged mtDNA
depletion led to defective mtDNA replication, decreased
proliferation, and stimulated the expression of OCT4 and SHH. |
|
|
|
|
|
|
|
|
| -Cells recovered
their mtDNA copy number after 7 days of depletion. |
|
| Sun and St John,
2018 | Australia | HSR-GBM-1 cell
line | qPCR | MT-RNR2 | HBB | Reverted | mtDNA content was
restored to pre-depletion levels without significant
differences. | (151) |
| Shen et al,
2020 | Australia | 60 high-grade
glioma tissues | qPCR | D-loop &
MT-CO2 | ACTB | Decreased | Low mtDNA content
led to tumorigenicity through increased cell migration and
invasion, as well as resistance to therapy. | (123) |
| Braun et al,
2021 | Germany | 48 tissues from 22
patients primary and recurrent gliomas | qPCR | D-loop | B2M | Increased | High mtDNA content
was observed in IDH mutant tumors. | (152) |
| Oliva et al,
2010 | USA | U251 cell line | Semi-qPCR | MT-CO1 | 18S rRNA | Decreased |
Temozolomide-resistant glioma cells
exhibited reduced mtDNA content. | (153) |
| Luna et al,
2015 | USA | 48 tissues from
pediatric brain tumor | qPCR | MT-ND1 | 18S rRNA | Increased | A high mtDNA copy
number increased the probability of brain tumor expansion in female
children by 51 times compared to controls. | (154) |
In 1996, Liang (137) initially documented that cDNA
homologous to mtDNA at positions 1,679-1,948 and 2,017-2,057 had
been applied to evaluate 15 low-grade glial tumors, demonstrating
an elevated mtDNA-CN in these tumors compared with in normal brain
tissue controls. In an additional study, Liang and Hays disclosed
that up to a 25-fold increase in mtDNA-CN had been observed in 39
out of 45 (87%) assessed glial tumor specimens, both low and high
grade, and asserted that this prevalence exceeded the erb-b gene
amplification found in only 18% of the tumors, implying that
changes in mtDNA were more widespread than alterations in
nuclear-encoded genes in malignant glioma (138).
Correia et al (139) revealed that diffusely
infiltrating astrocytoma exhibited a significant decrease in
mtDNA-CN compared with that in non-neoplastic brain tissues, with
the most pronounced depletion occurring in GBM as malignancy
progressed. This decrease, when associated with the overexpression
of TFAM and POLG, was linked to prolonged survival in patients with
GBM, suggesting that these factors may serve as possible indicators
of more favorable outcomes. Consistent findings by Soltész et
al (140), where brain tissue
DNA and plasma-derived exosomal DNA from 44 patients with GBM and
40 control individuals were examined, demonstrated that lower
mtDNA-CN was found in GBM cases compared with in the control
subjects.
In 2013, oncocytic changes in GBM were identified
through histology, immunohistochemistry and ultrastructural
observation, and high levels of mtDNA-CN were detected, as reported
by Marucci et al (141). A
series of GBM cases was analyzed to establish any association with
morphology and survival, resulting in the identification of 10
cases where most cells exhibited characteristic oncocytic traits at
histological, immunohistochemical and ultrastructural levels, and
nine of these cases showed significantly higher mtDNA content
compared with in control tissue (141).
A Chinese case-control epidemiological study
examined the link between leukocyte mtDNA content and glioma risk,
finding that patients with glioma had markedly higher median mtDNA
content than healthy controls, and that increased mtDNA content was
strongly associated with a higher risk of glioma (142). This finding aligns with a
case-control study from the USA, which showed that mtDNA-CN levels
in whole blood were significantly higher in patients with glioma
compared with in healthy controls, thereby reinforcing the role of
mtDNA-CN in glioma carcinogenesis (143). Conversely, Zhang et al
(144) demonstrated that an
increased mtDNA-CN was significantly inversely associated with
tumor grade, recurrence and cancer-related death, and critically,
higher mtDNA content was closely linked to prolonged survival in
patients with glioma. Additionally, Dardaud et al (136) corroborated these findings by
observing that a higher mtDNA-CN was significantly associated with
improved overall survival in young adult patients with GBM, which
corresponded with the findings of Sourty et al (145), who reported that high mtDNA-CN
was associated with longer survival in young adults but shorter
survival in older patients with GBM. More recently, these findings
were further supported by our previous study, which showed that
longer overall survival periods and notably better outcomes were
experienced by patients with higher mtDNA-CN, especially in
high-grade brain tumor cases (146). Aligned with findings from glial
tumors, Hua et al (147)
analyzed 87 WHO grade III meningioma samples, revealing that high
mtDNA content was associated with improved outcomes, while low
mtDNA content was associated with enhanced progression-free
survival in patients who received post-operative radiation therapy,
indicating that mtDNA content in tumors could function as an
indicator for anticipating the prognosis of patients with WHO grade
III meningioma.
A study conducted in India by Sravya et al
(148) indicated that improved
overall survival in GBM was associated with a high mtDNA-CN in
tumor tissue. In a distinct study conducted within the same year,
Sravya et al (149)
compared mtDNA-CN between newly diagnosed and recurrent GBM in
paired samples, revealing that poor prognosis in patients with GBM
was linked with low mtDNA content. Additionally, the research
showed that increased neurosphere formation, indicative of higher
stemness, and consequently, resistance to radiation and TMZ therapy
in malignant glioma cell lines, was caused by mtDNA depletion.
In a study of 336 patients with glioma, it was
reported that high mtDNA content was strongly linked to a poorer
prognosis in younger patients, those with high-grade gliomas or
those receiving adjuvant radiochemotherapy, and it was observed
that these patients had markedly reduced natural killer cell
frequencies and elevated concentrations of IL-2 and TNF-α,
indicating that an immunosuppression-associated process may be
engaged in mtDNA-mediated prognosis (150).
The research of Dickinson et al (122) on mtDNA content modulation in GBM
cell lines highlighted that partial depletion of mtDNA may restore
replication events and promote cell differentiation. However,
extended depletion compromised mtDNA replication, decreased cell
proliferation and triggered the expression of genes associated with
early developmental processes. A phenotype that could not
regenerate in vitro was produced by the gradual depletion of
mtDNA content in human GBM cells (122). However, when researchers
implanted these cells into immunocompromised mice, they observed
that tumor development was delayed and mitochondrial function was
recovered, depending on the extent of mtDNA copy reduction. In
another study, Sun and St John (151) demonstrated that restoring
mtDNA-CN during tumor development led to significant alterations in
the nuclear genome, causing variations in DNA methylation and gene
expression. These modifications enriched developmental processes
and key metabolic pathways linked with GBM. Moreover, the
interaction between the nuclear and mitochondrial genomes in
reinstating tumorigenic potential was emphasized by the changes in
nuclear-encoded mtDNA replication factors (151).
Notably, in 2020, Shen et al (123) made a novel discovery by
identifying a significant reduction in mtDNA content in most cases
of pediatric high-grade glioma (pHGG) compared with in normal brain
tissue, which was proposed as the molecular mechanism mediating the
Warburg effect. Kinase modulators have significantly reduced pHGG
viability by shifting glucose metabolism to mitochondrial
oxidation, and combining this approach with metformin, which
affects mitochondrial function, has disturbed tumor cell energy
balance, resulting in elevated DNA injury and enhanced apoptosis
(123).
A study conducted by Braun et al (152) investigated whether mitochondrial
biomass differed between IDH-mutant and IDH-wild-type diffuse
glioma, revealing that IDH-mutant tumors had higher mtDNA-CN
associated with unique metabolic and epigenetic profiles compared
with IDH-wild-type tumors, thereby highlighting that changes in
mtDNA-CN correspond with these distinct profiles and suggesting
that mtDNA levels could serve as valuable biomarkers for glioma
characterization and prognosis.
In 2010, Oliva et al (153) studied the effects of TMZ on mtDNA
and mitochondrial function in TMZ-resistant glioma cells and
xenografts, indicating that TMZ reduced mtDNA-CN, heightened
heteroplasmy, and disrupted mitochondrial ETC and bioenergetics,
with similar changes observed in patient biopsies after adjuvant
TMZ treatment, emphasizing their clinical importance. In a separate
study, Luna et al (154)
explored the impact of mtDNA-CN, oxidative damage and mtDNA
variants as risk factors for pediatric brain tumors, using Bayesian
network and Markov Chain Monte Carlo modelling. This analysis
showed that the combined presence of certain mtDNA variants,
oxidative damage and high mtDNA-CN significantly increased the
likelihood of developing brain tumors in female children, with a
51-fold higher risk compared to the normal incidence (154).
Variability in mtDNA-CN findings across GBM studies
is shaped by several factors inherent to the nature of the disease,
and is often constrained by limitations in methodology, sample
selection biases and study design inconsistencies. The complex and
heterogeneous nature of GBM, which includes diverse molecular,
genetic and metabolic characteristics, notably contributes to
discrepancies in mtDNA-CN observations. As mtDNA-CN can vary
considerably across different cell types, the cellular composition
of the tissue under investigation becomes a crucial factor to
consider.
Various molecular techniques, including qPCR,
digital PCR and other genomic methods, such as whole exome
sequencing and whole genome sequencing, have been employed to
determine relative mtDNA-CN values (155). Each of these methods offers
distinct advantages and limitations, and their application can
influence the accuracy and consistency of mtDNA-CN quantification
across different studies. Additionally, DNA extraction methods can
significantly impact the accuracy of measurements, with classic
procedures using phenol-chloroform or current commercial DNA
extraction kits based on spin-column technology both altering the
mtDNA:nuclear DNA ratio and potentially modifying the experimental
results (156).
Another important consideration is that tissue
composition can vary due to aging or disease, such as the neuronal
loss seen in neurodegenerative disorders or fibrotic changes in
aging tissues (156). In cancer,
tumor samples typically consist of various cell types, thus
complicating the analysis, although methods such as laser-capture
microdissection can successfully isolate specific cells to a
certain extent (157). However,
the widespread use of these techniques is constrained by time
limitations and the availability of the necessary technology,
particularly when dealing with large sample sizes.
Study design also serves a crucial role in ensuring
accuracy, requiring careful consideration of factors such as age,
sex and lifestyle when choosing cohorts. Research has highlighted
significant sex differences, with female individuals typically
having higher mtDNA-CN than male individuals (91). Lifestyle factors, such as
consistent aerobic exercise, can also increase mitochondrial mass
and mtDNA levels in skeletal muscle through adaptive metabolic
responses (156).
Integrating mtDNA-CN with GBM heterogeneity
and therapeutic resistance
GBM, a complex and treatment-resistant brain tumor,
is influenced by mtDNA-CN, with alterations contributing to its
heterogeneity and enhancing its resistance to therapy (158). It has been suggested that
variations in mtDNA-CN across different GBM subpopulations may
facilitate adaptive responses to environmental stressors, including
chemotherapy, radiation and metabolic changes.
Increased mtDNA-CN is necessary in the process of
carcinogenesis. For example, it is possible that higher mtDNA-CN
drives mitochondrial biogenesis to increase the mitochondrial
activity needed for the increase in macromolecular synthesis
required for cell growth and replication (159). Higher mtDNA-CN may be linked to
increased mitochondrial biogenesis and enhanced OXPHOS, driving
cellular energy production in the face of nutrient deprivation or
therapeutic intervention. Conversely, low mtDNA-CN may favor
glycolysis and other compensatory pathways that reduce reliance on
OXPHOS, conferring resistance in a distinct subset of tumor cells.
Mou et al (160)
reinforced the idea that mtDNA depletion may trigger aerobic
glycolysis and a reversible apoptosis-resistant phenotype in SW480
cells, with the Akt/mTOR pathway potentially serving a role in the
drug-induced resistance to apoptosis.
Tumor cells exhibit heightened glucose uptake
rates, even in oxygen-rich conditions, driven by a metabolic shift
to aerobic glycolysis, known as the Warburg effect (161). A hypothesis has suggested that
variations in mtDNA-CN may impact this shift in GBM cells, where
glycolysis is elevated despite the presence of oxygen. Reduced
mtDNA-CN levels have been reported to be associated with increased
oxidative stress due to higher ROS production (71), which may drive cells to rely on
glycolysis, shifting the metabolic burden to the cytoplasm and
providing an alternative energy source that promotes GBM
tumorigenesis and resistance to treatment. This is supported by
evidence indicating that mtDNA-CN is notably reduced in pHGG,
resulting in a glycolytic phenotype that is closely associated with
increased cell migration, invasion, resistance to therapy and
enhanced tumorigenicity in vivo (123).
mtDNA-CN serves a crucial role in regulating the
stem cell-like properties of GBM cells. A previous study revealed
that the HSR-GBM1 cancer stem cell line cannot increase its
mtDNA-CN during differentiation, which may lead to impaired
differentiation and abnormal expression of the astrocyte marker
GFAP (162). This failure to
expand mtDNA-CN likely prevents the cell from enhancing OXPHOS
capacity and generating enough ATP for full differentiation. As a
result, the inability of multipotent cells to differentiate appears
to be directly linked to the failure to increase mtDNA-CN (162).
GBM cells with higher mtDNA-CN may have better
self-renewal ability and resistance to chemotherapy through
mitochondrial-dependent mechanisms. This includes the activation of
pro-survival signaling pathways, such as PI3K/Akt/mTOR, which boost
cell proliferation, growth, and resistance to chemo- and
immunotherapy (163). By
contrast, GBM subpopulations with low mtDNA-CN may display
flexibility in mitochondrial function, allowing the cells to shift
between oxidative and glycolytic metabolism in response to
environmental stress (164). This
metabolic adaptability helps the cells survive in low-oxygen or
nutrient-deprived conditions, aiding in resistance to standard
treatments (164). Future
research on incorporating mtDNA-CN with GBM heterogeneity and
resistance has the potential to reveal novel therapeutic strategies
targeting the metabolic weaknesses of GBM.
Therapeutic strategies for restoring
mtDNA-CN and improving health outcomes
Compelling evidence has suggested that total mtDNA
levels have a crucial role in human pathology and aging, with
researchers recognizing changes in mtDNA-CN, whether increases or
decreases, as significant in cancer development and progression,
and linking these fluctuations to various types of cancer,
including breast, lung and colorectal cancer, and glioma (10). This has prompted the creation of
approaches that modify mtDNA levels, either directly or indirectly,
to mitigate or prevent disease progression, while ongoing research
into mitochondrial dysfunction in cancer has led to the growing
adoption of therapeutic methods targeting mtDNA-CN abnormalities,
which hold promise for enhancing the effectiveness of conventional
cancer treatments and improving patient outcomes. Researchers have
employed two approaches to boost OXPHOS capacity and revive
mitochondrial performance, either by altering total mitochondrial
mass or by selectively adjusting mtDNA to affect mtDNA-CN and/or
heteroplasmy levels; these methods, primarily aimed at managing
primary mitochondrial disorders, may also tackle common age-related
diseases if human trials prove effective (165,166).
Mitochondrial biogenesis, which expands
mitochondrial mass, is driven by environmental factors such as
exercise, calorie restriction, temperature changes, oxidative
stress, cell division, renewal and differentiation, and boosts
mtDNA-CN and metabolic enzyme subunits, enhancing metabolic
capacity; this necessitates coordinated expression of both nuclear
and mtDNA-encoded genes (167,168). Modulating mitochondrial
biogenesis to rectify mtDNA-CN imbalances has emerged as a leading
approach, with the enhancement of mitochondrial quantity through
this process serving as a practical solution to address
bioenergetic impairments resulting from mutations in either mtDNA
or the nuclear genome (169).
Experimental findings have indicated that the augmented
mitochondrial mass can partially offset the diminished respiratory
chain activity by sustaining overall ATP production in the skeletal
muscle of mice with myopathy, acting as a compensatory response to
respiratory chain deficiency and aiding in the enhancement of
energy balance in the affected tissue (170).
Peroxisome proliferator-activated receptor (PPAR)γ
coactivator 1α (PGC-1α) is a co-transcriptional regulator that
enhances mitochondrial production by interacting with nuclear
transcription factors, such as PPARs, nuclear respiratory factors
(NRFs, such as NRF-1 and NRF-2) and estrogen-related receptors, to
stimulate the expression of nucleus-encoded genes, including TFAM
and OXPHOS subunits. The activity of PGC-1α is regulated by sirtuin
1 and its deacetylation controlled by AMP-activated protein kinase
(AMPK) (171). Research has
revealed that elevating PGC-1α expression can increase mtDNA levels
and amplify the activity of mitochondrial respiratory chain
complexes in COX-deficient mouse models (172), while also improving aging-related
traits in mtDNA mutator mice (173), supporting the notion that the
effects of mitochondrial diseases may be mitigated by enhanced
mitochondrial biogenesis.
Reinforcing this perspective, research by Giordano
et al (174) on Leber's
hereditary optic neuropathy revealed that unaffected mutation
carriers exhibited significantly higher mtDNA-CN and mitochondrial
mass compared with their affected relatives and controls, implying
that increased mitochondrial biogenesis in these carriers could
potentially counteract some of the pathogenic effects of mtDNA
mutations. Recently, Wu et al (175) uncovered in a mouse model of
peritoneal dialysis fluid-induced peritoneal fibrosis that
stimulating the AMPK-PGC-1α pathway elevated the expression of
phosphorylated-AMPK, PGC-1α, NRF-1, NRF-2 and TFAM, increased mtDNA
content, enhanced mitochondrial morphology, prevented apoptosis of
peritoneal mesothelial cells, and reduced peritoneal fibrosis by
promoting mitochondrial biogenesis.
In 2020, human trials on mitochondrial biogenesis
enhancers, including niacin (a vitamin B3 derivative and
NAD+ precursor) were conducted. Scientists from the
University of Helsinki demonstrated that niacin effectively
increased NAD+ levels and promoted mitochondrial
biogenesis, thereby boosting muscle strength in patients with
mitochondrial myopathy and NAD+ depletion (176).
A previous study demonstrated that ZLN005, an
innovative transcriptional regulator of PGC-1α, enhanced PGC-1α and
downstream gene expression, boosted mitochondrial biogenesis and
metabolic maturation in human embryonic stem cell-derived
cardiomyocytes, and led to a higher mtDNA-CN in treated
cardiomyocytes compared with the controls (177).
Researchers have also shown that electroacupuncture
(EA) pretreatment safeguards mitochondria and enhances
mitochondrial biogenesis by elevating mtDNA levels, and increasing
mitochondrial volume and number through CB1R-dependent PGC-1α
activation and upregulation of NRF-1 and TFAM expression, thus
revealing a new mechanism for EA pretreatment-induced ischemic
tolerance (178).
Research has also confirmed that TFAM protein
levels precisely regulate mtDNA quantity, enabling the genetic
modulation of TFAM expression to alter mtDNA-CN (80). It has been observed that a slight
increase in TFAM levels in mice has led to an elevated mtDNA-CN
while leaving mitochondrial gene expression, mitochondrial mass and
respiratory chain capacity unaffected, thereby indicating that an
increase in mtDNA-CN can occur independently of a rise in
mitochondrial quantity (78,80).
In 2010, Nishiyama et al (179) conducted the initial study
demonstrating that overexpression of TFAM in a mouse model with
extensive mtDNA deletions elevated the total mtDNA-CN, mitigated
severe mitochondrial disease symptoms and extended lifespan.
Likewise, enhancing mtDNA quantities through TFAM overexpression in
a study of male infertility partially restored spermatogenesis and
OXPHOS functionality in the testes of mtDNA mutator mice,
subsequently recovering their fertility phenotype (180). Both findings were corroborated by
the work of Filograna et al (165), who experimentally manipulated
mtDNA levels by boosting TFAM expression in mice with the C5024T
mutation in the tRNAAla gene, which alleviated the
pathological outcomes of pathogenic mtDNA, emphasizing a potential
therapeutic approach for mitochondrial diseases. Notably, these
studies revealed that while the ratio of pathogenic mtDNA mutations
remained constant, preserving the mutational load, the levels of
wild-type mtDNA increased, which partially improved mitochondrial
performance even though mutant mtDNA continued to prevail (165,180). Beyond its relevance to
mitochondrial disease research, the advantageous impacts of TFAM
overexpression have been shown to mitigate the reduction in
mtDNA-CN and to maintain it at normal levels in cardiac tissues,
while also restoring respiratory chain enzymatic activity in a
myocardial infarction mouse model (181). Hayashi et al (182) previously reported that transgenic
overproduction of human TFAM can diminish age-related motor
learning and memory deficits, along with reducing 8-oxoguanine
accumulation in aged mice. Furthermore, in a separate study,
researchers from the same group revealed that TFAM may successfully
disrupt the mitochondria-driven vicious cycle in Alzheimer's
disease model neurons and mouse brains, leading to considerable
progress in Alzheimer's pathology, with declined amyloid β
accumulation and enhanced cognitive function (183). In contrast to stimulating the
entire mitochondrial biogenesis process, regulating mtDNA
quantities has been identified as a more targeted approach for
enhancing OXPHOS, although thorough evaluation of the extent of
TFAM expression remains crucial. Conversely, an in vivo
study reported that TFAM overexpression could increase the mtDNA
content, which was shown to have detrimental effects, such as
nucleoid enlargement, disrupted transcription, age-related mtDNA
deletions and impairments in the respiratory chain (184). Altering TFAM levels to regulate
mtDNA-CN could offer a promising approach for addressing
mitochondrial issues in both primary mitochondrial disorders and
other related conditions.
Research has uncovered that a lower mtDNA level is
linked to reduced chemotherapy efficacy in both clinical and
laboratory studies, with evidence indicating that mtDNA depletion
serves a role in triggering chemoresistance in various malignant
tumors (185,186). However, a decrease in mtDNA
levels may also enhance tumor cell sensitivity to chemotherapy in
certain types of cancer, such as nasopharyngeal cancer (187), highlighting the unclear
relationship between mtDNA-CN and chemotherapeutic resistance in
cancer.
Conclusion and future perspectives
mtDNA-CN alterations have been increasingly
recognized as significant factors in the development and
progression of GBM. These variations can markedly influence
mitochondrial function, energy metabolism and the behavior of tumor
cells. Research has indicated that fluctuations in mtDNA levels,
whether elevated or reduced, may disrupt OXPHOS, promoting the
generation of ROS, which not only drives tumor growth but also
affects how GBM cells respond to environmental challenges and
therapies. Understanding the implications of mtDNA-CN alterations
is essential for advancing therapeutic strategies. Although both
increased and decreased mtDNA levels have been observed in GBM,
their precise effects on tumor biology and treatment outcomes
remain to be fully elucidated.
From a clinical perspective, targeting mtDNA-CN
alterations offers a promising therapeutic strategy for improving
GBM treatment outcomes. Strategies aimed at restoring or modulating
mtDNA levels, such as stabilizing mtDNA-CN through mitochondrial
biogenesis enhancers or TFAM overexpression, have the potential to
boost mitochondrial function, optimize OXPHOS, and heighten tumor
cell sensitivity to chemotherapy or radiation. These interventions
could serve as valuable additions to current treatments by
addressing the metabolic vulnerabilities associated with mtDNA
alterations in GBM.
Future research initiatives should focus on
elucidating the fundamental mechanisms that connect mtDNA-CN
variations to GBM pathology. This may include examining how these
alterations interact with nuclear DNA mutations, epigenetic changes
and environmental factors, as well as their influence on
mitochondrial dynamics, ROS production and cellular metabolism in
shaping tumor progression and resistance to treatment. Exploring
these mechanisms could uncover novel biomarkers for prognosis or
therapeutic response, contributing to the advancement of more
personalized treatment strategies.
In conclusion, improving the understanding of
mtDNA-CN alterations in GBM has substantial potential to enhance
diagnostic and therapeutic strategies. By elucidating the roles of
mtDNA variations, it may be feasible to develop treatments that not
only optimize mitochondrial function but also counteract resistance
to conventional therapies. This area of research could ultimately
facilitate more effective management of GBM, improving patient
outcomes and providing potential options in the treatment of this
aggressive brain tumor.
Acknowledgements
Not applicable.
Funding
This review was supported by Universiti Sains Malaysia under the
Graduate Development Incentive-PhD (grant no.
311/PPSP/4404815).
Availability of data and materials
Not applicable.
Authors' contributions
AAMY contributed to the conceptualization, design
and drafting of the manuscript, and wrote most of the content.
SZNMK and SMAR performed the literature search and selection, wrote
the manuscript, designed the figures and prepared the tables. Data
authentication is not applicable. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of AI tools declaration
During the preparation of this work, AI tools
(Grammarly) were used to improve the readability and language of
the manuscript, and subsequently, the authors revised and edited
the content produced by the AI tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Libby CJ, McConathy J, Darley-Usmar V and
Hjelmeland AB: The role of metabolic plasticity in blood and brain
stem cell pathophysiology. Cancer Res. 80:5–16. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Morrison AJ: Cancer cell metabolism
connects epigenetic modifications to transcriptional regulation.
FEBS J. 289:1302–1314. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seker-Polat F, Pinarbasi Degirmenci N,
Solaroglu I and Bagci-Onder T: Tumor cell infiltration into the
brain in glioblastoma: From mechanisms to clinical perspectives.
Cancers (Basel). 14:4432022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pandey N, Anastasiadis P, Carney CP,
Kanvinde PP, Woodworth GF, Winkles JA and Kim AJ: Nanotherapeutic
treatment of the invasive glioblastoma tumor microenvironment. Adv
Drug Deliv Rev. 188:1144152022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Obrador E, Moreno-Murciano P,
Oriol-Caballo M, López-Blanch R, Pineda B, Gutiérrez-Arroyo JL,
Loras A, Gonzalez-Bonet LG, Martinez-Cadenas C, Estrela JM and
Marqués-Torrejón MÁ: Glioblastoma therapy: Past, present and
future. Int J Mol Sci. 25:25292024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
San-Millán I: The key role of
mitochondrial function in health and disease. Antioxidants (Basel).
12:7822023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang SF, Tseng LM and Lee HC: Role of
mitochondrial alterations in human cancer progression and cancer
immunity. J Biomed Sci. 30:612023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Leão Barros MB, Pinheiro DDR and Borges
BDN: Mitochondrial DNA alterations in glioblastoma (GBM). Int J Mol
Sci. 22:58552021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mohamed Yusoff AA: Role of mitochondrial
DNA mutations in brain tumors: A mini-review. J Cancer Res Ther.
11:535–544. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abd Radzak SM, Mohd Khair SZN, Ahmad F,
Patar A, Idris Z and Mohamed Yusoff AA: Insights regarding
mitochondrial DNA copy number alterations in human cancer (Review).
Int J Mol Med. 50:1042022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Denisenko TV, Gorbunova AS and Zhivotovsky
B: Mitochondrial involvement in migration, invasion and metastasis.
Front Cell Dev Biol. 7:3552019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang Z, Yang D, Zhou B, Luan Y, Yao Q,
Liu Y, Yang S, Jia J, Xu Y, Bie X, et al: Decrease of MtDNA copy
number affects mitochondrial function and involves in the
pathological consequences of ischaemic stroke. J Cell Mol Med.
26:4157–4168. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tuchalska-Czuroń J, Lenart J, Augustyniak
J and Durlik M: Is mitochondrial DNA copy number a good prognostic
marker in resectable pancreatic cancer? Pancreatology. 19:73–79.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Y, Yang B, Huang Y, Zhang Y, Jiang Y,
Ma L and Shen YQ: Mitochondrial DNA-targeted therapy: A novel
approach to combat cancer. Cell Insight. 2:1001132023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schaff LR and Mellinghoff IK: Glioblastoma
and other primary brain malignancies in adults: A review. JAMA.
329:574–587. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ostrom QT, Price M, Neff C, Cioffi G,
Waite KA, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: primary brain and other central nervous system tumors
diagnosed in the United States in 2016–2020. Neuro Oncol. 25 (12
Suppl 2):iv1–iv99. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thomas DL: 2021 Updates to the World
Health Organization classification of adult-type and pediatric-type
diffuse gliomas: A clinical practice review. Chin Clin Oncol.
12:72023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang P, Xia Q, Liu L, Li S and Dong L:
Current opinion on molecular characterization for GBM
classification in guiding clinical diagnosis, prognosis, and
therapy. Front Mol Biosci. 7:5627982020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Grochans S, Cybulska AM, Simińska D,
Korbecki J, Kojder K, Chlubek D and Baranowska-Bosiacka I:
Epidemiology of glioblastoma multiforme-literature review. Cancers
(Basel). 14:24122022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ostrom QT, Price M, Neff C, Cioffi G,
Waite KA, Kruchko C and Barnholtz-Sloan JS: CBTRUS statistical
report: Primary brain and other central nervous system tumors
diagnosed in the United States in 2015–2019. Neuro Oncol. 24 (Suppl
5):v1–v95. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Siegel RL, Giaquinto AN and Jemal A:
Cancer statistics, 2024. CA Cancer J Clin. 74:12–49. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Onciul R, Brehar FM, Toader C,
Covache-Busuioc RA, Glavan LA, Bratu BG, Costin HP, Dumitrascu DI,
Serban M and Ciurea AV: Deciphering glioblastoma: Fundamental and
novel insights into the biology and therapeutic strategies of
gliomas. Curr Issues Mol Biol. 46:2402–2443. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tan AC, Ashley DM, López GY, Malinzak M,
Friedman HS and Khasraw M: Management of glioblastoma: State of the
art and future directions. CA Cancer J Clin. 70:299–312. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yalamarty SSK, Filipczak N, Li X, Subhan
MA, Parveen F, Ataide JA, Rajmalani BA and Torchilin VP: Mechanisms
of resistance and current treatment options for glioblastoma
multiforme (GBM). Cancers (Basel). 15:21162023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ohka F, Natsume A and Wakabayashi T:
Current trends in targeted therapies for glioblastoma multiforme.
Neurol Res Int. 2012:8784252012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ohgaki H and Kleihues P: The definition of
primary and secondary glioblastoma. Clin Cancer Res. 19:764–772.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Louis DN, Perry A, Reifenberger G, von
Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD,
Kleihues P and Ellison DW: The 2016 World Health Organization
classification of tumors of the central nervous system: A summary.
Acta Neuropathol. 131:803–820. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO Classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Torrisi F, Alberghina C, D'Aprile S,
Pavone AM, Longhitano L, Giallongo S, Tibullo D, Di Rosa M, Zappalà
A, Cammarata FP, et al: The hallmarks of glioblastoma:
Heterogeneity, intercellular crosstalk and molecular signature of
invasiveness and progression. Biomedicines. 10:8062022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cancer Genome Atlas Research Network, .
Comprehensive genomic characterization defines human glioblastoma
genes and core pathways. Nature. 455:1061–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Verdugo E, Puerto I and Medina MÁ: An
update on the molecular biology of glioblastoma, with clinical
implications and progress in its treatment. Cancer Commun (Lond).
42:1083–1111. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marker DF, Agnihotri S, Amankulor N,
Murdoch GH and Pearce TM: The dominant TP53 hotspot mutation in
IDH-mutant astrocytoma, R273C, has distinctive pathologic features
and sex-specific prognostic implications. Neurooncol Adv.
4:vdab1822021.PubMed/NCBI
|
|
33
|
Dekker LJM, Verheul C, Wensveen N,
Leenders W, Lamfers MLM, Leenstra S and Luider TM: Effects of the
IDH1 R132H mutation on the energy metabolism: A comparison between
tissue and corresponding primary glioma cell cultures. ACS Omega.
7:3568–3578. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lan Z, Li X and Zhang X Glioblastoma: An
update in pathology, molecular mechanisms and biomarkers. Int J Mol
Sci. 25:30402024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mohd Khair SZN, Abd Radzak SM and Mohamed
Yusoff AA: The uprising of mitochondrial DNA biomarker in cancer.
Dis Markers. 2021:76752692021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jacobs LJHC and Riemer J: Maintenance of
small molecule redox homeostasis in mitochondria. FEBS Lett.
597:205–223. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nguyen TT, Wei S, Nguyen TH, Jo Y, Zhang
Y, Park W, Gariani K, Oh CM, Kim HH, Ha KT, et al:
Mitochondria-associated programmed cell death as a therapeutic
target for age-related disease. Exp Mol Med. 55:1595–1619. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Antonucci S, Di Lisa F and Kaludercic N:
Mitochondrial reactive oxygen species in physiology and disease.
Cell Calcium. 94:1023442021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Habbane M, Montoya J, Rhouda T, Sbaoui Y,
Radallah D and Emperador S: Human mitochondrial DNA:
Particularities and diseases. Biomedicines. 9:13642021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Bonekamp NA and Larsson NG: SnapShot:
Mitochondrial nucleoid. Cell. 172:388–388.e1. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brown TA, Tkachuk AN, Shtengel G, Kopek
BG, Bogenhagen DF, Hess HF and Clayton DA: Superresolution
fluorescence imaging of mitochondrial nucleoids reveals their
spatial range, limits, and membrane interaction. Mol Cell Biol.
31:4994–5010. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kukat C, Wurm CA, Spåhr H, Falkenberg M,
Larsson NG and Jakobs S: Super-resolution microscopy reveals that
mammalian mitochondrial nucleoids have a uniform size and
frequently contain a single copy of mtDNA. Proc Natl Acad Sci USA.
108:13534–13539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
He J, Cooper HM, Reyes A, Di Re M,
Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A,
et al: Mitochondrial nucleoid interacting proteins support
mitochondrial protein synthesis. Nucleic Acids Res. 40:6109–6121.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Han S, Udeshi ND, Deerinck TJ, Svinkina T,
Ellisman MH, Carr SA and Ting AY: Proximity biotinylation as a
method for mapping proteins associated with mtDNA in living cells.
Cell Chem Biol. 24:404–414. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Alam TI, Kanki T, Muta T, Ukaji K, Abe Y,
Nakayama H, Takio K, Hamasaki N and Kang D: Human mitochondrial DNA
is packaged with TFAM. Nucleic Acids Res. 31:1640–1645. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang YE, Marinov GK, Wold BJ and Chan DC:
Genome-wide analysis reveals coating of the mitochondrial genome by
TFAM. PLoS One. 8:e745132013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ngo HB, Lovely GA, Phillips R and Chan DC:
Distinct structural features of TFAM drive mitochondrial DNA
packaging versus transcriptional activation. Nat Commun.
5:30772014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Farge G, Mehmedovic M, Baclayon M, van den
Wildenberg SMJL, Roos WH, Gustafsson CM, Wuite GJ and Falkenberg M:
In vitro-reconstituted nucleoids can block mitochondrial DNA
replication and transcription. Cell Rep. 8:66–74. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Anderson S, Bankier AT, Barrell BG, de
Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA,
Sanger F, et al: Sequence and organization of the human
mitochondrial genome. Nature. 290:457–465. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Slone J and Huang T: The special
considerations of gene therapy for mitochondrial diseases. NPJ
Genom Med. 5:72020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Basu U, Bostwick AM, Das K,
Dittenhafer-Reed KE and Patel SS: Structure, mechanism, and
regulation of mitochondrial DNA transcription initiation. J Biol
Chem. 295:18406–18425. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ren B, Guan MX, Zhou T, Cai X and Shan G:
Emerging functions of mitochondria-encoded noncoding RNAs. Trends
Genet. 39:125–139. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Montoya J, Christianson T, Levens D,
Rabinowitz M and Attardi G: Identification of initiation sites for
heavy-strand and light-strand transcription in human mitochondrial
DNA. Proc Natl Acad Sci USA. 79:7195–7199. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zollo O, Tiranti V and Sondheimer N:
Transcriptional requirements of the distal heavy-strand promoter of
mtDNA. Proc Natl Acad Sci USA. 109:6508–6512. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tan BG, Gustafsson CM and Falkenberg M:
Mechanisms and regulation of human mitochondrial transcription. Nat
Rev Mol Cell Biol. 25:119–132. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yan B, Tzertzinis G, Schildkraut I and
Ettwiller L: Comprehensive determination of transcription start
sites derived from all RNA polymerases using ReCappable-seq. Genome
Res. 32:162–174. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tan BG, Mutti CD, Shi Y, Xie X, Zhu X,
Silva-Pinheiro P, Menger KE, Díaz-Maldonado H, Wei W, Nicholls TJ,
et al: The human mitochondrial genome contains a second light
strand promoter. Mol Cell. 82:3646–3660.e9. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Kummer E and Ban N: Mechanisms and
regulation of protein synthesis in mitochondria. Nat Rev Mol Cell
Biol. 22:307–325. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Menger KE, Rodríguez-Luis A, Chapman J and
Nicholls TJ: Controlling the topology of mammalian mitochondrial
DNA. Open Biol. 11:2101682021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Matkarimov BT and Saparbaev MK: DNA repair
and mutagenesis in vertebrate mitochondria: Evidence for asymmetric
DNA strand inheritance. Adv Exp Med Biol. 1241:77–100. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Falkenberg M, Larsson NG and Gustafsson
CM: Replication and transcription of human mitochondrial DNA. Annu
Rev Biochem. 93:47–77. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Liu Y, Liu H, Zhang F and Xu H: The
initiation of mitochondrial DNA replication. Biochem Soc Trans.
52:1243–1251. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Falkenberg M: Mitochondrial DNA
replication in mammalian cells: Overview of the pathway. Essays
Biochem. 62:287–296. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Peter B and Falkenberg M: TWINKLE and
other human mitochondrial DNA helicases: Structure, function and
disease. Genes (Basel). 11:4082020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Silva-Pinheiro P, Pardo-Hernández C, Reyes
A, Tilokani L, Mishra A, Cerutti R, Li S, Rozsivalova DH,
Valenzuela S, Dogan SA, et al: DNA polymerase gamma mutations that
impair holoenzyme stability cause catalytic subunit depletion.
Nucleic Acids Res. 49:5230–5248. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Posse V, Al-Behadili A, Uhler JP, Clausen
AR, Reyes A, Zeviani M, Falkenberg M and Gustafsson CM: RNase H1
directs origin-specific initiation of DNA replication in human
mitochondria. PLoS Genet. 15:e10077812019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fusté JM, Wanrooij S, Jemt E, Granycome
CE, Cluett TJ, Shi Y, Atanassova N, Holt IJ, Gustafsson CM and
Falkenberg M: Mitochondrial RNA polymerase is needed for activation
of the origin of light-strand DNA replication. Mol Cell. 37:67–78.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sarfallah A, Zamudio-Ochoa A, Anikin M and
Temiakov D: Mechanism of transcription initiation and primer
generation at the mitochondrial replication origin OriL. EMBO J.
40:e1079882021. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Picard M: Blood mitochondrial DNA copy
number: What are we counting? Mitochondrion. 60:1–11. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kozhukhar N, Fant A and Alexeyev MF:
Quantification of mtDNA content in cultured cells by direct droplet
digital PCR. Mitochondrion. 61:102–113. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Castellani CA, Longchamps RJ, Sun J,
Guallar E and Arking DE: Thinking outside the nucleus:
Mitochondrial DNA copy number in health and disease. Mitochondrion.
53:214–223. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Koller A, Filosi M, Weissensteiner H,
Fazzini F, Gorski M, Pattaro C, Schönherr S, Forer L, Herold JM,
Stark KJ, et al: Nuclear and mitochondrial genetic variants
associated with mitochondrial DNA copy number. Sci Rep.
14:20832024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rath SP, Gupta R, Todres E, Wang H,
Jourdain AA, Ardlie KG, Calvo SE and Mootha VK: Mitochondrial
genome copy number variation across tissues in mice and humans.
Proc Natl Acad Sci USA. 121:e24022911212024. View Article : Google Scholar
|
|
74
|
Shokolenko I and Alexeyev M: Mitochondrial
DNA: Consensuses and controversies. DNA (Basel). 2:131–148.
2022.PubMed/NCBI
|
|
75
|
Khozhukhar N, Spadafora D, Rodriguez Y and
Alexeyev M: Elimination of mitochondrial DNA from mammalian cells.
Curr Protoc Cell Biol. 78:20.11.1–20.11.14. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
O'Hara R, Tedone E, Ludlow A, Huang E,
Arosio B, Mari D and Shay JW: Quantitative mitochondrial DNA copy
number determination using droplet digital PCR with single-cell
resolution. Genome Res. 29:1878–1888. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Matsushima Y, Matsumura K, Ishii S,
Inagaki H, Suzuki T, Matsuda Y, Beck K and Kitagawa Y: Functional
domains of chicken mitochondrial transcription factor A for the
maintenance of mitochondrial DNA copy number in lymphoma cell line
DT40. J Biol Chem. 278:31149–31158. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ekstrand MI, Falkenberg M, Rantanen A,
Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM and Larsson
NG: Mitochondrial transcription factor A regulates mtDNA copy
number in mammals. Hum Mol Genet. 13:935–944. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kanki T, Ohgaki K, Gaspari M, Gustafsson
CM, Fukuoh A, Sasaki N, Hamasaki N and Kang D: Architectural role
of mitochondrial transcription factor A in maintenance of human
mitochondrial DNA. Mol Cell Biol. 24:9823–9834. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bonekamp NA, Jiang M, Motori E, Garcia
Villegas R, Koolmeister C, Atanassov I, Mesaros A, Park CB and
Larsson NG: High levels of TFAM repress mammalian mitochondrial DNA
transcription in vivo. Life Sci Alliance. 4:e2021010342021.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Matsushima Y, Goto Y and Kaguni LS:
Mitochondrial Lon protease regulates mitochondrial DNA copy number
and transcription by selective degradation of mitochondrial
transcription factor A (TFAM). Proc Natl Acad Sci USA.
107:18410–18415. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Aasumets K, Basikhina Y, Pohjoismäki JL,
Goffart S and Gerhold J: TFAM knockdown-triggered mtDNA-nucleoid
aggregation and a decrease in mtDNA copy number induce the
reorganization of nucleoid populations and mitochondria-associated
ER-membrane contacts. Biochem Biophys Rep. 28:1011422021.PubMed/NCBI
|
|
83
|
Matsuda T, Kanki T, Tanimura T, Kang D and
Matsuura ET: Effects of overexpression of mitochondrial
transcription factor A on lifespan and oxidative stress response in
Drosophila melanogaster. Biochem Biophys Res Commun.
430:717–721. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Maniura-Weber K, Goffart S, Garstka HL,
Montoya J and Wiesner RJ: Transient overexpression of mitochondrial
transcription factor A (TFAM) is sufficient to stimulate
mitochondrial DNA transcription, but not sufficient to increase
mtDNA copy number in cultured cells. Nucleic Acids Res.
32:6015–6027. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Brinckmann A, Weiss C, Wilbert F, von
Moers A, Zwirner A, Stoltenburg-Didinger G, Wilichowski E and
Schuelke M: Regionalized pathology correlates with augmentation of
mtDNA copy numbers in a patient with myoclonic epilepsy with
ragged-red fibers (MERRF-syndrome). PLoS One. 5:e135132010.
View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kozhukhar N and Alexeyev MF: Limited
predictive value of TFAM in mitochondrial biogenesis.
Mitochondrion. 49:156–165. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Lu B, Lee J, Nie X, Li M, Morozov YI,
Venkatesh S, Bogenhagen DF, Temiakov D and Suzuki CK:
Phosphorylation of human TFAM in mitochondria impairs DNA binding
and promotes degradation by the AAA+ Lon protease. Mol Cell.
49:121–132. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kühl I, Miranda M, Posse V, Milenkovic D,
Mourier A, Siira SJ, Bonekamp NA, Neumann U, Filipovska A, Polosa
PL, et al: POLRMT regulates the switch between replication primer
formation and gene expression of mammalian mtDNA. Sci Adv.
2:e16009632016. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Sitarz KS, Yu-Wai-Man P, Pyle A, Stewart
JD, Rautenstrauss B, Seeman P, Reilly MM, Horvath R and Chinnery
PF: MFN2 mutations cause compensatory mitochondrial DNA
proliferation. Brain. 135:e2191–3. –e220. 1–3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Vielhaber S, Debska-Vielhaber G, Peeva V,
Schoeler S, Kudin AP, Minin I, Schreiber S, Dengler R, Kollewe K,
Zuschratter W, et al: Mitofusin 2 mutations affect mitochondrial
function by mitochondrial DNA depletion. Acta Neuropathol.
125:245–256. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
van Leeuwen N, Beekman M, Deelen J, van
den Akker EB, de Craen AJM, Slagboom PE and 't Hart LM: Low
mitochondrial DNA content associates with familial longevity: The
leiden longevity study. Age (Dordr). 36:96292014. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Mengel-From J, Thinggaard M, Dalgård C,
Kyvik KO, Christensen K and Christiansen L: Mitochondrial DNA copy
number in peripheral blood cells declines with age and is
associated with general health among elderly. Hum Genet.
133:1149–1159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mizuno G, Yamada H, Tsuboi Y, Munetsuna E,
Yamazaki M, Ando Y, Kageyama I, Nouchi Y, Teshigawara A, Hattori Y,
et al: Low mitochondrial DNA copy number in peripheral blood
mononuclear cells is associated with future mortality risk: A
long-term follow-up study from Japan. J Nutr Health Aging.
28:1000132024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wang R, Lei H, Wang H, Qi L, Liu Y, Liu Y,
Shi Y, Chen J and Shen QT: Dysregulated inter-mitochondrial
crosstalk in glioblastoma cells revealed by in situ cryo-electron
tomography. Proc Natl Acad Sci USA. 121:e23111601212024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon
CJ, Yang Y, Martincorena I, Creighton CJ, Weinstein JN, et al:
Comprehensive molecular characterization of mitochondrial genomes
in human cancers. Nat Genet. 52:342–352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Watson DC, Bayik D, Storevik S, Moreino
SS, Sprowls SA, Han J, Augustsson MT, Lauko A, Sravya P, Røsland
GV, et al: GAP43-dependent mitochondria transfer from astrocytes
enhances glioblastoma tumorigenicity. Nat Cancer. 4:648–664. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Vidone M, Clima R, Santorsola M, Calabrese
C, Girolimetti G, Kurelac I, Amato LB, Iommarini L, Trevisan E,
Leone M, et al: A comprehensive characterization of mitochondrial
DNA mutations in glioblastoma multiforme. Int J Biochem Cell Biol.
63:46–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mohamed Yusoff AA, Mohd Nasir KN, Haris K,
Mohd Khair SZN, Abdul Ghani ARI, Idris Z and Abdullah JM: Detection
of somatic mutations in the mitochondrial DNA control region D-loop
in brain tumors: The first report in Malaysian patients. Oncol
Lett. 14:5179–5188. 2017.PubMed/NCBI
|
|
99
|
Radzak S, Khair Z, Ahmad F, Idris Z and
Yusoff A: Accumulation of mitochondrial DNA microsatellite
instability in Malaysian patients with primary central nervous
system tumors. Turk Neurosurg. 31:99–106. 2021.PubMed/NCBI
|
|
100
|
Yeung KY, Dickinson A, Donoghue JF,
Polekhina G, White SJ, Grammatopoulos DK, McKenzie M, Johns TG and
St John JC: The identification of mitochondrial DNA variants in
glioblastoma multiforme. Acta Neuropathol Commun. 2:12014.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Khair SZNM, Ab Radzak SM, Idris Z, Zin
AAM, Ahmad WMAW and Yusoff AAM: The effect of somatic mutations in
mitochondrial DNA on the survival of patients with primary brain
tumors. Croat Med J. 65:111–121. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Mohamed Yusoff AA, Zulfakhar FN, Mohd
Khair SZN, Wan Abdullah WS, Abdullah JM and Idris Z: Mitochondrial
10398A>G NADH-dehydrogenase subunit 3 of complex I is frequently
altered in intra-axial brain tumors in Malaysia. Brain Tumor Res
Treat. 6:31–38. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lloyd RE, Keatley K, Littlewood DT,
Meunier B, Holt WV, An Q, Higgins SC, Polyzoidis S, Stephenson KF,
Ashkan K, et al: Identification and functional prediction of
mitochondrial complex III and IV mutations associated with
glioblastoma. Neuro Oncol. 17:942–952. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Keatley K, Stromei-Cleroux S, Wiltshire T,
Rajala N, Burton G, Holt WV, Littlewood DTJ, Briscoe AG, Jung J,
Ashkan K, et al: Integrated approach reveals role of mitochondrial
germ-line mutation F18L in Respiratory Chain, oxidative
alterations, drug sensitivity, and patient prognosis in
glioblastoma. Int J Mol Sci. 20:33642019. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Mohamed Yusoff AA, Mohd Khair SZN, Abd
Radzak SM, Idris Z and Lee HC: Prevalence of mitochondrial DNA
common deletion in patients with gliomas and meningiomas: A first
report from a Malaysian study group. J Chin Med Assoc. 83:838–844.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Wang J, Qiu X, Huang J, Zhuo Z, Chen H,
Zeng R, Wu H, Guo K, Yang Q, Ye H, et al: Development and
validation of a novel mitophagy-related gene prognostic signature
for glioblastoma multiforme. BMC Cancer. 22:6442022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Xie Z, Hua W and Wang H: Comprehensive
analysis of mitochondrial dynamic-related genes on their functions
and prognostic values for glioblastoma multiforme. Genes Dis.
11:1010842023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Su J and Li Y, Liu Q, Peng G, Qin C and Li
Y: Identification of SSBP1 as a ferroptosis-related biomarker of
glioblastoma based on a novel mitochondria-related gene risk model
and in vitro experiments. J Transl Med. 20:4402022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Peng G, Feng Y, Wang X, Huang W and Li Y:
The mitochondria-related gene risk mode revealed p66Shc as a
prognostic mitochondria-related gene of glioblastoma. Sci Rep.
14:114182024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Porporato PE, Filigheddu N, Pedro JMB,
Kroemer G and Galluzzi L: Mitochondrial metabolism and cancer. Cell
Res. 28:265–280. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Raimondi V, Ciccarese F and Ciminale V:
Oncogenic pathways and the electron transport chain: A dangeROS
liaison. Br J Cancer. 122:168–181. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Franceschi S, Corsinovi D, Lessi F,
Tantillo E, Aretini P, Menicagli M, Scopelliti C, Civita P,
Pasqualetti F, Naccarato AG, et al: Mitochondrial enzyme GLUD2
plays a critical role in glioblastoma progression. EBioMedicine.
37:56–67. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Li C, Peng W, Song X, Wang Q and Wang W:
Anticancer effect of icaritin inhibits cell growth of colon cancer
through reactive oxygen species, Bcl-2 and cyclin D1/E signaling.
Oncol Lett. 12:3537–3542. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wang X, Liu J, Jiang L, Wei X, Niu C, Wang
R, Zhang J, Meng D and Yao K: Bach1 induces endothelial cell
apoptosis and cell-cycle arrest through ROS generation. Oxid Med
Cell Longev. 2016:62340432016. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhao J, Ma X, Gao P, Han X, Zhao P, Xie F
and Liu M: Advancing glioblastoma treatment by targeting
metabolism. Neoplasia. 51:1009852024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Zhang R, Wang C, Zheng X, Li S, Zhang W,
Kang Z, Yin S, Chen J, Chen F and Li W: Warburg effect-related risk
scoring model to assess clinical significance and immunity
characteristics of glioblastoma. Cancer Med. 12:20639–20654. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Chisari A, Golán I, Campisano S, Gélabert
C, Moustakas A, Sancho P and Caja L: Glucose and amino acid
metabolic dependencies linked to stemness and metastasis in
different aggressive cancer types. Front Pharmacol. 12:7237982021.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Iranmanesh Y, Jiang B, Favour OC, Dou Z,
Wu J, Li J and Sun C: Mitochondria's role in the maintenance of
cancer stem cells in glioblastoma. Front Oncol. 11:5826942021.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Nakhle J, Khattar K, Özkan T, Boughlita A,
Abba Moussa D, Darlix A, Lorcy F, Rigau V, Bauchet L,
Gerbal-Chaloin S, et al: Mitochondria transfer from mesenchymal
stem cells confers chemoresistance to glioblastoma stem cells
through metabolic rewiring. Cancer Res Commun. 3:1041–1056. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Qian W and Van Houten B: Alterations in
bioenergetics due to changes in mitochondrial DNA copy number.
Methods. 51:452–457. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Singh KK, Ayyasamy V, Owens KM, Koul MS
and Vujcic M: Mutations in mitochondrial DNA polymerase-gamma
promote breast tumorigenesis. J Hum Genet. 54:516–524. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Dickinson A, Yeung KY, Donoghue J, Baker
MJ, Kelly RD, McKenzie M, Johns TG and St John JC: The regulation
of mitochondrial DNA copy number in glioblastoma cells. Cell Death
Differ. 20:1644–1653. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Shen H, Yu M, Tsoli M, Chang C, Joshi S,
Liu J, Ryall S, Chornenkyy Y, Siddaway R, Hawkins C and Ziegler DS:
Targeting reduced mitochondrial DNA quantity as a therapeutic
approach in pediatric high-grade gliomas. Neuro Oncol. 22:139–151.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Ricci C, Pastukh V, Leonard J, Turrens J,
Wilson G, Schaffer D and Schaffer SW: Mitochondrial DNA damage
triggers mitochondrial-superoxide generation and apoptosis. Am J
Physiol Cell Physiol. 294:C413–C422. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Park JS, Sharma LK, Li H, Xiang R,
Holstein D, Wu J, Lechleiter J, Naylor SL, Deng JJ, Lu J and Bai Y:
A heteroplasmic, not homoplasmic, mitochondrial DNA mutation
promotes tumorigenesis via alteration in reactive oxygen species
generation and apoptosis. Hum Mol Genet. 18:1578–1589. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sadakierska-Chudy A, Kotarska A,
Frankowska M, Jastrzębska J, Wydra K, Miszkiel J, Przegaliński E
and Filip M: The alterations in mitochondrial DNA copy number and
nuclear-encoded mitochondrial genes in rat brain structures after
cocaine self-administration. Mol Neurobiol. 54:7460–7470. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Hu L, Yao X and Shen Y: Altered
mitochondrial DNA copy number contributes to human cancer risk:
Evidence from an updated meta-analysis. Sci Rep. 6:358592016.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Al-Kafaji G and Golbahar J: High
glucose-induced oxidative stress increases the copy number of
mitochondrial DNA in human mesangial cells. Biomed Res Int.
2013:7549462013. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Long S, Zheng Y, Deng X, Guo J, Xu Z,
Scharffetter-Kochanek K, Dou Y and Jiang M: Maintaining
mitochondrial DNA copy number mitigates ROS-induced oocyte decline
and female reproductive aging. Commun Biol. 7:12292024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Grady CI, Walsh LM and Heiss JD:
Mitoepigenetics and gliomas: Epigenetic alterations to
mitochondrial DNA and nuclear DNA alter mtDNA expression and
contribute to glioma pathogenicity. Front Neurol. 14:11547532023.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Liao S, Chen L, Song Z and He H: The fate
of damaged mitochondrial DNA in the cell. Biochim Biophys Acta Mol
Cell Res. 1869:1192332022. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Fu Y, Tigano M and Sfeir A: Safeguarding
mitochondrial genomes in higher eukaryotes. Nat Struct Mol Biol.
27:687–695. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Lee HC and Wei YH: Mitochondrial role in
life and death of the cell. J Biomed Sci. 7:2–15. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Fontana GA and Gahlon HL: Mechanisms of
replication and repair in mitochondrial DNA deletion formation.
Nucleic Acids Res. 48:11244–11258. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Reznik E, Miller ML, Şenbabaoğlu Y, Riaz
N, Sarungbam J, Tickoo SK, Al-Ahmadie HA, Lee W, Seshan VE, Hakimi
AA and Sander C: Mitochondrial DNA copy number variation across
human cancers. Elife. 5:e107692016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Dardaud LM, Bris C, Desquiret-Dumas V,
Boisselier B, Tabouret E, Mokhtari K, Figarella-Branger D, Rousseau
A and Procaccio V: High mitochondrial DNA copy number is associated
with longer survival in young patients with glioblastoma. Neuro
Oncol. 21:1084–1085. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Liang BC: Evidence for association of
mitochondrial DNA sequence amplification and nuclear localization
in human low-grade gliomas. Mutat Res. 354:27–33. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Liang BC and Hays L: Mitochondrial DNA
copy number changes in human gliomas. Cancer Lett. 105:167–173.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Correia RL, Oba-Shinjo SM, Uno M, Huang N
and Marie SKN: Mitochondrial DNA depletion and its correlation with
TFAM, TFB1M, TFB2M and POLG in human diffusely infiltrating
astrocytomas. Mitochondrion. 11:48–53. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Soltész B, Pös O, Wlachovska Z, Budis J,
Hekel R, Strieskova L, Liptak JB, Krampl W, Styk J, Németh N, et
al: Mitochondrial DNA copy number changes, heteroplasmy, and
mutations in plasma-derived exosomes and brain tissue of
glioblastoma patients. Mol Cell Probes. 66:1018752022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Marucci G, Maresca A, Caporali L, Farnedi
A, Betts CM, Morandi L, de Biase D, Cerasoli S, Foschini MP, Bonora
E, et al: Oncocytic glioblastoma: A glioblastoma showing oncocytic
changes and increased mitochondrial DNA copy number. Hum Pathol.
44:1867–1876. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Zhang J, Li D, Qu F, Chen Y, Li G, Jiang
H, Huang X, Yang H and Xing J: Association of leukocyte
mitochondrial DNA content with glioma risk: Evidence from a Chinese
case-control study. BMC Cancer. 14:6802014. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Shen J, Song R, Lu Z and Zhao H:
Mitochondrial DNA copy number in whole blood and glioma risk: A
case control study. Mol Carcinog. 55:2089–2094. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang Y, Qu Y, Gao K, Yang Q, Shi B, Hou P
and Ji M: High copy number of mitochondrial DNA (mtDNA) predicts
good prognosis in glioma patients. Am J Cancer Res. 5:1207–1216.
2015.PubMed/NCBI
|
|
145
|
Sourty B, Dardaud LM, Bris C,
Desquiret-Dumas V, Boisselier B, Basset L, Figarella-Branger D,
Morel A, Sanson M, Procaccio V and Rousseau A: Mitochondrial DNA
copy number as a prognostic marker is age-dependent in adult
glioblastoma. Neurooncol Adv. 4:vdab1912022.PubMed/NCBI
|
|
146
|
Ab Radzak SM, Mohd Khair SZN, Idris Z, Wan
Ahmad WMA, Patar A and Mohamed Yusoff AA: Mitochondrial
deoxyribonucleic acid copy number elevation as a predictor for
extended survival and favorable outcomes in high-grade brain tumor
patients: A Malaysian study. Eurasian J Med. 56:7–14. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Hua L, Juratli TA, Zhu H, Deng J, Wang D,
Sun S, Xie Q, Wakimoto H and Gong Y: High tumor mitochondrial DNA
content correlates with an improved patient's outcome in WHO grade
III Meningioma. Front Oncol. 10:5422942020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Sravya P, Krishna ASU, Santosh V and
Arivazhagan A: Mitochondrial DNA content in tumor tissue and blood
of patients with glioblastoma-A reliable biomarker? Int J
Neurooncol. 3:12–18. 2020. View Article : Google Scholar
|
|
149
|
Sravya P, Nimbalkar VP, Kanuri NN, Sugur
H, Verma BK, Kundu P, Rao S, Uday Krishna AS, Somanna S, Kondaiah
P, et al: Low mitochondrial DNA copy number is associated with poor
prognosis and treatment resistance in glioblastoma. Mitochondrion.
55:154–163. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Chen Y, Zhang J, Huang X, Zhang J, Zhou X,
Hu J, Li G, He S and Xing J: High leukocyte mitochondrial DNA
content contributes to poor prognosis in glioma patients through
its immunosuppressive effect. Br J Cancer. 113:99–106. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Sun X and St John JC: Modulation of
mitochondrial DNA copy number in a model of glioblastoma induces
changes to DNA methylation and gene expression of the nuclear
genome in tumours. Epigenetics Chromatin. 11:532018. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Braun Y, Filipski K, Bernatz S, Baumgarten
P, Roller B, Zinke J, Zeiner PS, Ilina E, Senft C, Ronellenfitsch
MW, et al: Linking epigenetic signature and metabolic phenotype in
IDH mutant and IDH wildtype diffuse glioma. Neuropathol Appl
Neurobiol. 47:379–393. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Oliva CR, Nozell SE, Diers A, McClugage SG
III, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie
GY, Landar A and Griguer CE: Acquisition of temozolomide
chemoresistance in gliomas leads to remodeling of mitochondrial
electron transport chain. J Biol Chem. 285:39759–39767. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Luna B, Bhatia S, Yoo C, Felty Q, Sandberg
DI, Duchowny M, Khatib Z, Miller I, Ragheb J, Prasanna J and Roy D:
Proteomic and mitochondrial genomic analyses of pediatric brain
tumors. Mol Neurobiol. 52:1341–1363. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Longchamps RJ, Castellani CA, Yang SY,
Newcomb CE, Sumpter JA, Lane J, Grove ML, Guallar E, Pankratz N,
Taylor KD, et al: Evaluation of mitochondrial DNA copy number
estimation techniques. PLoS One. 15:e02281662020. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Filograna R, Mennuni M, Alsina D and
Larsson NG: Mitochondrial DNA copy number in human disease: The
more the better? FEBS Lett. 595:976–1002. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Domazet B, Maclennan GT, Lopez-Beltran A,
Montironi R and Cheng L: Laser capture microdissection in the
genomic and proteomic era: Targeting the genetic basis of cancer.
Int J Clin Exp Pathol. 1:475–488. 2008.PubMed/NCBI
|
|
158
|
Kurdi M, Bamaga A, Alkhotani A, Alsharif
T, Abdel-Hamid GA, Selim ME, Alsinani T, Albeshri A, Badahdah A,
Basheikh M and Baeesa S: Mitochondrial DNA alterations in
glioblastoma and current therapeutic targets. Front Biosci
(Landmark Ed). 29:3672024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Chen J, Zheng Q, Hicks JL, Trabzonlu L,
Ozbek B, Jones T, Vaghasia AM, Larman TC, Wang R, Markowski MC, et
al: MYC-driven increases in mitochondrial DNA copy number occur
early and persist throughout prostatic cancer progression. JCI
Insight. 8:e1698682023. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Mou JJ, Peng J, Shi YY, Li N, Wang Y, Ke
Y, Zhou YF and Zhou FX: Mitochondrial DNA content reduction induces
aerobic glycolysis and reversible resistance to drug-induced
apoptosis in SW480 colorectal cancer cells. Biomed Pharmacother.
103:729–737. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Devic S: Warburg effect-a consequence or
the cause of carcinogenesis? J Cancer. 7:817–822. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Dickinson A, Yeung V and St. John J:
Abnormal regulation of mitochondrial DNA copy number in
glioblastoma multiforme cancer stem cells. Biol Reprod. 87 (Suppl
1):S5982012. View Article : Google Scholar
|
|
163
|
Wiese W, Barczuk J, Racinska O, Siwecka N,
Rozpedek-Kaminska W, Slupianek A, Sierpinski R and Majsterek I:
PI3K/Akt/mTOR signaling pathway in blood malignancies-new
therapeutic possibilities. Cancers (Basel). 15:52972023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Tufail M, Jiang CH and Li N: Altered
metabolism in cancer: Insights into energy pathways and therapeutic
targets. Mol Cancer. 23:2032024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Filograna R, Koolmeister C, Upadhyay M,
Pajak A, Clemente P, Wibom R, Simard ML, Wredenberg A, Freyer C,
Stewart JB and Larsson NG: Modulation of mtDNA copy number
ameliorates the pathological consequences of a heteroplasmic mtDNA
mutation in the mouse. Sci Adv. 5:eaav98242019. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Tian Q, Moore AZ, Oppong R, Ding J,
Zampino M, Fishbein KW, Spencer RG and Ferrucci L: Mitochondrial
DNA copy number and heteroplasmy load correlate with skeletal
muscle oxidative capacity by P31 MR spectroscopy. Aging Cell.
20:e134872021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Ding Q, Qi Y and Tsang SY: Mitochondrial
biogenesis, mitochondrial dynamics, and mitophagy in the maturation
of cardiomyocytes. Cells. 10:24632021. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Liu L, Li Y, Chen G and Chen Q: Crosstalk
between mitochondrial biogenesis and mitophagy to maintain
mitochondrial homeostasis. J Biomed Sci. 30:862023. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Popov LD: Mitochondrial biogenesis: An
update. J Cell Mol Med. 24:4892–4899. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Wredenberg A, Wibom R, Wilhelmsson H,
Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H and Larsson
NG: Increased mitochondrial mass in mitochondrial myopathy mice.
Proc Natl Acad Sci USA. 99:15066–15071. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Qian L, Zhu Y, Deng C, Liang Z, Chen J,
Chen Y, Wang X, Liu Y, Tian Y and Yang Y: Peroxisome
proliferator-activated receptor gamma coactivator-1 (PGC-1) family
in physiological and pathophysiological process and diseases.
Signal Transduct Target Ther. 9:502024. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Viscomi C, Bottani E, Civiletto G, Cerutti
R, Moggio M, Fagiolari G, Schon EA, Lamperti C and Zeviani M: In
vivo correction of COX deficiency by activation of the AMPK/PGC-1α
axis. Cell Metab. 14:80–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Dillon LM, Williams SL, Hida A, Peacock
JD, Prolla TA, Lincoln J and Moraes CT: Increased mitochondrial
biogenesis in muscle improves aging phenotypes in the mtDNA mutator
mouse. Hum Mol Genet. 21:2288–2297. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Giordano C, Iommarini L, Giordano L,
Maresca A, Pisano A, Valentino ML, Caporali L, Liguori R, Deceglie
S, Roberti M, et al: Efficient mitochondrial biogenesis drives
incomplete penetrance in Leber's hereditary optic neuropathy.
Brain. 137:335–353. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Wu J, Li J, Feng B, Bi Z, Zhu G, Zhang Y
and Li X: Activation of AMPK-PGC-1α pathway ameliorates peritoneal
dialysis related peritoneal fibrosis in mice by enhancing
mitochondrial biogenesis. Ren Fail. 44:1545–1557. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Pirinen E, Auranen M, Khan NA, Brilhante
V, Urho N, Pessia A, Hakkarainen A, Kuula J, Heinonen U, Schmidt
MS, et al: Niacin cures systemic NAD+ deficiency and
improves muscle performance in adult-onset mitochondrial myopathy.
Cell Metab. 31:1078–1090.e5. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Liu Y, Bai H, Guo F, Thai PN, Luo X, Zhang
P, Yang C, Feng X, Zhu D, Guo J, et al: PGC-1α activator ZLN005
promotes maturation of cardiomyocytes derived from human embryonic
stem cells. Aging (Albany NY). 12:7411–7430. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Sun S, Jiang T, Duan N, Wu M, Yan C, Li Y,
Cai M and Wang Q: Activation of CB1R-dependent PGC-1α is involved
in the improved mitochondrial biogenesis induced by
electroacupuncture pretreatment. Rejuvenation Res. 24:104–119.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Nishiyama S, Shitara H, Nakada K, Ono T,
Sato A, Suzuki H, Ogawa T, Masaki H, Hayashi J and Yonekawa H:
Over-expression of Tfam improves the mitochondrial disease
phenotypes in a mouse model system. Biochem Biophys Res Commun.
401:26–31. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Jiang M, Kauppila TES, Motori E, Li X,
Atanassov I, Folz-Donahue K, Bonekamp NA, Albarran-Gutierrez S,
Stewart JB and Larsson NG: Increased total mtDNA copy number cures
male infertility despite unaltered mtDNA mutation load. Cell Metab.
26:429–436.e4. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Ikeuchi M, Matsusaka H, Kang D, Matsushima
S, Ide T, Kubota T, Fujiwara T, Hamasaki N, Takeshita A, Sunagawa K
and Tsutsui H: Overexpression of mitochondrial transcription factor
a ameliorates mitochondrial deficiencies and cardiac failure after
myocardial infarction. Circulation. 112:683–690. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Hayashi Y, Yoshida M, Yamato M, Ide T, Wu
Z, Ochi-Shindou M, Kanki T, Kang D, Sunagawa K, Tsutsui H and
Nakanishi H: Reverse of age-dependent memory impairment and
mitochondrial DNA damage in microglia by an overexpression of human
mitochondrial transcription factor a in mice. J Neurosci.
28:8624–8634. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Oka S, Leon J, Sakumi K, Ide T, Kang D,
LaFerla FM and Nakabeppu Y: Human mitochondrial transcriptional
factor A breaks the mitochondria-mediated vicious cycle in
Alzheimer's disease. Sci Rep. 6:378892016. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Ylikallio E, Tyynismaa H, Tsutsui H, Ide T
and Suomalainen A: High mitochondrial DNA copy number has
detrimental effects in mice. Hum Mol Genet. 19:2695–2705. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Kleih M, Böpple K, Dong M, Gaißler A,
Heine S, Olayioye MA, Aulitzky WE and Essmann F: Direct impact of
cisplatin on mitochondria induces ROS production that dictates cell
fate of ovarian cancer cells. Cell Death Dis. 10:8512019.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Kubo Y, Tanaka K, Masuike Y, Takahashi T,
Yamashita K, Makino T, Saito T, Yamamoto K, Tsujimoto T, Harino T,
et al: Low mitochondrial DNA copy number induces chemotherapy
resistance via epithelial-mesenchymal transition by DNA methylation
in esophageal squamous cancer cells. J Transl Med. 20:3832022.
View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Mei H, Sun S, Bai Y, Chen Y, Chai R and Li
H: Reduced mtDNA copy number increases the sensitivity of tumor
cells to chemotherapeutic drugs. Cell Death Dis. 6:e17102015.
View Article : Google Scholar : PubMed/NCBI
|














