Introduction
Cancer is a complex disease involving multiple steps
for the transition from a normal state to a neoplastic growth
state, followed by the generation of malignant cells. The
‘hallmarks of cancer’ concept encompasses 14 elements required for
this process in human cancers (1),
and metabolic reprogramming is one of these elements (1,2).
Several characteristic metabolic mechanisms involving glucose and
amino acids (AAs) in cancer cells have been revealed, and new
insights continue to emerge (2).
Tumor tissues are exposed to stress through insufficient supply of
molecular oxygen and nutrients (3). Cancer cells must adapt to these
severe environmental conditions and change their mechanisms for
energy acquisition and immune evasion to achieve progression
(3). A recent review has revealed
that metabolic crosstalk between cancer cells and tumor stromal
cells, including immune cells and fibroblasts, can promote
symbiosis and malignancy (3).
Since the discovery of the Warburg effect, glucose
has been regarded as a primary nutrient for cancer cells through
aerobic glycolysis (2). However,
this adenosine triphosphate (ATP) production pathway is not always
predominant because oxidative phosphorylation remains active in
many cancer cells (2). Recent
studies have shown that cancer cells utilize glutamine and lipids
not only as ATP sources but also as precursors of plasma membrane
components and lipid droplets (LDs) (4,5).
Regarding lipids, long-chain fatty acids (LCFAs) are energy sources
and precursors of various phospholipids (5,6).
Indeed, endogenously and exogenously supplied fatty acids (FAs) are
sources for ATP production in cancer cells via oxidative
phosphorylation (5,6). Although cholesterol is not an energy
source, it is important for plasma membrane function, steroid
hormone generation, and cellular signaling (6).
Several studies have suggested that lipid metabolism
has potential as a therapeutic target in cancer treatment (4–6). A
recent study further revealed that inhibition of ATP citrate lyase
(ACLY), which is responsible for production of acetyl-CoA, a
lipogenesis precursor, can overcome cancer immunotherapy resistance
(7). Fatty acid synthase (FAS),
which is generally responsible for lipogenesis, can contribute to
many aspects of cancer progression (8). An intermediate compound of
cholesterol biosynthesis, 7-dehydrocholesterol, can act as a
natural inhibitor of ferroptosis in multiple cancer cells (9,10).
Thus, the development of new therapeutic strategies targeting these
various forms of lipid metabolism is ongoing.
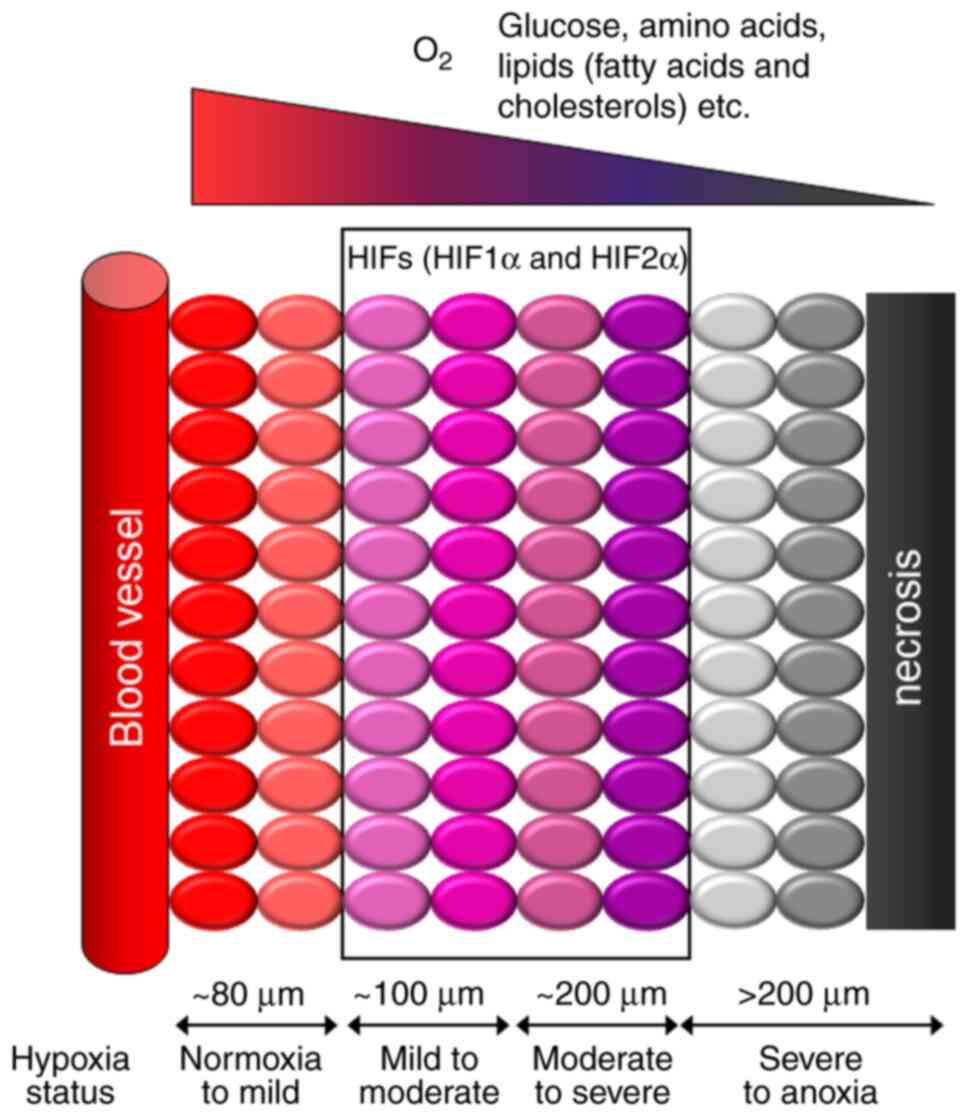
Hypoxia is a general condition in tumors that limits
the availability of not only molecular oxygen but also glucose,
AAs, and/or lipids from the bloodstream depending on the tumor's
distance from the vasculature (11,12)
(Fig. 1). Deficiency of these
elements in tumors is likely accelerated by the high catabolic
demand of cancer cells and vascular immaturity. Therefore,
adaptation to these harsh conditions is essential for cancer
progression. However, the published relationships among hypoxia,
glucose deficiency, AA deficiency, and lipid deficiency in cancer
cells have been poorly integrated. A therapeutic strategy targeting
these harsh tissue conditions may be beneficial because they are
expected to be characteristic of tumors, and such therapy is thus
likely to mitigate the toxic adverse effects associated with other
therapeutic approaches. The objective of the present review is to
summarize the latest understanding on this topic, with a particular
focus on lipid metabolism. The review mainly aims to provide a
comprehensive perspective on how cancer cells adapt to severe tumor
conditions and to discuss possible future research directions and
therapeutic applications from the perspective of lipid
metabolism.
Adaptive response mechanisms to glucose
deprivation
Relationship to hypoxia
Generally, cancer cells adapt to hypoxia and glucose
deficiency without relying on lipid metabolism, primarily through
the hypoxia-inducible factor (HIF) pathway (11,12)
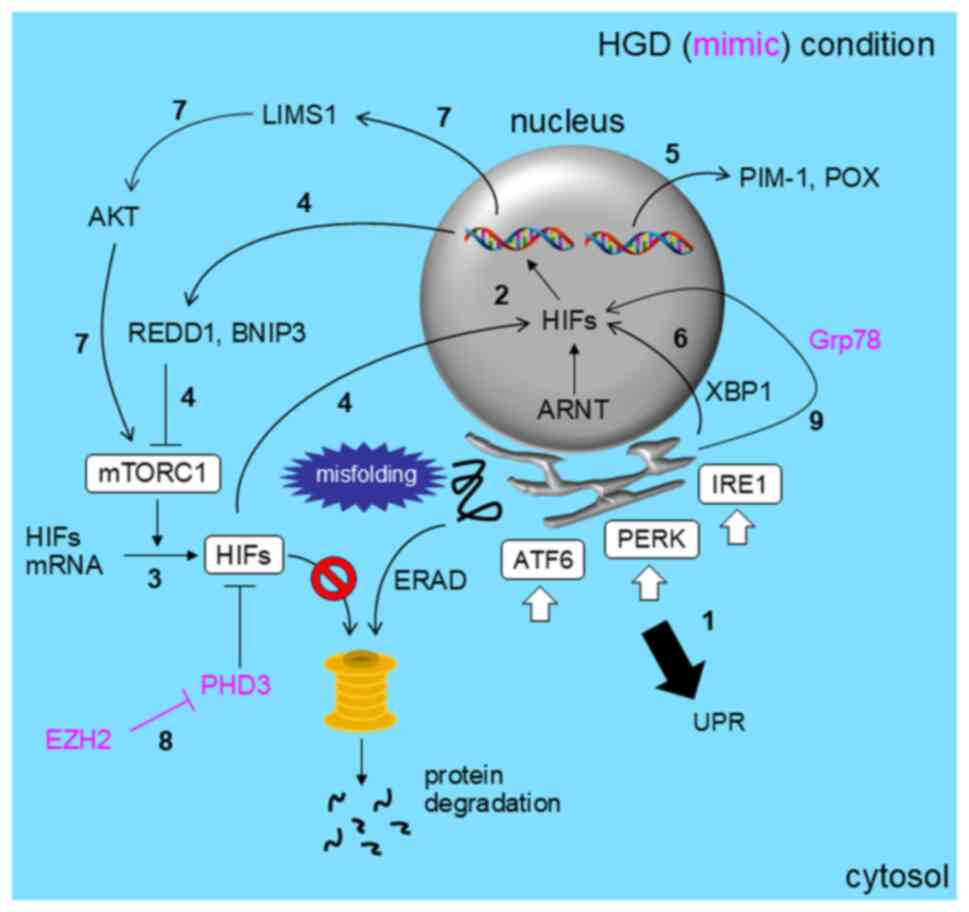
and the unfolded protein response (UPR) (13) (Fig.
2). The UPR involves endoplasmic reticulum (ER)-associated
degradation of incorrectly folded proteins produced under the above
stress conditions, preventing accumulation of toxic misfolded
proteins followed by ER stress. However, expression of HIF1α,
required for adaptation to hypoxia, is impaired in cancer cells
cultured under low-glucose conditions (14). The combination of hypoxia and
glucose deficiency (HGD) is cytotoxic to cancer cells. Indeed,
recent studies have shown that HGD induces cancer cell death in
association with CRE-binding protein (15) and overproduction of poly
(ADP-ribose) polymer (16).
However, cancer cells can synergistically respond to HGD and
activate genes required for adaptation to this harsh condition
(17).
The common adaptive response mechanism in cancer
cells exposed to HGD may involve both HIF pathway activation
(18–20) and the UPR (13,15,16,19,20).
Hypoxia induces the UPR through activation of the ER stress sensor
molecules activating transcription factor 6, inositol-requiring
protein 1 (IRE1), and PKR-like ER kinase (PERK) (1 in Fig. 2) (13,19),
which helps cancer cells tolerate low-oxygen conditions. These
mechanisms can be induced under hypoxia and/or glucose deficiency.
The HIF pathway is mediated by hypoxia-inducible transcription
factors HIF1α and HIF2α, which are mainly induced in mildly to
moderately hypoxic tumor regions (Figs. 1 and 2), along with the constitutively
expressed aryl hydrocarbon receptor nuclear translocator (ARNT) (2
in Fig. 2) (12).
Mammalian target of rapamycin (mTOR) plays a role in
adaptation to HGD. mTOR complex 1 (mTORC1) promotes the translation
of proteins, including HIFα proteins, to maintain energy
homeostasis (3 in Fig. 2)
(19,20). However, suppression of mTORC1
signaling can also be important for adapting to hypoxia by enabling
appropriate mRNA translation and activating the UPR (19,20).
mTOR can be inhibited through the HIF1α-REDD1 and HIF1α-BNIP3
pathways under hypoxia, suggesting a negative feedback loop between
HIFα and mTOR (route 4 in Fig. 2)
(19). Thus, HIFα, the UPR, and
mTOR are interconnected under HGD (13,19).
Several studies have revealed the effects of HGD on
cancer cell survival. Resistance to apoptosis induced under HGD is
mediated by the serine/threonine kinase proviral integration site
for Moloney murine leukemia virus 1 (PIM-1) in some cancer cells
(21). Meanwhile, proline oxidase
(POX) expression is increased under HGD in an AMP-activated protein
kinase (AMPK)-dependent manner (22). Expression of both PIM-1 and POX is
HIF-independent (5 in Fig. 2)
(21,22). Survival of cancer cells exposed to
HGD may be dependent on POX because proline oxidation results in
ATP production under HGD (22).
Thus, POX is a potential target of cancer therapy. HIF1α and the
UPR cooperate to enhance the stemness of breast cancer cells via
HIF1α binding to XBP1 under HGD (6 in Fig. 2) (23). HIF1α-driven expression of LIMS1 not
only facilitates glucose uptake but also enhances HIF1α translation
via the AKT-mTOR pathway in pancreatic cancer cells exposed to HGD
(route 7 in Fig. 2), resulting in
a cell survival advantage (24).
Recent studies have shown that GD generates an
HGD-mimicking condition because even under normoxia, expression of
HIF1α can be induced by GD to augment cancer cell survival
(25–27). This induction of HIF1α expression
occurs through EZH2-dependent suppression of PHD3 expression (8 in
Fig. 2) (25). This simple GD-driven pseudo-HGD
condition with HIF1α expression through inhibition of its
degradation augments the aggressiveness of lung adenocarcinoma
cells (25). GD under normoxia
also increases HIF1α expression via ER stress-inducible molecular
chaperone GRP78 in pancreatic cancer cells to augment
chemoresistance (9 in Fig. 2)
(26). The GRP78-HIF1α complex
binds to the regulatory region of the HIF1A gene to promote
transcription. Thus, GRP78 can induce HIF1α expression at the mRNA
level (26). In summary, GD can
enhance tumor hypoxia by upregulating HIF1α expression at both the
protein and mRNA levels. Meanwhile, lysophosphatidic acid receptors
were shown to contribute to chemoresistance in pancreatic cancer
cell line PANC-1 under HGD (28).
Expression of immune checkpoint receptor PD-1 and TIGIT can be
synergistically increased in esophageal cancer cells under HGD and
is responsible for immune tolerance (29). The roles of HIFs and the UPR were
not examined in these two studies (28,29).
Relationship to lipid metabolism
Glucose uptake and subsequent catabolism are
activated in cancer cells. Recent studies have shown that GD
influences lipid metabolism in cancer cells to support their
survival, although its relationship to hypoxia has not yet been
established. Indeed, some glioma cells have these characteristics
and become susceptible to GD in culture (30). GD in cancer cells is associated
with multiple lipid metabolism pathways, as summarized in Table I and Fig. 2. GD together with serum deficiency
was also reported to drive the use of extracellular glutamine and
lactate in glycerophospholipid synthesis for biomembrane generation
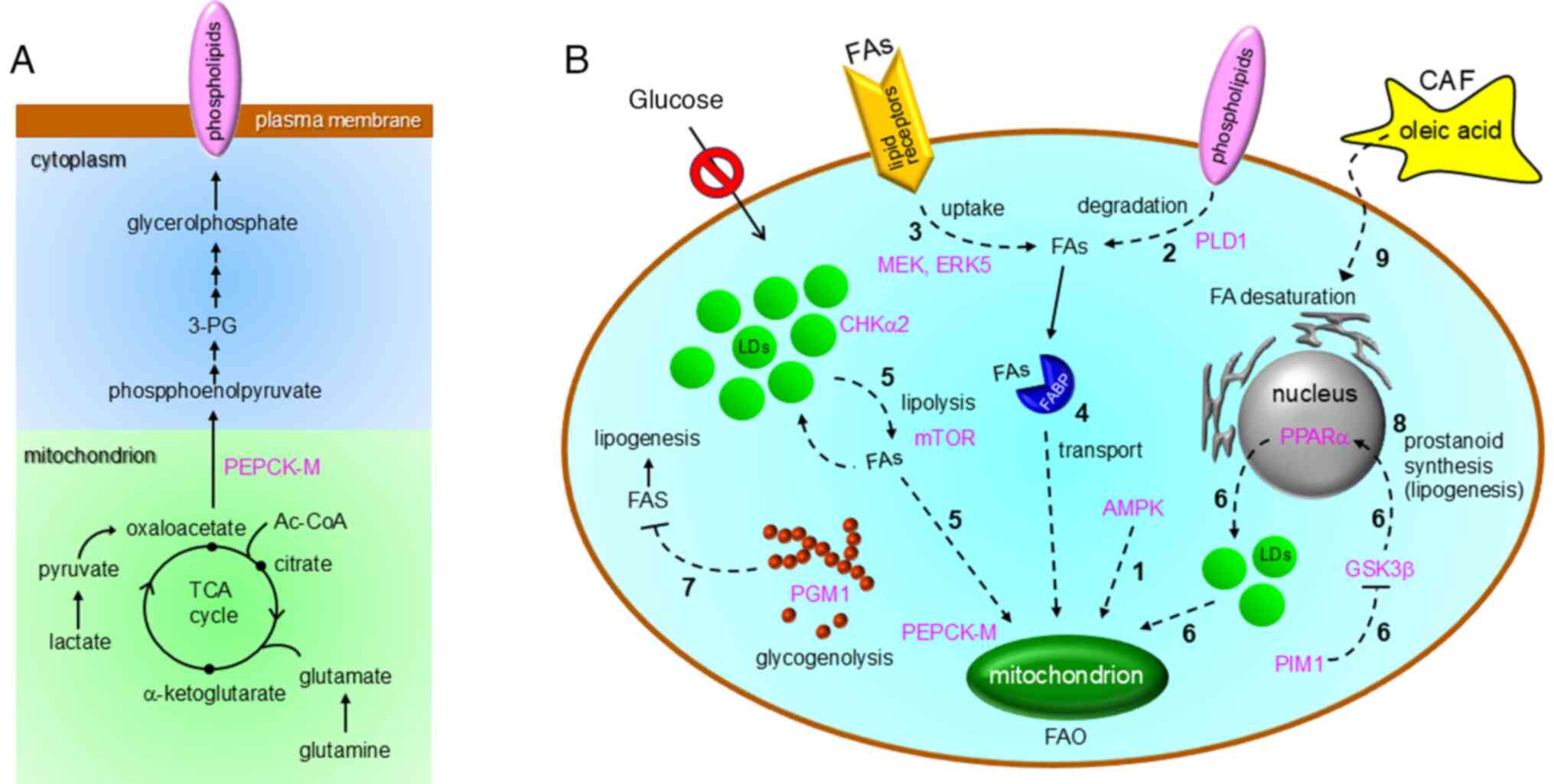
via oxaloacetate-phosphoenolpyruvate conversion (Fig. 3A) in lung cancer cells (31). Mitochondrial phosphoenolpyruvate
carboxykinase (PEPCK-M) plays key roles in this process (Fig. 3A) (31). Pharmacological activation of AMPK,
a cellular energy sensor molecule, activates fatty acid oxidation
(FAO) to acquire ATP in Akt-transformed cells under GD (30) (1 in Fig. 3B). Collectively, these studies have
revealed novel mechanisms for cancer cell adaptation to severe
nutrient deficiency.
 | Table I.Reported relationships between
glucose deprivation and lipid metabolism pathways associated with
cancer progression. |
Table I.
Reported relationships between
glucose deprivation and lipid metabolism pathways associated with
cancer progression.
| Adaptive lipid
metabolism | Key factor and
mechanism responsible for adaptive lipid metabolism
[reference] | Examined cancer
cell (histological type) [reference] |
|---|
| FAO | AMPK activation
(30), PLD1 (32), MEK5-ERK5 pathway activation
(33), FABP7 (34), mTOR activation (35), lipophagy (35), CHKα2 (36), chaperone-mediated lipophagy
(36), PIM1-GSK3β-PPARα pathway
inactivation (37), PGM1 (38), FAS (38), CAF (42), oleic acid (42), autophagosome maturation (42), cancer stemness (42) | LN18, LN229 (brain)
(30), MDA-MB-231 (breast)
(32), MCF-7 (breast) (32), RCC4 (renal) (32), HCT116 (colon) (32), OCI-AML3 (lymphocyte) (33), BCL-P2 (lymphocyte) (33), HepG2 (liver) (33), HuH-7 (liver) (33), Primary cells (brain) (34), LN229 (brain) (35), GaMg (brain) (35), U87MG (brain) (35), A172 (brain) (35), HuH7 (liver) (36), U87 (brain) (36), GP06 (brain) (36), GP08 (brain) (36), PC3 (prostate) (37), PC3LN4 (prostate) (37), DU145 (prostate) (37), BGC-823 (gastric) (38), MKN-28 (gastric) (38), NCI-H460 (lung) (42) |
| Glycerophospholipid
synthesis | PEPCK-M (31), glyceroneogenesis (31) | A549 (lung)
(31), H23 (lung) (31) |
| Membrane lipid
degradation | Autophagy (32), PLD1 (32) | MDA-MB-231
(32), MCF-7 (breast) (32), RCC4 (renal) (32), HCT116 (colon) (32) |
| FA transport | FABP7 (34) | Primary cells
(brain) (34) |
| FA uptake | CD36 (33), LRP1 (33) | OCI-AML3
(lymphocyte) (33), BCL-P2
(lymphocyte) (33), HepG2 (liver)
(33), HuH-7 (liver) (33) |
| LD catabolism | mTOR activation
(35), lipophagy (35) CHKα2 (36), chaperone-mediated lipophagy
(36), PIM1-GSK3β-PPARα pathway
activation (37) | LN229 (brain)
(35), GaMg (brain) (35), U87MG (brain) (35), A172 (brain) (35), HuH7 (liver) (36), U87 (brain) (36), GP06 (brain) (36), GP08 (brain) (36), PC3 (prostate) (37), PC3LN4 (prostate) (37), DU145 (prostate) (37) |
| Lipogenesis | PGM1 (38), glycogenolysis (38), FAS (38) | BGC-823 (gastric)
(38), MKN-28 (gastric) (38) |
| Alteration of
membrane lipid composition |
Glycerophospholipids (31), phospholipids
(phosphatidylethanolamine, cardiolipin, etc.) (39), cholesterol (39) | A549 (lung)
(31), H23 (lung) (31), Caco-2 (colon) (39) |
| Prostaglandin E2
synthesis | UPR (41), AMPK activation (41), COX-2 (41) | HT29 (colon)
(41), RG/C2 (colon) (41), AA/C1/SB/10C (colon) (41), SW480 (colon) (41) |
| Stearoyl-CoA
desaturase expression | CAF (42), oleic acid supply (42), autophagy activation (42), F-actin polymerization-YAP nuclear
translocation pathway (42),
cancer stemness (42) | NCI-H460 (lung)
(42) |
Membrane phospholipids can be substrates for
phospholipase D1-mediated autophagy in multiple cancer cell types
under GD (32). FAs generated
during this process (2 in Fig. 3B)
can be used for FAO to sustain cell survival (32). In hepatocellular carcinoma and
leukemia cells, GD activates MEK-ERK5 signaling to increase FA
uptake (3 in Fig. 3B), followed by
ATP generation through existing FAO activity (33). The importance of FAO under GD was
also demonstrated in drug-resistant slow-cycling glioblastoma
subpopulations (34). The same
study showed that FA transport (4 in Fig. 3B) by fatty acid-binding protein
(FABP)-7 is crucial for survival of mitochondria (oxidative
phosphorylation)-active glioma cells (34). LD catabolism by lipophagy, followed
by existing FAO activity (route 5 in Fig. 3B), also contributes to the survival
of glucose-starved glioblastoma cells (35,36).
This involves hyperactivation of mTOR (35) (Fig.
3B). Meanwhile, choline kinase 2 (CHKα2) is responsible for
phosphorylation of LD-associated perilipins, finally resulting in
chaperone-mediated autophagy-driven degradation of LDs to generate
FAs (35) (route 5 in Fig. 3B). FAO under GD is also important
for prostate cancer cells, in which LD accumulation via the
PIM1-GSK3β-PPARα axis can be activated under nutrient stress
conditions for FA generation (37)
(route 6 in Fig. 3B).
Glycogenolysis serves as an alternative pathway for
cellular glucose supply under GD. Phosphoglucomutase PGM1, a key
enzyme for glycogenolysis followed by glycolysis, contributes to
the viability of gastric cancer cells exposed to GD by suppressing
lipogenesis (38) (7 in Fig. 3B). FAS can compensate for the
reduced cell viability caused by PGM1 inhibition under GD
conditions (38). Thus, combined
inhibition of interconversion of the phosphate position within
glucose molecules and FAS may be promising for gastric cancer
treatment (38). A study using
colon cancer cell line Caco-2 revealed that the composition of
phospholipids, cholesterol, and unsaturated FAs in the cell
membrane is considerably altered under GD, leading to metabolic
adaptations that augment cell survival (39). GD also increases prostaglandin E2
synthesis at the nuclear membrane (40) (8 in Fig. 3B) by repressing
15-hydroxyprostaglandin dehydrogenase expression in colon cancer
cells, resulting in a survival advantage for these cells (41). The stemness of lung cancer cells
exposed to GD may be enhanced by oleic acid supplied by surrounding
cancer-associated fibroblasts (CAFs), followed by stearoyl-CoA
desaturase expression (9 in Fig.
3B), nuclear translocation of polymerized actin, and
yes-associated protein (42). This
process can be inhibited by treatment with an FAO inhibitor
(42).
In summary, this section has revealed that various
cancer cells generally circumvent GD through FAO (Table I). Additionally, the biosynthesis
of glycerophospholipids and prostaglandins can contribute to their
adaption to GD.
Adaptive response mechanisms to amino acid
deprivation through lipid metabolism and their correlation with
hypoxia
In addition to their role as protein components,
AAs, such as glucose, can serve as major energy sources. Numerous
studies have demonstrated the effects of AA deficiencies on cancer
phenotypes, similar to those observed in GD. However, there is
limited knowledge on their correlations with hypoxia and lipid
metabolism. This section will primarily address this issue. Studies
have generally demonstrated that AA deficiencies are associated
with multiple lipid metabolism pathways (Table II) and hypoxia in cancer
cells.
| Table II.Reported relationships between amino
acid deficiencies and lipid metabolism pathways in cancer
cells. |
Table II.
Reported relationships between amino
acid deficiencies and lipid metabolism pathways in cancer
cells.
| Deprived amino
acid | Responsive lipid
metabolism | Key factor and
mechanism responsible for adaptive lipid metabolism
[reference] | Examined cancer
cell line (histological type) [reference] |
|---|
| Glutamine | Suppression of
lipogenesis | Loss of reductive
carboxylationa
(44), LD-lipophagy, followed by
suppression of cholesterol synthesis (53) | PRC3 (kidney)
(44), 786-O (kidney) (44), UMRC2 (kidney) (44), HepG2 (liver) (53), HuH6 (liver) (53), Huh7 (liver) (53) |
|
| LPO | Impairment of
glutathione synthesisa (46), ferroptosisa (46) | RCC-4 (kidney)
(46), 786-O (kidney) (46) |
|
| FAO activation | Sestrin2-mTORCs
pathway activation (47), CHTM1
(48), HRD1 (51), CPT2 degradation (51), PI3K-C2γ pathway inactivation
(52), glutamine auxotroph
(52), AMPK-CHKα2-LD-ATGL pathway
activation (50) | H358 (lung)
(47), H1299 (lung) (47), H460 (Lung) (47), MCF-7 (breast) (48), RKO (colon) (48), UACC-62 (skin) (48), MDA-MB-231 (breast) (51), Capan1 (pancreas) (52), MiaPaca2 (pancreas) (52), Panc1 (pancreas) (52), H322 (lung) (50), H358 (lung) (50) |
|
| Lipogenesis | CHTM1 (48), PKC-CREB-PGC-1α pathway activation
(48), SREBP1-ACC1-LD pathway
pathway activation (49),
SREBP1-glutamine synthesis activation (49), | MCF-7 (breast)
(48), RKO (colon) (48), UACC-62 (skin) (48), HepG2 (liver) (49) |
|
| Lipolysis | AMPK-CHKα2-LD-ATGL
pathway activation (50) | H322 (lung)
(50), H358 (lung) (50) |
|
| Maintenance of
cellular lipid balance | TRIAP-1-p53
interaction (54), phospholipids
and sterols homeostasis (54) | HCT-116 (colon)
(54) |
| Serine
(Glycine) | LPO inhibition | Glutathione
synthesis (59), p53-p21 pathway
activation (59) | HCT116 (colon)
(59), RKO (colon) (59) |
|
| FAO activation | Pyruvate transfer
to the TCA cycle (59) | HCT116 (colon)
(59), RKO (colon) (59) |
|
| FAO
suppression | Impairment of
ceramide synthesisa
(58), mitochondrial
dysfunctiona
(58) | HCT116 (colon)
(58), HT29 (colon) (58) |
|
| Deoxyceramide
synthesis | Serine to alanine
substitution within ceramide moleculea (60, 61) | HCT116 (colon) (60,
61), MOLT-4 (lymphocyte) (61),
HEK293T (kidney) (61) |
|
| Accumulation of
cellular sphingosine | SK1 degradation
(61), autonomous serine synthesis
(61) | HCT116 (colon)
(61), MOLT-4 (lymphocyte)
(61), HEK293T (kidney) (61) |
| Cysteine | Suppression of
per-oxidized lipid accumulation | Activation of FA
metabolism (64), GPX4 expression
(64), glutathione synthesis
(67,68), NRF2-CBS pathway (67), macropinocytosis of albumin
(68), iron storage (68, 69),
ATM-MTF1-ferritin/FPN1 pathway (69), CISD3 (70), glutaminolysis (70), conversion of peroxidized lipid to
lipid alcohol (71), anti-oxidant
tryptophan metabolites (71) | HL60 (bone marrow)
(64), MOLM13 (bone marrow)
(64), SKOV3 (ovary) (67), OVCA429 (ovary) (67), HT-1080 (fibroblast) (68,69),
A375 (skin) (68), T98G (brain)
(68), U2OS (bone) (68), PaTu8988T (pancreas) (68), GS187 (brain) (68), MDA-MB-231 (breast) (69), RCC4 (kidney) (69), HEK293T (kidney) (69), HL60 (bone marrow) (70), 786-O (kidney) (71), AsPC-1 (pancreas) (71), CFPAC-1 (pancreas) (71), PANC-1 (pancreas) (71), U251 (brain) (71), Be2C, (neuroblast) (71) |
|
| Lipid
peroxidation | Suppression of
glutathione synthesisa (46,64),
ferroptosisa
(46,64) | RCC-4 (kidney)
(46), 786-O (kidney) (46), HL60 (bone marrow) (64), MOLM13 (bone marrow) (64) |
| Arginine | Suppression of
FAO | Arginine auxotroph
(73), impairment of mitochondria
functiona (73), cytotoxic autophagya (73) | MDA-MB-231 (breast)
(73), T47-D (breast) (73) |
|
|
| LD accumulation
(73) |
|
|
| Lipid
peroxidation | Arginine auxotroph
(74), inhibition of
mTOR-SREBP1-SCD5 pathwaya (74), ferroptosisa (74) | H1299 (lung)
(74), HCC827 (lung) (74) |
|
| Suppression of
lipogenesis | Arginine auxotroph
(75,76), activation of MEK-ERK-cMyc-ASS1
pathway (75), reduction of ACLY
(76), ACC1 (76), and FAS (76) synthesis | SKLMS-1 (vulva)
(75), A2058 (skin) (76), SK-Mel-2 (skin) (76) |
| Methionine | Phospholipid
metabolism | Increase of
cellular glycerophospholipids (78,79). | B16 (mouse skin)
(78), HepG2 (liver) (79) |
|
|
| Decrease of choline
(78) and phosphatidylcholine
(79). |
|
|
| Impairment of
cholesterol biosynthesis, facilitation of cholesterol
excretion | Suppression of
SREBP2-FOXM1 axisa
(80), inhibition of
S-adenosylmethionine synthesisa (80) | In-house human
glioma initiating cell lines (brain) (80) |
|
| Lipid
peroxidation | Suppression of
glutathione synthesisa (65), ferroptosisa (65) | MG1-4 (mouse brain)
(65), TS543 (brain) (65), KNS42 (brain) (65) |
Glutamine deprivation
Glutamine is another primary nutrient for cancer
cells in addition to glucose. The levels of non-essential AAs,
including glutamine, in pancreatic cancer tissues are considerably
lower than those in adjacent benign tissues (43). In cells with an inactivated von
Hippel Lindau (VHL) gene under hypoxia, glutamine is used to
generate α-ketoglutarate, which is then used to produce the citrate
required for de novo LCFA synthesis through reductive
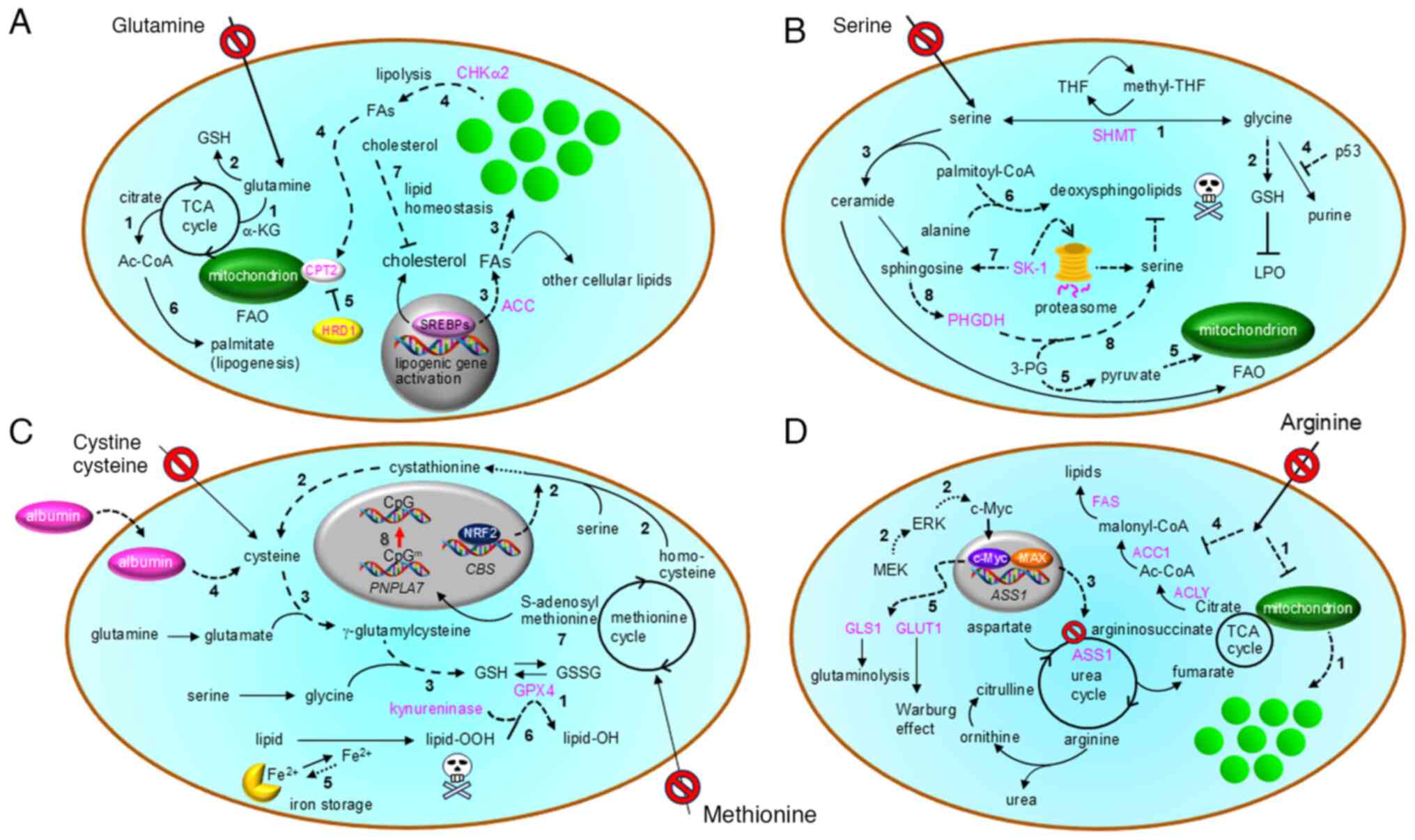
carboxylation (44,45) (route 1 in Fig. 4A) and the glutathione (GSH)
synthesis (2 in Fig. 4A) necessary
for suppression of lipid peroxide accumulation, which is
responsible for ferroptosis (46).
Thus, clear cell renal cell carcinoma (ccRCC) cells lacking
functional VHL are susceptible to glutamine deficiency
(GlnD) caused by glutaminase inhibition (44) or glutamine withdrawal from the
culture medium (45) (Table II).
Cancer cells can adapt to stressful GlnD conditions
through multiple metabolic mechanisms. A recent study using lung
cancer cell lines showed that differential regulation of mTORC1 and
mTORC2 via Sestrin2, followed by FAO, contributes to cell survival
under GlnD (47) (Table II). Under both GD and GlnD,
mitochondrial protein coiled-coil helix tumor and metabolism 1 can
facilitate LCFA synthesis and FAO via LD formation in multiple
cancer cells (48) (routes 3 and 4
in Fig. 4A and Table II). GlnD likely activates the
SREBF1 gene, which encodes the lipogenic enzyme SREBP1
(49). SREBF1 activation, mediated
by O-linked N-acetylglucosaminylated transcription factor
specificity protein 1 (Sp1) and followed by acetyl-CoA carboxylase
(ACC) expression, contributes to LD biosynthesis in various cancer
cells (49) (route 3 in Fig. 4A). Similar to the previously
described glioblastoma cell response to GD (35), CHKα2 plays a key role in LD
lipolysis in lung cancer cells exposed to GlnD (50) (route 4 in Fig. 4A and Table II). In this context, neutral
lipolysis and lipophagy likely contribute to LD catabolism (route 4
in Fig. 4A). Notably, the same
study demonstrated that phosphorylation of CHKα2 at S279, which is
involved in neutral lipolysis, is associated with a worse prognosis
in patients with non-small-cell lung cancer (50).
HMG-CoA reductase degradation protein 1 (HRD1) was
found to suppress the proliferation of breast cancer cell line
MDA-MB-231 through inhibition of FAO (51). HRD1 can interact and ubiquitinate
FA transporter protein carnitine palmitoyltransferase 2 (CPT2) (5
in Fig. 4A), resulting in blockade
of mitochondrial LCFA transport through its degradation (51). Under GlnD, this mechanism can be
abolished to activate FAO, resulting in resistance to glutaminase
inhibitors. Thus, simultaneous inhibition of FAO and glutamate
production may be therapeutically promising, and high HRD1
expression potentially predicts better efficacy of glutaminase
inhibitors (51).
PI3K-C2γ expression is inactivated to activate mTOR
in cancer cells from pancreatic tumor tissues. These cells depend
on exogenous glutamine to activate lipogenesis (52) (6 in Fig. 4A). Thus, treatment with mTOR and
glutaminase inhibitors may be therapeutically beneficial. GlnD
induces lipophagy-driven LD degradation to increase cellular
cholesterol levels in hepatocellular carcinoma cells (53) (7 in Fig. 4A). Cholesterol inhibits SREBP2
maturation, leading to decreased cholesterol synthesis, which helps
maintain redox balance and provides a survival advantage to cancer
cells under GlnD conditions (53).
Interaction between p53 and TP53-regulated inhibitor of apoptosis 1
(TRIAP1) may also be important for survival of colon cancer cells
under GlnD (54) (Table II). TRIAP1 affects lipid
homeostasis through multiple lipids, including glycerolipids,
sphingolipids, and cholesterols, in HCT116 cells (Table II). The TRIAP1-p53 interaction is
important for glutamine metabolism. Indeed, under GlnD, p53 can
compensate for TRIAP1 function to overcome metabolic stress
(54). Collectively, GlnD can
couple to multiple lipid metabolism pathways (Table II). Cancer cells likely acquire
ATP through the activation of FAO via LD metabolism under this
nutrient stress (Fig. 4A).
Maintaining cellular lipid homeostasis is also important (Fig. 4A).
GlnD associated with hypoxia is known to trigger
expression of UPR-executor transcription factor ATF4 (19,20)
in glioblastoma cells (55). This
UPR-mediated stress response is responsible for resistance to
temozolomide treatment (55).
Hypoxia with GlnD can also promote cancer progression through
epigenetic mechanisms. In melanoma tissue, the glutamine
concentration in the hypoxic tumor core region is considerably
lower than that in the tumor periphery (56). Hypoxia with GlnD can cause
hypermethylation of histones through inhibition of demethylation
and promote cancer cell dedifferentiation, resulting in resistance
to BRAF inhibitor treatment (56).
The implications for lipid metabolism involvement in these hypoxia-
and GlnD-associated features observed in tumor tissues remain to be
determined.
Serine and glycine deprivation
Serine and glycine can be enzymatically
interconverted through a single reaction mediated by serine
hydroxymethyltransferase, involving the conversion of
tetrahydrofolate to its methylated form (1 in Fig. 4B), and they contribute to common
metabolic pathways such as GSH synthesis (57,58)
(2 in Fig. 4B). Thus, studies have
tended to consider the effects of these AAs together (Fig. 4B, Table II).
Studies using colon cancer cell line HCT116 showed
that serine/glycine deficiency (SGD) can suppress FAO through
impaired mitochondrial function (58). Specifically, the ceramide level is
decreased in serine-depleted cells (3 in Fig. 4B), leading to mitochondrial
dysfunction (58). Thus,
restriction of the serine supply is promising in treatment of
p53-deficient cancers. However, SGD causes metabolic reprogramming
with increased oxidative phosphorylation activity (59). p53-dependent synthesis of GSH with
suppression of purine synthesis (4 in Fig. 4B), followed by scavenging of
reactive oxygen species, can contribute to resistance of cancer
cells against this metabolic stress (59). This process is accompanied by an
increase in pyruvate transfer via 3-phosphoglycerate (3-PG) to the
mitochondria, which activates FAO (route 5 in Fig. 4B). Furthermore, SGD facilitates the
biosynthesis of toxic deoxysphingolipid, in which serine residues
are substituted with alanine residues (6 in Fig. 4B), in cancer cells (60). Thus, dietary restriction and
pharmacological targeting of the serine supply pathway can lead to
tumor regression. This effect was prominent for growth of
alanine-rich spheroid cancer cells (60). A more recent study further
demonstrated that deoxysphingolipid (deoxysphinganine) generation
is non-toxic and important for adaptation of cancer cells to SGD
(61). This response mechanism
involves accumulation of sphingosine in cancer cells due to the
proteasomal degradation of sphingosine kinase 1 (7 in Fig. 4B), leading to increased expression
of phosphoglycerate dehydrogenase (PHGDH) and subsequent de
novo serine synthesis (route 8 in Fig. 4B) to overcome SGD (61). In summary, cancer cells can adapt
to serine deficiency through inhibition of lipid peroxidation,
activation of FAO, and modulation of sphingolipid biosynthesis.
Regarding the relationship between SGD and hypoxia,
glioblastoma cells were reported to activate the AMPK-HIF1α pathway
and induce a pseudo-hypoxia condition under SGD to bypass this
metabolic stress (62). The
effects of lipid metabolism involvement in this adaptation
mechanism remain to be elucidated.
Cysteine deprivation
The major source of cellular cysteine is dietary
cystine, which is metabolically associated with glutamine (63). Cysteine is also produced by de
novo synthesis from methionine (63). The major effect of cysteine
deficiency (CD) on cancer cells is impaired GSH synthesis,
resulting in ferroptosis through accumulation of toxic peroxidized
phospholipids (Fig. 4C, Table II) (46,64).
Thus, CD may be therapeutically beneficial (46,64–66).
The mechanisms underlying how cancer cells
circumvent CD fall into a single category (Table II): ferroptosis resistance through
suppression of peroxidized lipid accumulation. Multiple pathways
may be involved in this process in various cancer cells (Table II). For example, acute myeloid
leukemia cells are auxotrophic for cysteine and can acquire CD
resistance through overexpression of glutathione peroxidase 4 (1 in
Fig. 4C) and microsomal
glutathione-S-transferase 1, which may be associated with
activation of FA metabolism (64)
(Table II). In
VHL-defective ccRCC cells, generation of peroxidized lipids
is suppressed by FAO inhibition and GSH synthesis (46). In ovarian cancer cells,
transcription factor NRF2 enhances cystathionine β-synthase
(CBS) gene expression and activates autonomous synthesis of
cysteine (route 2 in Fig. 4C),
resulting in resistance to CD-induced ferroptosis through GSH
synthesis (route 3 in Fig. 4C)
(67). A recent study showed that
albumin can be an extracellular source of cysteine under CD
(68). Albumin incorporated into
cancer cells via macro-pinocytosis in association with mTOR
inhibition undergoes lysosomal degradation (68). Cysteine released into the cytoplasm
also contributes to GSH synthesis (4 and route 3 in Fig. 4C), thereby preventing ferroptosis
of cancer cells (68). This
mechanism is especially important under spheroid culture conditions
associated with tumor-like stress conditions (68).
Deprivation of cellular free iron ions is an
alternative mechanism of ferroptosis resistance. Expression of
genes involved in cellular iron storage can be increased via the
ATM-MTF1 axis under CD. As a result, the balance between iron
storage and release is expected to shift toward storage (5 in
Fig. 4C), leading to ferroptosis
resistance in multiple cancer cells (69). Expression of iron-sulfur cluster
protein CISD3 is also increased in cancer cells and contributes to
iron storage under CD, followed by ferroptosis resistance (70).
Tryptophane metabolites contribute to ferroptosis
resistance under CD through a detoxification mechanism.
Specifically, serotonin and 3-hydroxy-anthranilic acid produced by
kynureninase act as radical trapping agents and reduce peroxidized
lipids to non-toxic lipid alcohols (71) (6 in Fig. 4C). Overall, cancer cells utilize
multiple molecular defense mechanisms against CD-driven ferroptosis
(Table II).
In the context of CD and hypoxia, CD-induced death
of MDA-MB-231 cells can be alleviated by hypoxia through inhibition
of ATF4 expression (72). Thus,
hypoxia may reduce CD-mediated cytotoxicity in certain cancer
cells.
Arginine deprivation
Cancer cells are also sensitive to arginine
deficiency (AD), and this vulnerability is often associated with
lipid metabolism (Table II).
Expression of argininosuccinate synthetase 1 (ASS1), an enzyme in
the urea cycle (Fig. 4D) that also
catalyzes arginine biosynthesis, is low in >60% of clinical
breast cancer samples (73). Thus,
many cancer cells are arginine auxotrophs (Table II). AD induces cytotoxic autophagy
in breast cancer cells, leading to cell death due to impaired
mitochondrial function (Table
II). In such cases, FAO suppression is followed by a metabolic
shift to increase LDs (73) (route
1 in Fig. 4D). Meanwhile,
ASS1-high non-small-cell lung cancer cells can confer ferroptosis
resistance through monounsaturated FA synthesis. To facilitate this
process, ASS1-driven synthesis of arginine activates the
mTOR-SREBP1-SCD5 pathway to cause unsaturation of autonomously
synthesized FAs, leading to inhibition of peroxidized lipid
generation (74). This mechanism
provides additional insight into why arginine auxotroph lung cancer
cells are susceptible to AD-induced cell death. These observations
suggest that arginine-deficient diets may be a therapeutic strategy
through induction of ferroptosis. However, cancer cells can
circumvent this stress condition. For example, AD can activate the
MEK-ERK signaling pathway (route 2 in Fig. 4D) to induce c-Myc-Max transcription
factor-driven ASS1 gene activation (3 in Fig. 4D) in ASS1-negative vulvar
leiomyosarcoma (SKLMS-1) cells (75) (Table
II). This cellular response causes adaptive metabolic
reprogramming, including reduced ACLY expression, followed by
suppression of de novo FA synthesis (75) (4 in Fig. 4D). Moreover, reports have described
resistance to pharmacological AD in melanoma cells with ASS1
overexpression. In these cells, de novo lipogenesis is also
inhibited through suppression of ACLY (Fig. 4D), acetyl-CoA carboxylase 1 (ACC1
in Fig. 4D), and FAS (Fig. 4D) to promote the Warburg effect
(76). In this scenario, c-Myc
contributes to AD resistance not only through ASS1 expression but
also by enhancing glycolysis and glutaminolysis (5 in Fig. 4D) via the expression of glucose
transporter 1 and glutaminase, respectively. Collectively, cancer
cells can evade the effects of AD by autonomously synthesizing
arginine, with the Warburg effect, rather than FAO, likely playing
a key role in supporting cell survival under AD.
Under hypoxia, pharmacological AD inhibits
expression of HIFs, inducible nitric oxide synthase, and ASS1 in
HCT116 cells to inhibit tumor growth (77). This effect is associated with UPR
induction. ASS1-deficient bladder cancer cell line UMUC3 shows
greater sensitivity to hypoxia under AD (77). Thus, hypoxic cancer cells are
vulnerable to AD.
Methionine deprivation
Deprivation of methionine, an essential AA, from
culture medium likely affects lipid metabolism in cancer cells
(Fig. 4C, Table II). Methionine deprivation (MD) is
linked to GSH synthesis through the methionine cycle and cysteine
synthesis pathway (routes 2 and 3 in Fig. 4C). Consequently, MD can impair GSH
synthesis (65), leading to
ferroptosis, similar to the effect of CD. However, acute myeloid
leukemia cells are cysteine auxotrophs as indicated by the fact
that methionine supplementation does not rescue them from CD-driven
ferroptosis (64). MD also affects
cellular lipids by increasing phosphorylethanolamine and decreasing
choline (Table II), thereby
contributing to a synergistic therapeutic effect with
chloroethylnitrosourea treatment (78). Meanwhile, methionine is a precursor
of S-adenosylmethionine (7 in Fig.
4C), a key methyl donor for biomolecules. Thus, MD affects
methylation-driven lipid metabolism pathways. For example, MD
causes S-adenosylmethionine deficiency in HepG2 cells and enhances
glycerophosphocholine synthesis through demethylation of CpGs
within the PNPLA7 promoter region (79) (8 in Fig. 4C). This adaptation mechanism
potentially promotes cancer progression via activation of
mitochondrial oxidative phosphorylation (79). MD may also be therapeutically
promising for glioma-initiating cells because it impairs
cholesterol synthesis and increases cholesterol excretion, thereby
suppressing key glioma-initiating cell functions (80).
Adaptive response mechanisms to simultaneous
deprivation of O2 and lipids
Cancer cells can utilize FAs obtained through
lipogenesis and uptake from extracellular spaces under glucose and
AA deficiency. However, the exogenous supply of lipids is
restricted in cancer cells within poorly vascularized tumor
tissues. This section will discuss recently revealed adaptive
response mechanisms to restricted supply of both lipids and
molecular oxygen, although the observations are currently limited
to certain cancer types.
Adaptive responses to LCFA starvation
and hypoxia in cancer cells
In general, tumor tissues are poorly vascularized.
Thus, many blood components, including lipids, are poorly supplied.
Impairment of lipogenesis may also enhance lipid insufficiency in
cancer cells exposed to hypoxia with limited exogenous lipid
supply. The effect of simultaneous deprivation of lipids and
molecular oxygen on cancer cells was first demonstrated by
experiments examining how serum starvation and hypoxia (SSH)
affects their phenotype in relation to mTOR activity (81). The same study using mouse embryonic
fibroblast cells and various cancer cells, including kidney cancer
cells, showed that mTOR activation under SSH augments ER stress,
resulting in UPR-mediated cell death (Table III) (81). This is likely due to abnormal ER
expansion induced by increased protein synthesis and insufficient
unsaturated LCFA supply because cell death was alleviated by
supplementation of albumin-conjugated oleic acid (81).
| Table III.Reported effects of hypoxia and lipid
starvation on cancer cell response. |
Table III.
Reported effects of hypoxia and lipid
starvation on cancer cell response.
| Deprived
lipids | Related cellular
events [reference] | Examined cancer
cells (histological origin) [reference] |
|---|
| Exogenous FA | ER
expansiona (81), UPR-induced cell deatha (81) | MCF7 (breast)
(81), RCC10 (kidney) (81), U251 (brain) (81), RT4 (bladder) (81), A498 (kidney) (81) |
|
| UPR (93), ICAM-1-driven resistance to
apoptosis (95) | OVSAYO (ovary)
(93,95), OVISE (ovary) (93,95) |
|
| Neutral
lipase-mediated LD catabolism (88), oleate-mediated suppression of
lipotoxicity (88) | A498 (kidney)
(88) |
|
| Lipophagy-mediated
LD catabolism (98) | OVSAYO (ovary)
(98), OVISE (ovary) (98) |
|
| EMT (96) | OVSAYO (ovary)
(96), OVISE (ovary) (96) |
| Endogenous FA | Impairment of
NAD+ productiona (82), induction of lipid auxotroph through
inhibition of lipogenesis (82) | Hela (uterus)
(82) |
| Cholesterol | Secretion of
procoagulant extracellular vesicles (99) | OVSAYO (ovary)
(99), OVISE (ovary) (99) |
|
| SREBP1-driven
lipogenic gene expression (100),
apoptosis (100), impairment of
spheroid growth (100) | U87 (brain)
(100), U251 (brain) (100) |
|
| SREBP2-driven
ACSS2 gene expression (101), palmitate synthesis (101), phospholipid synthesis (101) | BT474 (breast)
(101), DU145 (prostate)
(101) |
Hypoxia and delipidated serum cell culture
conditions, characterized by poor supply of molecular oxygen as an
electron acceptor and poor supply of extracellular lipids, was also
shown to reduce regeneration of NAD+, a cofactor
required for lipogenic citrate production in cancer cells, followed
by cell proliferation (82). Thus,
cancer cells exposed to SSH are likely to become lipid auxotrophs
(Table III). However,
NAD+-independent metabolism of exogenous acetate to
acetyl-CoA followed by lipogenesis in cancer cells can rescue this
auxotrophy (82).
ccRCC is a histological subtype of most kidney
cancers (70–80%). Most of these cancer cells lack VHL function,
resulting in constitutive HIF expression. Consequently, these
cancer cells exhibit hypoxia-mimicking phenotypes (83,84).
The phenotypes of ccRCC cells, such as ER homeostasis (83), motility (85), invasiveness (85), and anti-apoptosis (84), are highly dependent on LD
biogenesis rather than LD catabolism (83–85).
The kidney is a well-perfused organ (85), and studies on this cancer type
under true hypoxic conditions are limited because ccRCC cells
express HIF even in normoxic environments. However, hypoxia is a
general tumor condition. Indeed, hypoxic regions exist within the
normal renal medulla (86) and
renal tumors (87), and adaptation
of cancer cells to SSH may contribute to kidney cancer progression
(Table III).
The HIF2α-perilipin 2 pathway contributes to ER
homeostasis-mediated survival of ccRCC cells under SSH (1 in
Fig. 5 and Table III) (83). LD catabolism also contributes to
ccRCC cell phenotypes under LCFA starvation and hypoxia. LDs in
cancer cells undergo hormone-sensitive lipase-driven degradation
under SSH to maintain the cellular unsaturated FA (oleic acid)
level, thereby suppressing the effect of toxic saturated FAs
synthesized under hypoxia (2 in Fig.
5 and Table III) (88). Thus, the roles of LDs in malignancy
are context-dependent.
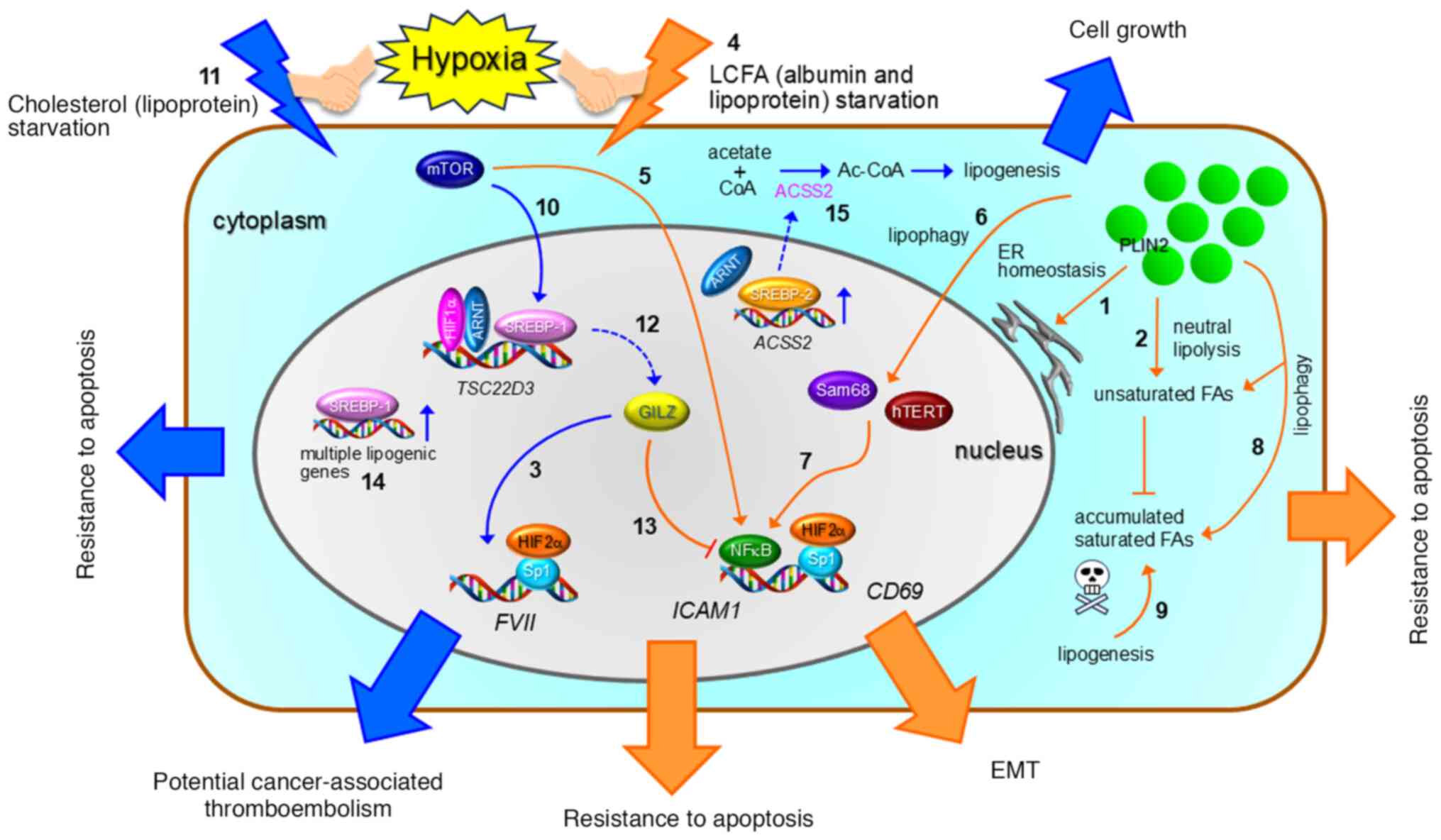 | Figure 5.Overview of lipid starvation and
hypoxia-driven cancer cell phenotypes. Cancer cells adapt to lipid
(LCFA and cholesterol) deprivation and hypoxia through various
resistance mechanisms. These malignant phenotypes are mediated by
lipolysis and lipogenesis. Transcription factors such as HIFs,
ARNT, SREBPs, NFκB, and Sp1, along with cofactors like GILZ, Sam68,
and hTERT, play key roles in these processes. Handshake symbols
between hypoxia and orange or blue lightning symbols represent LCFA
starvation + hypoxia and cholesterol starvation + hypoxia,
respectively. Plain arrows, dashed arrows, and T-bars indicate
activation processes, protein expression, and inhibition processes,
respectively. See the text for further details on the numbered
items. Orange and blue symbols correspond to processes induced by
LCFA and cholesterol starvation, respectively. Bold orange and blue
arrows represent phenotype expressions resulting from LCFA and
cholesterol starvation, respectively. EMT, epithelial-mesenchymal
transition; PLIN2, perilipin 2. |
Ovarian clear cell carcinoma (OCCC) has
morphological and biological similarities to ccRCC (89). As described above, ccRCC cells
exhibit hypoxia-mimicking phenotypes. This characteristic may also
be true for OCCC cells because the HIF pathway is more active in
this cancer subtype than in other histological subtypes of
epithelial ovarian cancer (90–92).
The effects of SSH on gene expression and
phenotypes of OCCC cells have been examined, revealing that
multiple genes can be synergistically activated in certain OCCC
cells exposed to SSH stress. We first discovered this phenomenon
for the FVII gene (93) (3
in Fig. 5), which is responsible
for initiation of the physiological blood coagulation cascade and a
potential contributor to cancer-associated thromboembolism
(Fig. 5 and Table III) (94). Unlike typical hypoxia response
genes, such as VEGF, this transcriptional activation
involves an Sp1-HIF2α interaction on the FVII gene promoter
(Fig. 5) (93). Subsequent studies revealed that
multiple genes, including ICAM1 (95) and CD69 (96), exhibit the same SSH-driven
transcriptional activation (Fig.
5) and have much higher synergism than the FVII gene
(95,96). We found that unlike hypoxia alone
and serum starvation alone, SSH has UPR involvement (Table III) (93). However, the UPR does not contribute
to the synergistic expression of FVII and ICAM1
(93,95).
Albumin serves as a major LCFA transporter in the
blood and is important for LCFA uptake by cells (97). Addition of albumin-LCFA complex was
found to abolish the SSH-driven ICAM1 and CD69
expression, suggesting that LCFA starvation is responsible for the
synergistic transcriptional activation under hypoxia (95,96)
(4 in Fig. 5). Indeed, addition of
low-density lipoproteins, including LCFAs and cholesterol as their
esterified form, also abolished the SSH-driven ICAM1
expression (4 in Fig. 5), whereas
addition of cholesterol alone did not (98). mTOR is involved in this
ICAM1 expression via NFκB (5 in Fig. 5) (95). Further studies revealed that
lipophagy is induced under SSH in OCCC cells and is responsible for
the synergistic ICAM1 and CD69 expression (6 in
Fig. 5) (96,98).
Lipophagy enhances binding of pro-inflammatory transcription factor
NFκB to the ICAM1 promoter region via Sam68 and hTERT (7 in
Fig. 5) (98). Currently, ICAM-1 is considered to
suppress the lipotoxicity-mediated apoptosis induced by
lipophagy-driven LD degradation (8 and 9 in Fig. 5 and Table III) (98). Meanwhile, CD69 causes
epithelial-mesenchymal transition in a fibronectin-dependent manner
(Fig. 5 and Table III), leading to OCCC cell
survival in vitro and in vivo (96).
Adaptive response mechanisms to
cholesterol starvation and hypoxia in cancer cells
The effect of SSH on synergistic transcriptional
activation in OCCC cells is mediated not only by LCFA starvation
but also by cholesterol deficiency. The synergistic FVII
gene expression under SSH is mediated through activation of the
mTOR-SREBP1 axis (10 in Fig. 5)
under cholesterol deprivation (11 in Fig. 5) (99). SREBP1 and HIF1α-ARNT complex
indirectly promotes FVII expression through transcriptional
activation of glucocorticoid-induced leucine zipper (GILZ) protein
(12 in Fig. 5) to generate
procoagulant extracellular vesicles, which are potentially
responsible for cancer-associated thromboembolism (Fig. 5 and Table III) (94,99).
It is noteworthy that GILZ suppresses SSH-driven ICAM1
expression because this anti-inflammatory transcriptional regulator
binds and inhibits NFkB (13 in Fig.
5) (99).
Glioblastoma cells have been found to exhibit
synergistic gene expression mediated by SREBP under both hypoxia
and lipoprotein-deficient medium conditions (100). These SSH conditions
synergistically enhance transcriptional activation of the lipogenic
stearoyl-CoA desaturase, FABP-3, and FABP-4 genes (14 in Fig. 5) to suppress apoptosis and promote
spheroid growth (Fig. 5 and
Table III). An SREBP-dependent
gene signature involving these genes predicts a poor survival rate
for patients with glioblastoma (100). Lipogenesis via acetate also
supports the survival of cancer cells under SSH, similar to the
effect observed with exogenous acetate supply in HeLa cells
(82,101). In this process, SREBP2 activates
ACSS2 gene expression to enhance acetyl-CoA production (15
in Fig. 5 and Table III) (101). Additionally, HIFs may contribute
to this mechanism because ARNT can upregulate ACSS expression under
SSH (Fig. 5 and Table III) (101).
Conclusion and perspectives
In recent years, a wealth of knowledge regarding
cancer cell metabolism has been accumulated, leading to the
proposal of various therapeutic approaches. However, the current
understanding of the combined effects of hypoxia and nutrient
deprivation on cancer cell phenotypes remains insufficient, and
further research is needed to enhance clinical applications. The
conclusion of this review is that cancer cells can adapt to severe
oxygenation and nutrient supply conditions through diverse lipid
metabolism pathways. Activation of these pathways results in
increased malignant phenotypes, including stemness, drug
resistance, immune tolerance, and resistance to apoptosis. Thus, in
addition to the accumulated information on metabolic reprogramming
in cancer, we propose that a detailed understanding of the
adaptation mechanisms to both hypoxia and various nutrient
starvation conditions provides a platform for exploring promising
therapeutic strategies targeting finely reprogrammed metabolisms in
cancer. For instance, the suppression of lipid peroxidation
correlates with an insufficient supply of multiple AAs, including
cysteine, suggesting a potential pro-ferroptosis strategy for
treating cancers prone to cysteine starvation. Furthermore, cancer
cells rely on multiple lipid metabolism pathways to adapt to GlnD.
Thus, combination therapy targeting glutamine supply and lipid
metabolism may be promising. Additionally, therapeutic approaches
targeting both severe hypoxia and tissue nutrient insufficiency may
be beneficial because these harsh tissue conditions are
characteristic of tumors. Specific targeting of these tissue
conditions is expected to avoid the unwanted adverse effects
associated with therapeutic strategies that target hypoxia
alone.
Given the contents of the present review, knowledge
of the effects of simultaneous deprivation of molecular oxygen and
AAs on lipid metabolism in cancer cells has been scarce. Therefore,
we propose three new research directions: identification of
unaddressed effects caused by depletion of both AAs and lipids,
which accelerate malignancy; exploration of cancer types that are
highly dependent on lipid metabolism when molecular oxygen and/or
nutrients are in poor supply; and identification of biomarkers for
such cancer cells and types, which may be vulnerable to lipid
metabolism-targeted therapy. Therefore, this preclinical research
field currently leaves the door open for greater understanding of
tumor biology.
In summary, we have presented a new viewpoint that
cancer cells can bypass both hypoxia and nutrient-poor tumor
conditions through various types of lipid metabolism. We expect the
information in this review to offer insights into how cancer cells
adapt to harsh hypoxia and nutrient-deficient conditions, which
partially mimic complex tumor microenvironments. Combined with
previous studies and future experimental investigations, these
efforts are likely to improve the development of new treatment
approaches for aggressive cancers, including the identification of
novel diagnostic markers and generation of new therapeutic
strategies targeting metabolic pathways characteristic to cancer
cells exposed to simultaneous insufficiency of molecular oxygen,
AAs, and/or lipids.
Acknowledgements
The authors would like to thank Dr Alison Sherwin
for editing a draft of this manuscript.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
SK conceived and wrote the manuscript. YM
contributed to drafting the manuscript. Data authentication is not
applicable. Both authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AA
|
amino acid
|
|
ACC1
|
acetyl-CoA carboxylase 1
|
|
ACLY
|
ATP citrate lyase
|
|
AD
|
arginine deficiency
|
|
AMPK
|
AMP-activated protein kinase
|
|
ASS1
|
argininosuccinate synthetase 1
|
|
ARNT
|
aryl hydrocarbon receptor nuclear
translocator
|
|
ATGL
|
adipose triglyceride lipase
|
|
ATM
|
ataxia-telangiectasia mutated
|
|
ATP
|
adenosine triphosphate
|
|
CAF
|
cancer-associated fibroblast
|
|
CBS
|
cystathionine β-synthase
|
|
ccRCC
|
clear cell renal cell carcinoma
|
|
CD
|
cysteine deficiency
|
|
CD36
|
cluster of differentiation 36
|
|
CHTM1
|
coiled-coil helix tumor and
metabolism 1
|
|
CHKα2
|
choline kinase α2
|
|
CISD3
|
CDGSH iron sulfur domain 3
|
|
c-Myc
|
MYC proto-oncogene, bHLH
transcription factor
|
|
CPT2
|
carnitine palmitoyltransferase 2
|
|
CREB
|
CRE-binding protein
|
|
EMT
|
epithelial-mesenchymal transition
|
|
ER
|
endoplasmic reticulum
|
|
FA
|
fatty acid
|
|
FABP
|
fatty acid binding protein
|
|
FAO
|
fatty acid oxidation
|
|
FAS
|
fatty acid synthase
|
|
FOXM1
|
forkhead box M1
|
|
FPN1
|
ferroportin
|
|
GD
|
glucose deficiency
|
|
GILZ
|
glucocorticoid-induced leucine
zipper
|
|
GlnD
|
glutamine deficiency
|
|
GSH
|
glutathione
|
|
GSK3β
|
glycogen synthase kinase 3β
|
|
HGD
|
hypoxia and glucose deficiency
|
|
HRD1
|
HMG-CoA reductase degradation protein
1
|
|
HIF
|
hypoxia-inducible factor
|
|
IRE1
|
inositol-requiring protein 1
|
|
LCFA
|
long-chain fatty acid
|
|
LD
|
lipid droplet
|
|
LPO
|
lipid peroxidation
|
|
LRP1
|
lipoprotein receptor-related protein
1
|
|
MD
|
methionine deficiency
|
|
MTF1
|
metal regulatory transcription factor
1
|
|
mTOR
|
mammalian target of rapamycin
|
|
NRF2
|
nuclear factor-erythroid 2-related
factor-2
|
|
OCCC
|
ovarian clear cell carcinoma
|
|
PEPCK-M
|
phosphoenolpyruvate
carboxykinase-M
|
|
PERK
|
PKR-like ER kinase
|
|
3-PG
|
3-phosphoglycerate
|
|
PGC-1α
|
peroxisome proliferator-activated
receptor γ, coactivator-1α
|
|
PGM1
|
phosphoglucomutase 1
|
|
PHGDH
|
phosphoglycerate dehydrogenase
|
|
PI3K-C2γ
|
phosphoinositide 3-kinase-C2γ
|
|
PIM1
|
proviral integration site for Moloney
murine leukemia virus 1
|
|
PKC
|
protein kinase C
|
|
PLD1
|
phospholipase D1
|
|
POX
|
proline oxidase
|
|
PPARα
|
peroxisome proliferator-activated
receptor α
|
|
Sp1
|
specificity protein 1
|
|
SREBP1
|
sterol regulatory element binding
protein-1
|
|
SREBP2
|
sterol regulatory element binding
protein-2
|
|
SSH
|
serum starvation and hypoxia
|
|
SCD5
|
stearoyl-CoA desaturase 5
|
|
SGD
|
serine/glycine deficiency
|
|
TRIAP-1
|
TP53-regulated inhibitor of apoptosis
1
|
|
UPR
|
unfolded protein response
|
|
VHL
|
von Hippel Lindau
|
References
|
1
|
Hanahan D: Hallmarks of cancer: New
dimensions. Cancer Discov. 12:31–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pavlova NN, Zhu J and Thompson CB: The
hall marks of cancer metabolism: Still emerging. Cell Metab.
34:355–377. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li F and Simon MC: Cancer Cells don't live
alone: Metabolic communication within tumor microenvironments. Dev
Cell. 54:183–195. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koizume S and Miyagi Y: Lipid droplets: A
key cellular organelle associated with cancer cell survival under
normoxia and hypoxia. Int. J Mol Sci. 17:14302016. View Article : Google Scholar
|
|
5
|
Broadfield LA, Pane AA, Talebi A, Swinnen
JV and Fendt SM: Lipid metabolism in cancer: New perspectives and
emerging mechanisms. Dev Cell. 56:1363–1393. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Riscal R, Skuli N and Simon MC: Even
cancer cells watch their cholesterol! Mol Cell. 76:220–231. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xiang W, Lv H, Xing F, Sun X, Ma Y, Wu L,
Lv G, Zong Q, Wang L, Wu Z, et al: Inhibition of ACLY overcomes
cancer immunotherapy resistance via polyunsaturated fatty acids
peroxidation and cGAS-STING activation. Sci Adv. 9:eadi24652023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vanauberg D, Schulz C and Lefebvre T:
Involvement of the pro-oncogenic enzyme fatty avid synthase in the
hallmarks of cancer: A promising target in anti-cancer therapies.
Oncogenesis. 12:162023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Freitas FP, Alborzinia H, dos Santos AF,
Nepachalovich P, Pedrera L, Zilka O, Inague A, Klein C, Aroua N,
Kaushal K, et al: 7-dehydrocholesterol is endogenous suppressor of
ferroptosis. Nature. 626:401–410. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li Y, Ran Q, Duan Q, Jin J, Wang Y, Yu L,
Wang C, Zhu Z, Chen X, Weng L, et al: 7-Dehydrocholesterol dictates
ferroptosis sensitivity. Nature. 626:411–418. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Denko NC: Hypoxia, HIF1 and glucose
metabolism in the solid tumor. Nat Rev Cancer. 8:705–713. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Macklin PS, Yamamoto A, Browning L, Hofer
M, Adam J and Pugh CW: Recent advances in the biology of tumor
hypoxia with relevance to diagnostic practice and tissue-based
research. J Pathol. 250:593–611. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haga N, Saito S, Tsukumo Y, Sakurai J,
Furuno A, Tsuruo T and Tomida A: Mitochondria regulate the unfolded
protein response leading to cancer cell survival under glucose
deprivation conditions. Cancer Sci. 101:1125–1132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kwon SJ and Lee YJ: Effect of low
glutamine/glucose on hypoxia-induced elevation of hypoxia-inducible
factor-1a in human pancreatic cancer MiaPaCa-2 and human prostatic
cancer DU-145 cells. Clin. Cancer Res. 11:4694–4700. 2005.
|
|
15
|
Kikuchi D, Tanimoto K and Nakayama K: CREB
is activated by ER stress and modulates the unfolded protein
response by regulating the expression of IRE1α and PERK. Biochem.
Biophys. Res Commun. 469:243–250. 2016.PubMed/NCBI
|
|
16
|
Wang HF, Wang ZQ, Ding Y, Piao MH, Feng
CS, Chi GF, Luo YN and Ge PF: Endoplasmic reticulum stress
regulates oxygen-glucose deprivation-induced parthanatos in human
SH-SY5Y cells via improvement of intracellular ROS. CNS Neurosci
Ther. 24:29–38. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Natsuizaka M, Ozasa M, Darmanin S,
Miyamoto M, Kondo S, Kamada S, Shindoh M, Higashino F, Suhara W,
Koide H, et al: Synergistic up-regulation of Hexokinase-2, glucose
transporters and angiogenic factors in pancreatic cancer cells by
glucose deprivation and hypoxia. Exp Cell Res. 313:3337–3348. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Keith B, Johnson RS and Simon MC: HIF1α
and HIF2α: Sibling rivalry in hypoxic tumor growth and progression.
Nat Rev Cancer. 12:9–22. 2012. View Article : Google Scholar
|
|
19
|
Wouters BG and Koritzinsky M: Hypoxia
signalling through mTOR and the unfolded protein response in
cancer. Nat Rev Cancer. 8:851–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lee P, Chandel N and Simon MC: Cellular
adaptation to hypoxia through hypoxia inducible factors and beyond.
Nat Rev Mol Cell Biol. 21:268–283. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen J, Kobayashi M, Darmanin S, Qiao Y,
Gully C, Zhao R, Kondo S, Wang H, Wang H, Yeung SC, et al:
Hypoxia-mediated up-regulation of Pim-1 contributes to solid tumor
formation. Am J Pathol. 175:400–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liu W, Glunde K, Bhujwalla ZM, Raman V,
Sharma A and Phang JM: Proline oxidase promotes tumor cell survival
in hypoxic tumor microenvironments. Cancer Res. 72:3677–3686. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen X, Ilipoulos D, Zhang Q, Tang Q,
Greenblatt MB, Hatziapostolou M, Lim E, Tam WL, Ni M, Chen Y, et
al: XBP1 promotes triple-negative breast cancer by controlling the
HIF1a pathway. Nature. 508:103–107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Huang C, Li Y, Li Z, Xu Y, Li N, Ge Y,
Dong J, Chang A, Zhao T, Wang X, et al: LIMS1 promotes pancreatic
cancer cell survival under oxygen-glucose deprivation conditions by
enhancing HIF1A protein translation. Clin Cancer Res. 25:4091–4103.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Saggese P, Pandey A, Alcaraz M, Fung E,
Hall A, Yanagawa J, Rodriguez EF, Grogan TR, Giurato G, Nassa G, et
al: Glucose deprivation promotes pseudohypoxia and
de-differentiation in lung adenocarcinoma. Cancer Res. 84:305–327.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhao T, Jiang T, Li X, Chang S, Sun Q,
Kong F, Kong X, Wei F, He J, Hao J, et al: Nuclear GRP78 promotes
metabolic reprogramming and therapeutic resistance in pancreatic
ductal adenocarcinoma. Clin Cancer Res. 29:5183–5195. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nishimoto A, Kugiyama N, Hosoyama T, Enoki
T, Li TS and Hamano K: HIF-1α activation under glucose deprivation
plays a central role in the acquisition of anti-apoptosis in human
colon cancer cells. Int J Oncol. 44:2077–2084. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Takai M, Takauchi M, Kuribayashi M and
Tsujiuchi T: LPA receptor-mediated signaling regulates cell
motility and survival to anticancer drug of pancreatic cancer cells
under glucose-deprived and hypoxic conditions. Biochem Biophys Res
Commun. 661:21–27. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Davern M, Fitzgerald MC, Buckley CE,
Heeran AB, Donlon NE, McGrath J, O'Connel F, Deshpande MR, Hayes C,
MacDonald J, et al: PD-1 and TIGIT blockade differentially affect
tumour cell survival under hypoxia and glucose deprived conditions
in oesophageal adenocarcinoma; implications for overcoming
resistance to PD-1 blockade in hypoxic tumors. Transl Oncol.
19:1013812022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Buzzai M, Bauer DE, Jones RG, DeBerardinis
RJ, Hatzivassiliou G, Elstrom RL and Tompson CB: The glucose
dependence of Akt-transformed cells can be reversed by
pharmacologic activation of fatty acid β-oxidation. Oncogene.
24:4165–4173. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leithner K, Triebl A, Trotzmuller M,
Hinteregger B, Leko P, Wieser BI, Grasmann G, Bertsch AL, Züllig T,
Stacher E, et al: The glycerol backbone of phospholipids derives
from noncarbohydrate precursors in staved lung cancer cells. Proc
Natl Acad Sci USA. 115:6225–6230. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cai M, He J, Xiong J, Tay LWR, Wang Z, Rog
C, Wang J, Xie Y, Wang G, Banno Y, et al: Phospholipase
D1-regulated autophagy supplies free fatty acids to counter
nutrient stress in cancer cells. Cell Death Dis. 7:e24482016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Khan AUH, Salehi H, Alexia C, Valdivielso
JM, Bozic M, Lopez-Mejia IC, Fajas L, Gerbal-Chaloin S,
DaujatChavanieu M, Gitenay D, et al: Glucose starvation or pyruvate
dehydrogenase activation induce a broad, ERK5-mediated, metabolic
remodeling leading to fatty acid oxidation. Cells. 11:13922022.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hoang-Minh LB, Siebzehnrubl FA, Yang C,
Suzuki-Hatano S, Dajac K, Loche T, Andrews N, Massari MS, Patel J,
Amin K, et al: Infiltrative and drug-resistant slow-cycling cells
support metabolic heterogeneity in glioblastoma. EMBO J.
37:e987722018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang C, Haas MA, Yeo SK, Paul R, Yang F,
Vallabhapurapu S, Qi X, Plas DR and Guan JL: Autophagy mediated
lipid catabolism facilitates glioma progression to overcome
bioenergetic crisis. Br J Cancer. 124:1711–1723. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu R, Lee JH, Li J, Yu R, Tan L, Xia Y,
Zheng Y, Bian XL, Lorenzi PL, Chen Q, et al: Choline kinase alpha 2
acts as a protein kinase to promote lipolysis of lipid droplets.
Mol Cell. 81:2722–2735. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chauhan SS, Casillas AL, Vizzerra AD, Liou
H, Clements AN, Flores CE, Prevost CT, Kashatus DF, Snider AJ,
Snider JM, et al: PIM1 drives lipid droplet accumulation to promote
proliferation and survival in prostate cancer. Oncogene.
43:406–419. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Cao B, Deng H, Cui H, Zhao R, Li H, Wei B
and Chen L: Knockdown of PGM1 enhances anticancer effects of
orlistat in gastric cancer under glucose deprivation. Cancer Cell
Int. 21:4812021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Monteiro-Cardoso VF, Silva AM, Oliveira
MM, Peixoto F and Videira RA: Membrane lipid profile alterations
are associated with the metabolic adaptation of the Caco-2 cells to
a glycemic nutritional condition. J Bioenerg Biomembr. 46:45–57.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spencer AG, Woods JW, Arakawa T, Singer II
and Smith WI: Subcellular localization of prostaglandin
endoperoxide H synthase-1 and −2 by immunoelectron microscopy. J
Biol Chem. 273:9886–9893. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Roberts HR, Smartt HJM, Greenhough A,
Moore AE, Williams AC and Paraskeva C: Colon tumor cells increase
PGE2 by regulating COX-2 and 15-PGDH to promote survival during the
microenvironmental stress of glucose deprivation. Carcinogenesis.
32:1741–1747. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hwang SH, Yang Y, Jung JH and Kim Y: Oleic
acid from cancer-associated fibroblast promotes cancer cell
stemness by stearoyl-CoA desaturase under glucose-deficient
condition. Cancer Cell Int. 22:4042022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kamphorst JJ, Nofal M, Commisso C, Hackett
SR, Lu W, Grabocka E, Heiden MGV, Miller G, Drebin JA, Bar-Sagi D,
et al: Human pancreatic cancer tumors are nutrient poor and tumor
cells actively scavenge extracellular protein. Cancer Res.
75:544–553. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gameiro PA, Yang J, Metelo AM, Perez-Carro
R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, López-Larrubia
P, et al: In vivo HIF-mediated reductive carboxylation is regulated
by citrate levels and sensitizes VHL-deficient cells to glutamine
deprivation. Cell Metab. 17:372–385. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kamphorst JJ, Cross JR, Fan J, de
Stanchina E, Mathew R, White EP, Thompson CB and Rabinowitz JD:
Hypoxic and Ras-transformed cells support growth by scavenging
unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci
USA. 110:8882–8887. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Miess H, Dankworth B, Gouw AM, Rosenfeldt
M, Schmitz W, Jiang M, Saunders B, Howell M, Downward J, Felsher
DW, et al: The glutathione redox system is essential to prevent
ferroptosis caused by impaired lipid metabolism in clear cell renal
cell carcinoma. Oncogene. 37:5435–5450. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Byun JK, Choi YK, Kim JH, Jeong JY, Jeon
HJ, Kim MK, Hwang I, Lee SY, Lee YM, Lee IK, et al: A positive
feedback loop between Sestrin 2 and mTORC2 is required for the
survival of glutamine-depleted lung cancer cells. Cell Rep.
20:586–599. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Babbar M, Huang Y, An J, Landas SK and
Sheikh MS: CHTM1, a novel metabolic marker deregulated in human
malignancies. Oncogene. 37:2052–2066. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Juh JW, Yan JB, Lin ZH, Lin SC and Peng
IC: SREBP1-induced glutamine synthetase triggers a feedforward loop
to upregulate SREBP1 through Sp1 O-GlcNAcylation and augments lipid
droplet formation in cancer cells. Int J Mol Sci. 22:98142021.
View Article : Google Scholar
|
|
50
|
Zhu R, Yang Y, Shao F, Wang J, Gao Y, He J
and Lu Z: Choline kinase alpha2 promotes lipid droplet lipolysis in
non-small-cell lung carcinoma. Front Oncol. 22:8484832022.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Guo X, Wang A, Wang W, Wang Y, Chen H, Liu
X, Xia T, Zhang A, Chen D, Qi H, et al: HRD1 inhibits fatty acid
oxidation and tumorigenesis by ubiquitinating CPT2 in triple
negative breast cancer. Mol Oncol. 15:642–656. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
De Santis MC, Gozzelino L, Margaria JP,
Costamagna A, Ratto E, Gulluni F, Gregorio ED, Mina E, Lorito N,
Bacci M, et al: Lysosomal lipid switch sensitises to nutrient
deprivation and mTOR targeting in pancreatic cancer. Gut.
72:360–371. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kong Y, Wu M, Wan X, Sun M, Zhang Y, Wu Z,
Li C, Liang X, Gao L, Ma C, et al: Lipophagy-mediated cholesterol
synthesis inhibition is required for the survival of hepatocellular
carcinoma under glutamine deprivation. Redox Biol. 63:1027322023.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nedara K, Reinhardt C, Lebraud E, Arena G,
Gracia C, Buard V, Pioche-Durieu C, Castelli F, Colsch B, Bénit P,
et al: Relevance of the TRIAP1/p53 axis in colon cancer cell
proliferation and adaptation to glutamine depletion. Front Oncol.
12:9581552022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lorenz NI, Sittig ACM, Urban H, Luger AL,
Engel AL, Munch C, Steinbach JP and Ronellenfitsch MW: Activating
transcription factor 4 mediates adaptation of human glioblastoma
cells to hypoxia and temozolomide. Sci Rep. 11:141612021.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Pan M, Reid MA, Lowman XH, Kulkarni RP,
Tran TQ, Liu X, Yang Y, Hernandez-Davies JE, Rosales KK, Li H, et
al: Regional glutamine deficiency in tumors promotes
dedifferentiation through inhibition of histone demethylation. Nat
Cell Biol. 18:1090–1101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jain M, Nilsson R, Sharma S, Madhusudhan
N, Kiytami T, Souza AL, Kafri R, Kirschner MW, Clish CB and Mootha
VK: Metabolite profiling identifies a key role for glycine in rapid
cancer cell proliferation. Science. 336:1040–1044. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gao X, Lee K, Reid MA, Sanderson SM, Qiu
C, Li S, Liu J and Locasale JW: Serine availability influences
mitochondrial dynamics and function through lipid metabolism. Cell
Rep. 22:3507–3520. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Maddocks OD, Berkers CR, Mason SM, Zheng
L, Blyth K, Gottlieb E and Vousden KH: Serine starvation induces
stress and p53-dependent metabolic remodelling in cancer cells.
Nature. 493:542–546. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Muthusamy T, Cordes T, Handzlik MK, You L,
Lim EW, Gengatharan J, Pinto AFM, Badur MG, Kolar MJ, Wallace M, et
al: Serine restriction alters sphingolipid diversity to constrain
tumour growth. Nature. 586:790–795. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Truman JP, Ruiz CF, Montal E,
Garcia-Barros M, Mileva I, Snider AJ, Hannun YA, Obeid LM and Mao
C: 1-Deoxysphinganine initiates adaptive responses to serine and
glycine starvation in cancer cells via proteolysis of sphingosine
kinase. J Lipid Res. 63:1001542022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yun HJ, Li M, Guo D, Jeon SM, Park SH, Lim
JS, Lee SB, Liu R, Du L, Kim SH, et al: AMPK-HIF-1α signaling
enhances glucose-derived de novo serine biosynthesis to promote
glioblastoma growth. J Exp Clin Cancer Res. 42:3402023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Bonifacio VDB, Pereira SA, Serpa J and
Vicente JB: Cysteine metabolic circuitries: Druggable targets in
cancer. Br J Cancer. 124:862–879. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cunningham A, Oudejans LL, Geugien M,
Pereira-Martins DA, Wierenga ATJ, Erdem A, Sternadt D, Huls G and
Schuringa JJ: The nonessential amino acid cysteine is required to
prevent ferroptosis in acute myeloid leukemia. Blood Adv. 8:56–69.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Upadhyayula PS, Higgins DM, Mela A, Banu
M, Dovas A, Zandkarimi F, Patel P, Mahajan A, Humala N, Nguyen TTT,
et al: Dietary restriction of cysteine and methionine sensitizes
gliomas to ferroptosis and induces alterations in energetic
metabolism. Nature Commun. 14:11872023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kerimoglu B, Lamb C, McPherson RD, Ergen
E, Stone EM and Ooi A: Cysteinase-rapamycin combination induces
ferroptosis in both in vitro and in vivo models of hereditary
leiomyomatosis and renal cell carcinoma. Mol Caner Ther.
21:419–426. 2022. View Article : Google Scholar
|
|
67
|
Liu N, Lin X and Huang C: Activation of
the reverse transsulfuration pathway through NRF2/CBS confers
erastin-induced ferroptosis resistance. Br J Cancer. 122:279–92.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Armenta DA, Laqtom NN, Alchemy G, Dong W,
Morrow D, Poltorack CD, Nathanson DA, Abu-Remaileh M and Dixon S:
Ferroptosis inhibition by lysosome-dependent catabolism of
extracellular protein. Cell Chem Biol. 29:1588–1600. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Chen PH, Wu J, Ding CKC, Lin CC, Pan S,
Bossa N, Xu Y, Yang WH, Mathey-Prevot B and Chi JT: Kinome screen
of ferroptosis reveals a novel role of ATM in regulating iron
metabolism. Cell Death Differ. 27:1008–1022. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Li Y, Wang X, Huang Z, Zhou Y, Xia J, Hu
W, Wang X, Du J, Tong X and Wang Y: CISD3 inhibition drives
cysteine-deprivation induced ferroptosis. Cell Death Dis.
12:8392021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Liu D, Liang C, Huang B, Zhuang X, Cui W,
Yang L, Yang Y, Zhang Y, Fu X, Zhang X, et al: Tryptophan
metabolism acts as a new anti-ferroptotic pathway to mediate tumor
growth. Adv Sci. 10:22040062023. View Article : Google Scholar
|
|
72
|
Hong SE, Kim MR, Jang SK, Seong MK, Kim
HA, Noh WC, Jin HO and Park IC: Hypoxia suppresses cysteine
deprivation-induced cell death via ATF4 regulation in MDA-MB-231
breast cancer cells. Anticancer Res. 40:1387–1394. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Qiu F, Chen YR, Liu X, Chu CY, Shen LJ, Xu
J, Gaur S, Forman HJ, Zhang H, Zheng S, et al: Arginine starvation
impairs mitochondrial respiratory function in ASS1-deficient breast
cancer cells. Sci Signal. 7:ra312014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hu Q, Dai J, Zhang Z, Yu H, Zhang J, Zhu
X, Qin Y, Zhang L and Zhang P: ASS1-mediated reductive
carboxylation of cytosolic glutamine confers ferroptosis resistance
in cancer cells. Cancer Res. 83:1646–1665. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Brashears CB, Barlin M, Ehrhardt WR,
Rathore R, Schultze M, Tzeng SC, Tine BAV and Held JM: Systems
level profiling of arginine starvation reveals MYC and ERK adaptive
metabolic reprogramming. Cell Death Dis. 11:6622020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Long Y, Tsai WB, Wangpaichitr M, Tsukamoto
T, Savaraj N, Feun LG and Kuo MT: Arginine deiminase resistance in
melanoma cells is associated with metabolic reprogramming, glucose
dependence, and glutamine addiction. Mol Cancer Ther. 12:2581–2590.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Burrows N, Cane G, Robson M, Gaude E,
Howat WJ, Szlosarek PW, Pedley RB, Frezza C, Ashcroft M and Maxwell
PH: Hypoxia-induced nitric oxide production and tumour perfusion is
inhibited by pegylated arginine deiminase (ADI-PEG20). Sci Rep.
6:229502016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Guenin S, Morvan D, Thivat E, Stepien G
and Demidem A: Combined methionine deprivation and
chloroethylnitrosourea have time-dependent therapeutic synergy on
melanoma tumors that NMR spectroscopy-based metabolomics explains
by methionine and phospholipid metabolism reprogramming. Nutr
Cancer. 61:518–529. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Harada S, Taketomi Y, Aiba T, Kawaguchi M,
Hirabayashi T, Uranbileg B, Kurano M, Yatomi Y and Murakami M: The
lisophospholipase PNPLA7 controls hepatic choline and methionine
metabolism. Biomolecules. 13:4172023. View Article : Google Scholar
|
|
80
|
Yokogami K, Kikuchi T, Watanabe T,
Nakatake Y, Yamashita S, Mizuguchi A and Takeshima H: Methionine
regulates self-renewal pluripotency, and cell death of GIC through
cholesterol-rRNA axis. BMC Cancer. 22:13512022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Young RM, Ackerman D, Quinn ZL, Mancuso A,
Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA,
Keith B, et al: Dysregulated mTORC1 renders cells critically
dependent on desaturated lipids for survival under tumor-like
stress. Genes Dev. 27:1115–1131. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li Z, Ji BW, Dixit PD, Tchourine K, Lien
EC, Hosios AM, Abbott KL, Rutter JC, Westermark AM, Gorodetsky EF,
et al: Cancer cells depend on environmental lipids for
proliferation when electron acceptors are limited. Nat Metab.
4:711–723. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Qiu B, Ackermann D, Sanchez DJ, Li B,
Ochocki JD, Grazioli A, Bobrovnikova-Marjon E, Diehl A, Keith B and
Simon C: HIF2α-dependent lipid storage promotes endoplasmic
reticulum homeostasis in clear-cell renal cell carcinoma. Cancer
Discov. 5:652–667. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Du W, Zhang L, Brett-Morris A, Aguila B,
Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, et
al: HIF drives lipid deposition and cancer in ccRCC via repression
of fatty acid metabolism. Nat Commun. 8:17692017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen C, Zhao W, Lu X, Ma Y, Zhang P, Wang
Z, Cui Z and Xia Q: AUP1 regulates lipid metabolism and induces
lipid accumulation to accelerate the progression of renal clear
cell carcinoma. Cancer Sci. 113:2600–2615. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Brezis M and Rosen S: Hypoxia of the renal
medulla-Its implications for disease. N Eng J Med. 332:647–655.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Little RA, Jamin Y, Boult JKR, Naish JH,
Watson Y, Cheung S, Holliday KF, Lu H, McHugh DJ, Irlam J, et al:
Mapping hypoxia in renal carcinoma with oxygen-enhanced MRI.
Radiology. 288:739–747. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Ackerman D, Tumanov S, Qiu B,
Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC and Kamphorst
JJ: Triglycerides promote lipid homeostasis during hypoxic stress
by balancing fatty acid saturation. Cell Rep. 24:2596–2605. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ji JX, Wang YK, Cochrane DR and Huntsman
DG: Clear cell carcinomas of the ovary and kidney: Clarity through
genomics. J Pathol. 244:550–564. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lee S, Garner EIO, Welch WR, Berkowitz RS
and Mok SC: Over-expression of hypoxia-inducible factor 1 alpha in
ovarian clear cell carcinoma. Gynecol Oncol. 106:311–317. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Anglesio MS, George J, Kulbe H,
Friedlander M, Rischin D, Lemech C, Power J, Coward J, Cowin PA,
House CM, et al: IL6-STAT3-HIF signaling and therapeutic response
to the angiogenesis inhibitor sunitinib in ovarian clear cell
carcinoma. Clin Cancer Res. 17:2538–2548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Spowart JE, Townsend KN, Huwait H, Eshragh
S, West NR, Ries JN, Kalloger S, Anglesio M, Gorski SM, Watson PH,
et al: The autophagy protein LC3A correlates with hypoxia and is a
prognostic marker of patient survival in clear cell ovarian cancer.
J Pathol. 228:437–447. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Koizume S, Ito S, Miyagi E, Hirahara F,
Nakamura Y, Sakuma Y, Osaka H, Takano Y, Ruf W and Miyagi Y:
HIF2α-Sp1 interaction mediates a deacetylation-dependent FVII-gene
activation under hypoxic condition in ovarian cancer cells. Nucleic
Acid Res. 40:5389–5401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Koizume S and Miyagi Y: Tissue factor in
cancer-associated thromboembolism: Possible mechanisms and clinical
applications. Br J Cancer. 127:2099–2107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Koizume S, Ito S, Nakamura Y, Yoshihara M,
Furuya M, Yamada R, Miyagi E, Hirahara F, Takano Y and Miyagi Y:
Lipid starvation and hypoxia synergistically activate ICAM1 and
multiple genes in an Sp1-dependent manner to promote the growth of
ovarian cancer. Mol Cancer. 14:772015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Koizume S, Kanayama T, Kimura Y, Hirano H,
Takahashi T, Ota Y, Miyazaki K, Yoshihara M, Nakamura Y, Yokose T,
et al: Cancer cell-derived CD69 induced under lipid and oxygen
starvation promotes ovarian cancer progression through fibronectin.
Cancer Sci. 114:2485–2498. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Trigatti BL and Gerber GE: A direct role
for serum albumin in the cellular uptake of long-chain fatty acids.
Biochem. J. 308:155–159. 1995.PubMed/NCBI
|
|
98
|
Koizume S, Takahashi T, Nakamura Y,
Yoshihara M, Ota Y, Sato S, Tadokoro H, Yokose T, Kato H, Miyagi E,
et al: Lipophagy-ICAM-1 pathway associated with fatty acid and
oxygen deficiencies is involved in poor prognoses of ovarian clear
cell carcinoma. Br J Cancer. 127:462–473. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Koizume S, Takahashi T, Yoshihara M,
Nakamura Y, Ruf W, Takenaka K, Miyagi E and Miyagi Y: Cholesterol
starvation and hypoxia activate the FVII gene via the SREBP1-GILZ
pathway in ovarian cancer cells to produce procoagulant
microvesicles. Thromb Haemost. 119:1058–1071. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lewis CA, Peck B, Bensaad K, Griffiths B,
Mitter R, Chakravarty P, East P, Dankworth B, Alibhai D, Harris AL
and Schulze A: SREBP maintains lipid biosynthesis and viability of
cancer cells under lipid- and oxygen-deprived conditions and
defines a gene signature associated with poor survival in
glioblastoma multiforme. Oncogene. 34:5128–5140. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Schug ZT, Peck B, Jones DT, Zhang Q,
Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K,
et al: Acetyl-CoA synthetase 2 promotes acetate utilization and
maintains cancer cell growth under metabolic stress. Cancer Cell.
27:57–71. 2015. View Article : Google Scholar : PubMed/NCBI
|