Introduction
Chronic obstructive pulmonary disease (COPD) is the
third most common cause of mortality globally and is associated
with a high burden of disease (1).
The pathogenesis of COPD is complex, involving genetic factors, air
pollution and other factors such as oxidative stress, immunologic
factors, systemic inflammation, cell senescence, apoptosis and
autophagy (2,3). Cigarette smoke (CS) is a preventable
cause of COPD (4). It is estimated
that 20–25% of smokers eventually develop COPD (5). A single puff of CS is composed of
thousands of chemicals, including polycyclic aromatic hydrocarbons
(6). Furthermore, it also contains
4,000-7,000 different types of oxidant compounds (7). These oxides and free radicals can
elevate the levels of oxidative stress, inactivate the antioxidant
defense ability, and cause lung and systemic oxidative stress
(8). Through chemical modification
of proteins, lipids, carbohydrates and nucleic acids, oxidative
distress causes damage to these macromolecules, thereby impairing
their function (9).
Oxidative stress is a key factor triggering the
occurrence of COPD and its progression, as well as amplifying acute
exacerbations (10). Increased
oxidative stress in the lungs of patients with COPD leads to
chronic inflammation by activating intracellular signaling
pathways, reducing anti-inflammatory effects of corticosteroids,
accelerating cellular senescence and lung ageing, inducing
autoimmunity based on autoantibodies to carbonylated proteins, and
damaging DNA (10,11). Oxidative stress ultimately leads to
emphysema by reducing the activity of antiproteases, and promoting
extracellular matrix proteolysis, small airway fibrosis and mucus
hypersecretion (10). Therefore,
antioxidant therapy is a potential approach for the treatment of
COPD.
N-acetyl-L-cysteine (NAC) is considered a
well-tolerated and safe drug that has been used for decades for the
treatment of various medical conditions such as
ischemia-reperfusion injury in brain and lethal endotoxemia
(12). NAC is a potent antioxidant
and is capable of reducing oxidants (13). There are three potential
explanations for the mechanism of NAC: i) Breaking disulfide bonds;
ii) direct antioxidant activity [scavenging reactive oxygen species
(ROS)]; and iii) indirect antioxidant activity [replenishing
depleted glutathione (GSH)] (13,14).
Although a number of studies have revealed that NAC already has a
well-defined mechanism of action (15–17),
other studies have revealed that that the mechanism of action of
NAC is still unclear and remains to be understood (13,14,18).
Thus, the present study aimed to observe the effect of CS in the
lungs of rats and rat alveolar epithelial cells by establishing a
rat model of COPD and a CS extract (CSE)-induced cell injury model.
The present study aimed to determine whether CS could induce
oxidative stress in alveolar epithelial cells and in rat lungs and
induce apoptosis. The present study also aimed to determine whether
NAC could protect against oxidative injury by replenishing depleted
intracellular GSH.
Materials and methods
Reagents and antibodies
NAC, 2′,7′-dichlorofluorescin diacetate (DCFH-DA)
and annexin V-FITC apoptosis detection kits were purchased from
Sigma-Aldrich; Merck KGaA. The GSH and GSH disulfide (GSSG)
detection kits were purchased from Beyotime Institute of
Biotechnology. Cigarettes were purchased from Gansu Tobacco
Industrial Co., Ltd. ELISA kits for malondialdehyde (MDA) (cat. no.
SP30131) and superoxide dismutase (SOD) (Rat SOD ELISA Kit,
SP12914) were purchased from Wuhan Saipei Biotechnology Co., Ltd.
The Rabbit Streptavidin-Peroxidase Immunohistochemical staining kit
(SP assay kit) and the 3,3-diaminobenzidine (DAB) detection kit
were ordered from Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.
The rabbit anti-8-oxo-7,8-dihydro-2′-deoxyguanosine
(8-OHdG) (bs1278R) and rabbit anti-caspase3 p12 (bs0087R)
antibodies were purchased from Beijing Biosynthesis Biotechnology
Co., Ltd. Other primary antibodies, including heme oxygenase-1
(HO-1) (ab68477), p53 (cat. no. ab26), BAX (ab32503), Bcl-2
(ab59348) and β-actin (ab8227) antibodies, were purchased from
Abcam. Other products such as RIPA Lysis Buffer, BCA protein assay
kits and phenylmethylsulfonyl fluoride were purchased from Beijing
Solarbio Science & Technology Co., Ltd.
Animal models
A total of 32 male 8-week-old Wistar rats weighing
~220 g were purchased from Experimental Animal Center of Lanzhou
University (Lanzhou, China). All procedures and animal experiments
were carried out in accordance with the protocol approved by the
Ethics Committee of the Institutional Animal Care of Lanzhou
University (Lzujcyxy20240106; Lanzhou, China). The animals were
provided with food and water ad libitum and were kept at
22–25°C and 50–60% relative humidity under a 12-h light/dark cycle.
The rats were randomly assigned to the following four groups with
eight rats per group: i) Control; ii) CS-exposed group; iii) NAC
treatment group; and iv) CS + NAC-exposed group. CS and CS + NAC
rats were exposed to CS for 4 weeks using 10 cigarettes twice a day
with a 4–6 h interval, 5 days a week. The rats in the NAC group and
in the CS+ NAC group were administered NAC dissolved in 0.9% saline
solution intragastrically at a dose of 150
mg/kg−1/day−1. Body weight was recorded every
week. Finally, rats were anesthetized with 1% pentobarbital sodium
at a dose of 40 mg/kg by intraperitoneal injection. Subsequently,
the rats were sacrificed by exsanguination (~3 ml blood was
collected simultaneously) whilst still under anesthesia, and the
lungs were removed immediately following euthanasia. Serum was
separated by centrifugation at 670 × g for 10 min at 4°C and stored
at −80°C for biochemical assays. A portion of the lungs was
snap-frozen at ~-196°C in liquid nitrogen for western blotting,
while another portion was fixed in 4% paraformaldehyde at room
temperature for 24 h for subsequent evaluation of histopathological
changes.
ELISA
Serum samples were immediately separated by
centrifugation at 670 × g for 10 min at 4°C. Serum levels of MDA
and SOD were detected using highly sensitive rat ELISA kits. The
assay was performed according to the manufacturer's instructions,
and absorbance was measured at 450 nm using a microplate reader
(BIO-RAD680; Bio-Rad Laboratories, Inc.).
H&E staining
Following dehydration with ascending ethanol
solution, xylene was used to remove the ethanol, and lung tissues
were embedded in paraffin wax and cut into 5-µm-thick sections.
These sections were finally stained with hematoxylin and eosin at
room temperature for 5 min for histopathological observation under
a light microscope (magnification, ×100).
Immunohistochemistry
Lung tissues were fixed overnight in 4%
paraformaldehyde solution at room temperature, dehydrated using
ethanol, embedded in paraffin and sectioned (5 µm) using a
microtome. The tissue sections were deparaffinized in xylene and
rehydrated using descending concentrations of ethanol solutions
(100, 95 and 75% diluted in water). Antigen retrieval was carried
out by immersing slides in sodium citrate buffer and heating at
95°C for 5 min. Slides were then incubated with 3% hydrogen
peroxide solution for 10 min and blocked in 10% normal goat serum
(Beijing Solarbio Science & Technology Co., Ltd.) for 10–15 min
at room temperature. Next, the primary antibodies, 8-OHdG (1:200)
and caspase-3 p12 (1:300), were applied to the slides, and slides
were incubated overnight at 4°C, and then rinsed three times for 3
min each in PBS. Subsequently, 100 µl biotin-labeled secondary
antibody (SP-9001, Beijing Zhongshan Golden Bridge Biotechnology
Co., Ltd.) was applied for 15 min at room temperature and then
slides were rinsed three times for 3 min each in PBS. Furthermore,
100 µl horseradish peroxidase-labeled streptavidin (cat. no.
ZB-2402, 1:300, Beijing Zhongshan Golden Bridge Biotechnology Co.,
Ltd.) working solution was incubated at room temperature for 15
min, and then sections were rinsed three times for 3 min each in
PBS. DAB was used as a chromogen and the slides were counterstained
with hematoxylin for 5 min at room temperature. After dehydration
and clearing, the slides were mounted using neutral gum on
coverslips. The slides were observed under a light microscope, and
images were analyzed using ImageJ software (version 1.52a; National
Institutes of Health).
Cell culture and treatment
Rat lung epithelial-6-T-antigen negative (RLE-6TN)
cells were purchased from Central South University (Changsha,
China) and cultured in Hyclone RPMI 1640 (Cytiva) with 10% fetal
bovine serum (Biological Industries; Sartorius AG) at 37°C in
humidified air containing 5% CO2. CS from three
cigarettes was dissolved in 20 ml RPMI 1640. After being adjusted
to pH 7.4 and filtered using a 0.22-µm filter to remove bacteria,
the solution was regarded as 100% CSE. The cells were then treated
with control, 5% CSE and 5% CSE + NAC (1 mM) for 24 h at 37°C. A
solution of 5% CSE was prepared (1:20; 1 part stock solution and 19
parts RPMI 1640 to obtain a total volume of diluted solution equal
to 20 times that of the stock solution).
Detection of ROS levels
Measurement of intracellular ROS levels in RLE-6TN
cells was based on the ROS-mediated conversion of non-fluorescent
DCFH-DA into fluorescent DCF. Briefly, RLE-6TN cells were treated
as aforementioned, then washed three times with PBS and incubated
in 10 µM DCFH-DA for 30 min at 37°C in the dark. Cells were
harvested by trypsinization, washed again with PBS and resuspended
in 1 ml PBS. The fluorescence intensity was measured by flow
cytometry (Epics XL; Beckman Coulter, Inc.) and analyzed using
FlowJo™ Software v10 (Becton, Dickinson & Company).
GSH assay
To detect the cellular GSH content, GSH and GSSG
detection kits (Beyotime Institute of Biotechnology) were used.
Briefly, fresh cells were digested with trypsin-EDTA and
centrifuged for precipitation at 167 × g for 5 min at 4°C. Reagent
M solution at a volume of three times the pellet was added to
remove protein and samples were vortexed vigorously. Next, two
rapid freeze-thaws using liquid nitrogen were carried out using a
37°C water bath. After incubation at 4°C for 10 min, the samples
were centrifuged at 10,000 × g for 10 min at 4°C. The supernatant
was removed and kept at 4°C for total GSH analysis. Then, 1/5
volume diluted GSH Removal Buffer was added to sample lysate and
samples were mixed immediately by vortexing. Subsequently, 1/25
volume GSH removal reagent working solution was added and samples
were mixed immediately by vortexing. The mixture was incubated at
25°C for 60 min and GSH was removed. Reactions were prepared in
96-well plates by mixing samples with 150 µl total GSH assay
working solution, which were incubated at room temperature for 5
min. Subsequently, 50 µl of 0.5 mg/ml NADPH solution was added per
well and samples were mixed by pipetting. Measurements were
performed 25 min after the NADPH solution was added. Based on the
A412, total GSH or GSSG content was determined from the
standard curve, which was plotted with different concentrations of
GSSG. The GSH content was determined by subtracting the GSSG
content from the total GSH content as follows: GSH=total GSH-GSSG
×2.
Apoptosis analysis
Apoptosis was detected using the annexin V-FITC
apoptosis detection kit. Briefly, cells were seeded in a 6-well
plate at a density of 1×105 cells/well for 24 h and then
the cells were treated as aforementioned. Subsequently, cells were
harvested as aforementioned and washed twice with Dulbecco's
Phosphate-Buffered Saline). The cells were resuspended in 1X
binding buffer at a concentration of 1×106 cells/ml, and
5 µl annexin V-FITC conjugate and 10 µl propidium iodide solution
were added to the cell suspension. After a 10-min incubation at
room temperature, the cells were analyzed by flow cytometry (Epics
XL; Beckman Coulter, Inc.) and analyzed using FlowJo™ Software v10
(Becton, Dickinson & Company).
Western blotting
Total protein from lung tissue or RLE-6TN cells was
isolated using RIPA Lysis Buffer with 1% protease inhibitor. The
protein concentration of each group was determined using BCA
protein assay kits. The samples were separated by 12% SDS-PAGE
loaded as 40 µg/ml per lane and transferred to a methanol-activated
PVDF membrane. The membrane was blocked with 5% BSA (Beijing
Solarbio Science & Technology Co., Ltd) in TBS with 0.1% Tween
20 (TBST) for 1.5 h at room temperature and subsequently incubated
with primary antibodies overnight at 4°C. The primary antibodies
were: HO-1 (1:10,000; Abcam), anti-p53 (1:200; Abcam), anti-Bcl2
(1:500; Abcam), anti-Bax (1:1,000; Abcam), anti-β-actin (1:5,000;
Abcam), anti-β-tubulin (1:5,000; Abcam) and anti-GAPDH (1:10,000;
ImmunoWay Biotechnology Company). Membranes were washed three times
for 8 min each with TBST, incubated with DyLight 800 Labeled
secondary antibody (1:5,000, KPL, Inc.) at room temperature for 1.5
h, washed three times for 8 min each and bands were detected using
an Odyssey Infrared Imaging System (LI-COR Biosciences) and
analyzed with ImageJ software version 1.50i (National Institutes of
Health, Bethesda, Maryland, USA).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from RLE-6TN cells using
RNAiso Plus (Takara Bio, Inc.) and reverse transcription was
carried out using a PrimeScript™ RT reagent Kit with gDNA Eraser
(Takara Bio, Inc.) according to the manufacturer's instructions.
qPCR was conducted using TB Green® Premix Ex Taq II
(Takara Bio, Inc.). The thermocycling conditions were as follows:
Initial denaturation at 95°C for 30 sec, followed by 50 cycles of
95°C for 5 sec and 60°C for 34 sec. β-actin was used as the
internal control and the expression levels of the target gene were
calculated using the 2−ΔΔCq method (19). The primers used were: β-actin
forward, 5′-GGAGATTACTGCCCTGGCTCCTA-3′ and reverse,
5′-GACTCATCGTACTCCTGCTTGCTG-3′; and HO-1 forward,
5′-AGGTGCACATCCGTGCAGAG-3′ and reverse,
5′-CTTCCAGGGCCGTATAGATATGGTA-3′.
Statistical analysis
Statistical analysis was carried out using SPSS
(version 22; IBM Corp.). The experimental data are presented as the
mean ± SD of ≥3 independent experiments. Statistical significance
was determined using one-way analysis of variance and comparisons
among groups were performed using Fisher's Least Significant
Difference test or the Bonferroni test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Body weight changes of Wistar rats
during the experimental procedure
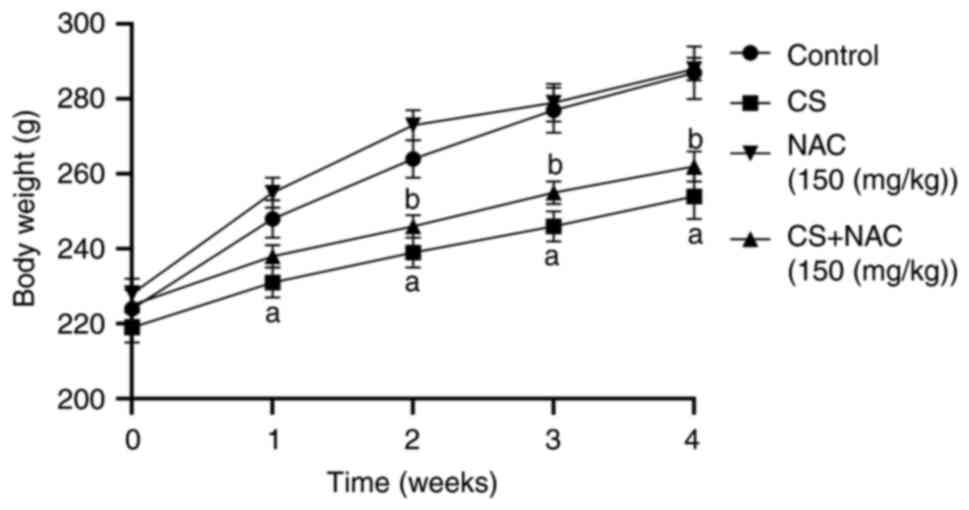
The body weight of the rats is shown in Fig. 1. The initial body weight was not
significantly different among the three groups. Rats treated with
normal saline exhibited the highest body weight gain. The weight of
the CS group was decreased compared with that of the control group
at each week (P<0.01). Compared with the CS group, the CS + NAC
group exhibited an increase in body weight from the end of week 2
to week 4 (P<0.05). These results indicated that NAC intake may
protect rats from CS-induced weight reduction.
Histopathological alterations of rat
lung tissue exposed to CSE
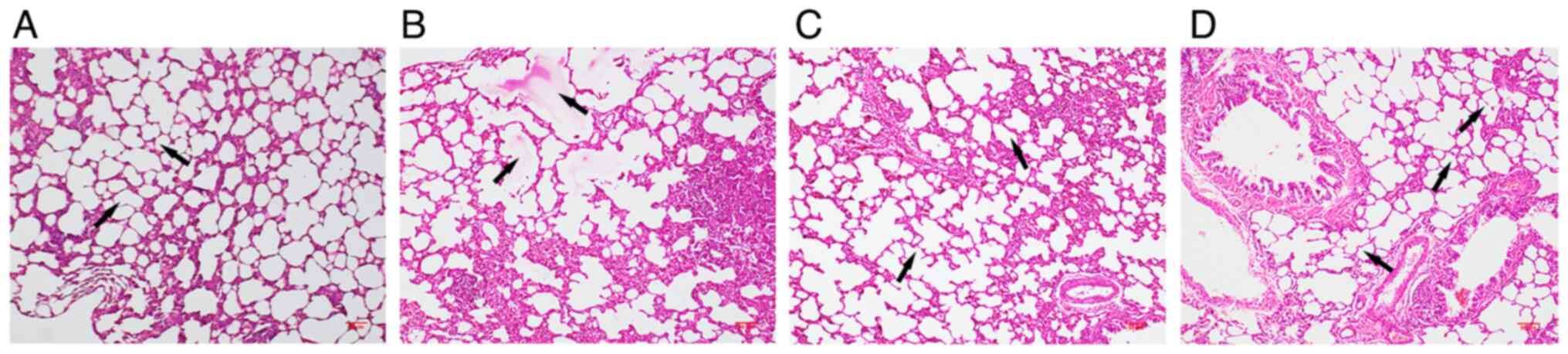
H&E staining results are presented in Fig. 2. The structures of the lung tissues
were normal in the control group. By contrast, the lung tissues of
rats exposed to CS exhibited interalveolar septa widening,
infiltration of inflammatory cells, edema fluid in airspaces and
abnormal enlargement of airspaces. These characteristics were
attenuated in the CS + NAC group. These results indicated that NAC
could protect rat lungs from CS-induced injury.
CS-induced oxidative stress in
rats
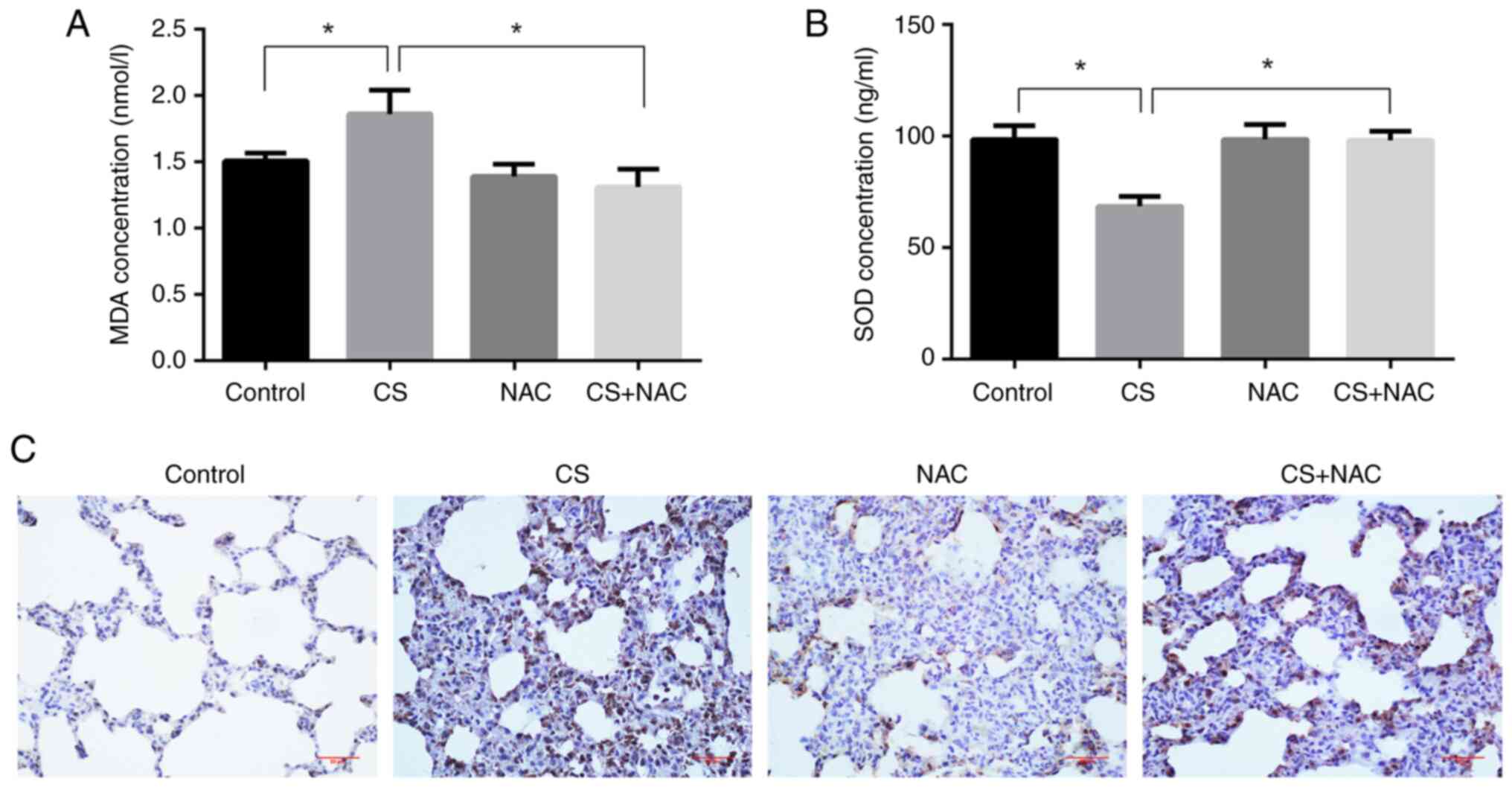
MDA is a classic representative of lipid
peroxidation products and 8-OHdG is a marker of DNA damage; these
reflect the levels of oxidative stress (20). SOD is an antioxidant enzyme, which
mitigates oxidative stress (21).
Therefore, the effects of CS + NAC on oxidative stress were
examined by measuring MDA and SOD levels in the serum, and 8-OHdG
levels in lung tissues. The results shown in Fig. 3 indicated increased levels of MDA
and decreased levels of SOD in the serum of Wistar rats exposed to
CS compared with the control group (P<0.05). NAC treatment
significantly prevented these changes compared with the CS group
(P<0.05). The immunohistochemistry results revealed that the
positive expression of 8-OHdG in CS-exposed rat lungs was increased
compared with that in the control group; however, this was
decreased following simultaneous administration of NAC. These
results indicated that CS stimulated changes in the expression of
oxidative stress-related markers, and that NAC effectively
antagonized the oxidative injury.
Effects of NAC on CS-induced apoptosis
in rat lungs
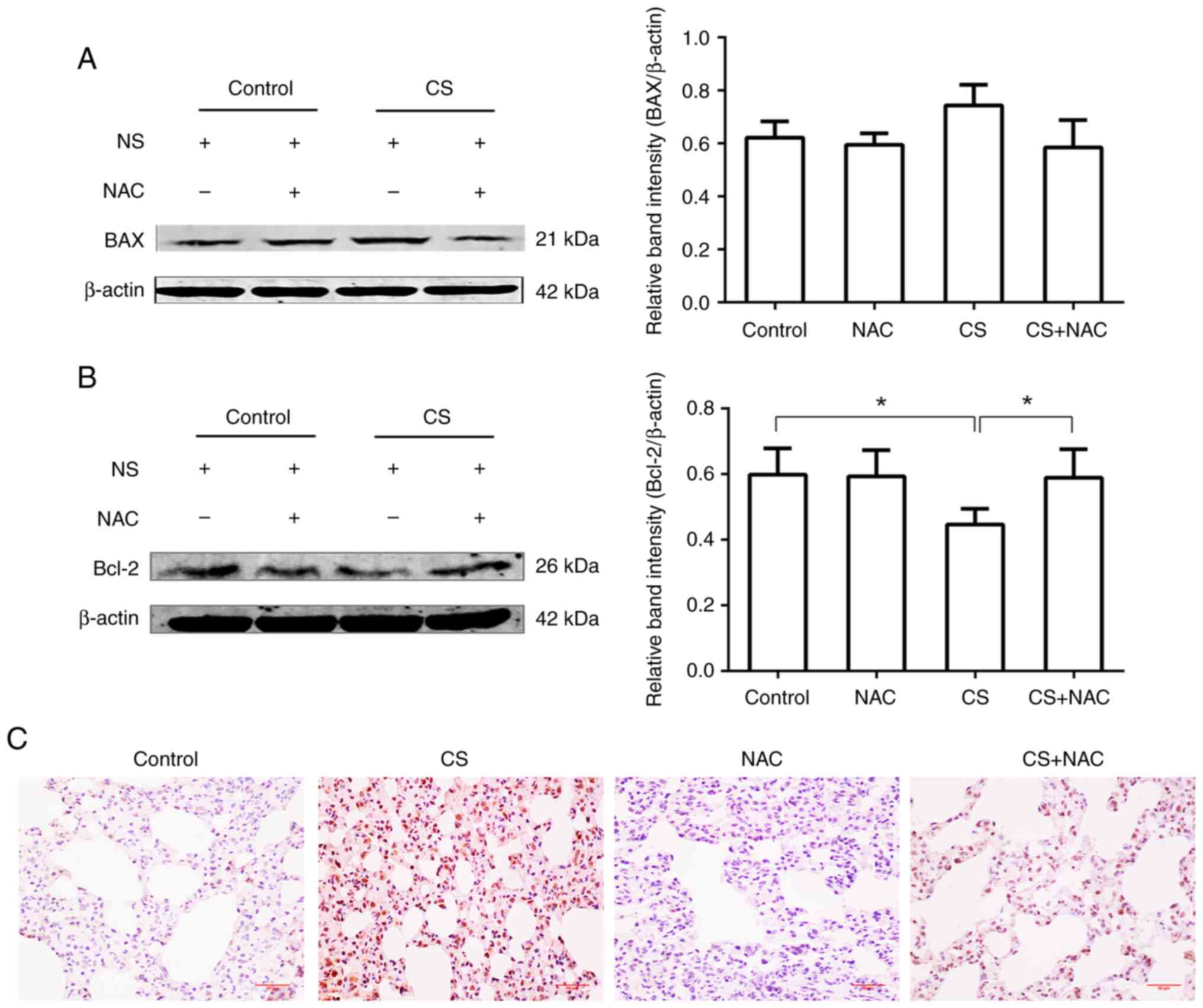
Although there was no significant change in the
expression levels of BAX in the lungs of CS-exposed rats compared
with the control group, Bcl-2 expression was significantly
decreased compared with that in the control group (P<0.05).
Additionally, the CS + NAC group exhibited increased expression
levels of Bcl-2 compared with the CS-exposed group (P<0.05).
Correspondingly, the results of immunohistochemical staining
revealed that the expression levels of Caspase-3 p12 in lung tissue
were increased following CS exposure, and this was antagonized by
the administration of NAC. Therefore, CS may stimulate apoptosis in
lung tissue, whereas NAC may protect against apoptosis (Fig. 4).
Effects of NAC on CSE-induced
oxidative stress in RLE-6TN cells
After incubation of RLE-6TN cells with DCFH-DA,
intracellular ROS levels were detected. Compared with those in the
control group, CSE significantly increased the average fluorescence
intensity as well as the percentage of fluorescent cells in the CSE
group. Co-treatment with NAC resulted in a significant decrease in
these parameters compared with the CSE group (Figs. 5A and B, S1 and S2). HO-1 mRNA and protein expression
under CSE stimulation was subsequently investigated. Analysis
revealed that both mRNA and protein levels of HO-1 were
significantly upregulated by CSE compared with the control group.
Co-treatment with NAC significantly prevented the induction of HO-1
compared with the CSE group (Fig. 5C
and D). The upregulation of HO-1 in CSE-treated cells may be in
response to CSE-induced oxidative stress, since HO-1 is an
antioxidant.
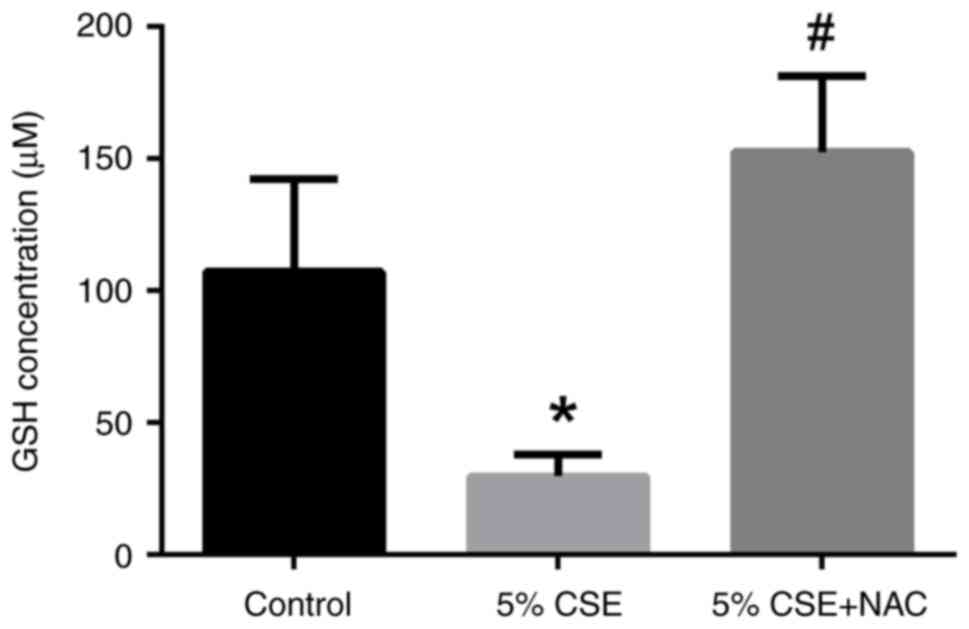
GSH content alterations in RLE-6TN
cells by CSE and/or NAC
The concentration of GSH in the 5% CSE-treated group
was reduced compared with that in the control group (P<0.05;
Fig. 6). Co-treatment with NAC
resulted in increased levels of GSH compared with the CSE-treated
group (P<0.05). In summary, CSE-induced oxidative stress could
oxidize or exhaust intracellular GSH, and NAC may protect alveolar
epithelial cells by replenishing the lost GSH.
Effects of NAC on CSE-induced
apoptosis in RLE-6TN cells
p53 expression was upregulated in alveolar
epithelial cells following 5% CSE administration (Fig. 7A), and correspondingly
anti-apoptotic protein Bcl2 was significantly downregulated
(Fig. 7B) compared with the
control group; however, the expression levels of the pro-apoptotic
protein BAX did not change (Fig.
S3). At the same time, co-treatment with NAC could prevent the
aforementioned alterations. As shown by flow cytometry analysis
(Fig. 7C), in the control group,
the percentage of cells in Q2 + Q3 was 13.43%. In cells exposed to
5% CSE, this increased to 66.6%. In CSE-treated RLE-6TN cells
co-treated with NAC, the percentage of cells in Q2 + Q3 decreased
to 35.2%. These data indicated that CSE may induce apoptosis of
alveolar epithelial cells, while NAC prevented CSE-induced
oxidative injury.
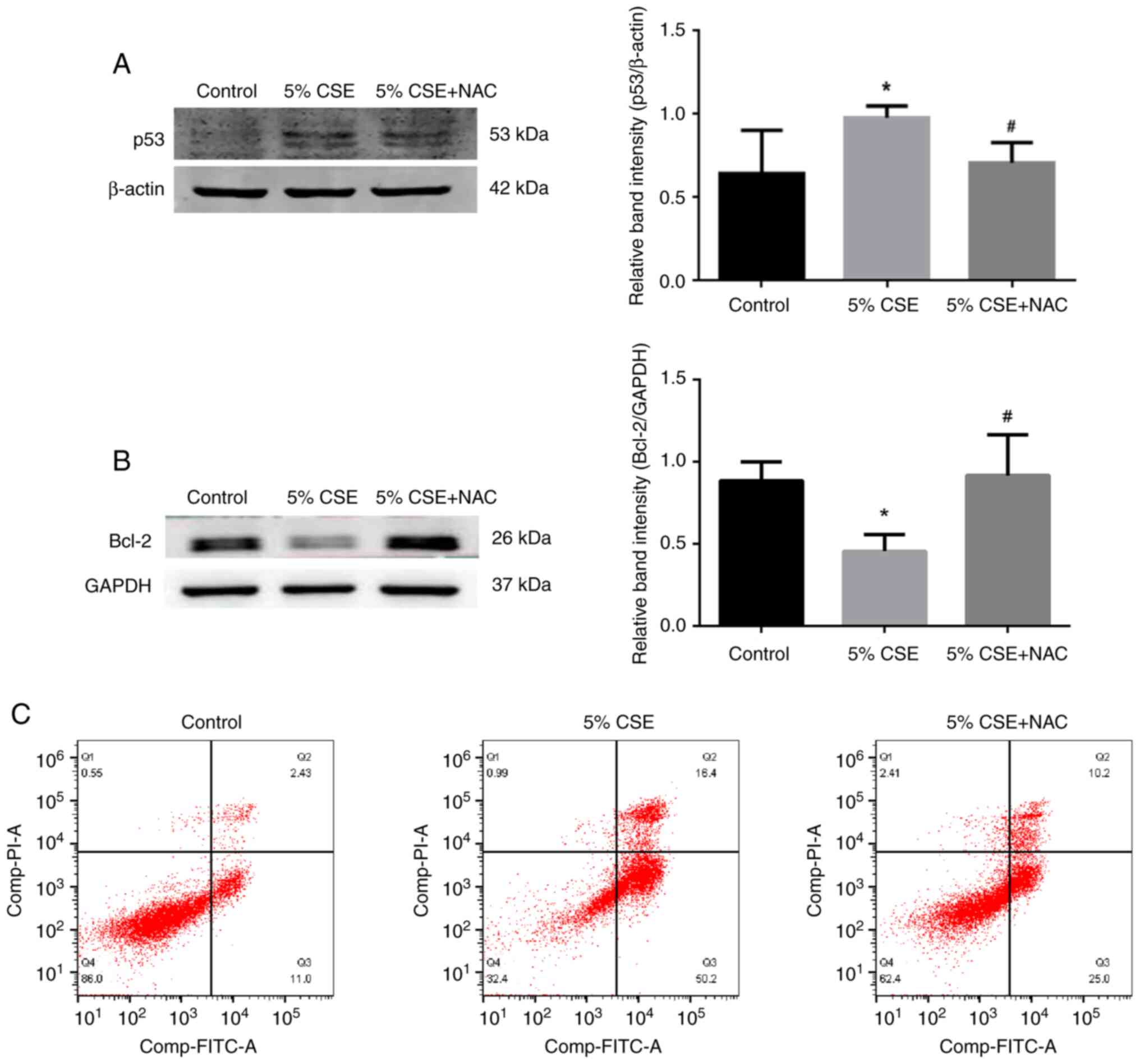 | Figure 7.Effects of NAC on CSE-induced
apoptosis. (A) p53 protein expression in RLE-6TN cells was
upregulated by CSE stimulation; however, NAC could prevent the
upregulation. (B) Protein expression levels of Bcl-2, an
anti-apoptotic protein, in RLE-6TN cells were downregulated in the
CSE-induced group; however, NAC could prevent the downregulation.
Data are presented as the mean ± SD (n=3). *P<0.05 vs. control
group; #P<0.05 vs. 5% CSE group. (C) Flow cytometry
analysis of apoptosis. Q1 represents annexin
V−/PI+ cells, Q2 represents annexin
V+/PI+ cells, Q3 represents annexin
V+/PI− cells and Q4 represents annexin
V−/PI− cells. In the control group, the cell
percentage in Q2 + Q3 was 13.43%. Upon exposure to 5% CSE for 24 h,
it was increased to 66.6%. By contrast, following co-treatment with
NAC, the proportion was decreased to 35.2%. These data indicated
that NAC could antagonize the apoptosis induced by CSE.
Representative plots of at least three independent experiments are
shown. CSE, cigarette smoke extract; NAC, N-acetyl-L-cysteine;
RLE-6TN, rat lung epithelial-6-T-antigen negative. |
Discussion
COPD is a global health problem that is increasing
in incidence and mortality. It is estimated that the global
prevalence is 9–10% in adults >40 years (22). CS is an important preventable risk
factor for human health, not only for smokers, but also for
non-smokers who bear tobacco-associated disease (23), accounting for ~27% of
tobacco-related mortalities in smokers and non-smokers (24).
To induce emphysema-like changes and airway
remodeling, animals are exposed to CS for at least several days
(25). In the present study,
Wistar rats were exposed to CS for 4 weeks, and using H&E
staining, the presence of characteristics such as airspace
enlargement, chronic inflammation and airway remodeling, including
interalveolar septa widening, was observed. The presence of these
characteristics was attenuated when NAC was simultaneously applied
alongside CS exposure.
COPD is considered to be a disease that involves
multiple systems, including skeletal muscle wasting, muscle
dysfunction and weight loss (26).
Nicotine can induce a decrease in appetite, impacting food intake
and body weight by influencing the hypothalamic melanocortin system
(27). CS contains ≥6,000
components that may directly or indirectly affect energy
expenditure (28), and in a
subchronic cigarette exposure model of mice, it has been reported
that 4-week CS exposure could markedly decrease body weight, food
intake, fat mass and the plasma leptin concentration by
fundamentally altering hypothalamic appetite regulation in the
central nervous system (27).
Consistently, the results of the present study revealed that the
body weight of rats in the CS group was significantly decreased
compared with that in the control group at each week. Co-treatment
with NAC resulted in a trend of increased body weight from the end
of week 2 until week 4 compared with the CS group.
MDA and 4-hydroxynonenal are biomarkers of lipid
peroxidation, and oxidative damage to DNA focuses on oxidation of
guanine to 8-OHdG (29).
Therefore, the relative contents of SOD, MDA and 8-OHdG are often
used as markers of oxidative stress in various lung diseases
(11). In the present study, the
content of SOD in serum was significantly decreased, while MDA
content in the serum and 8-OHdG content in lung tissue were
increased in the CS group. Furthermore, Bcl-2 expression was
significantly decreased, while caspase-3 p12 expression was
increased in the CS group compared with the control group. Under
normal conditions, caspase-3 has no activity and is stored in the
form of a zymogen in the cytoplasm; however, when activated by
upstream caspases, caspase-3 generates activated caspase-3 p17 and
caspase-3 p12 subunits, which serve a role in apoptosis (30). The results indicated that CS could
induce oxidative stress and apoptosis in the rat lungs.
Co-treatment with NAC prevented the aforementioned changes,
suggesting that NAC may protect against CS-induced oxidative
damage.
The alveolar epithelium is the initial defense
against inhaled particles and gases, as a progenitor for new
alveolar epithelial type 1 (ATI) and alveolar epithelial type 2
(ATII) cells during lung repair (31). Following injury of ATI cells,
adjacent ATII cells are stimulated to multiply and
transdifferentiate into ATI cells (32). In order to study the contribution
of ATII cells in the pathogenesis of COPD, the changes in RLE-6TN
cells under the stimulation of CSE were observed in vitro.
The results revealed that CSE induced apoptosis of RLE-6TN cells,
produced high levels of intracellular ROS, increased HO-1
expression and exhausted GSH. It also increased p53 expression and
decreased Bcl-2 expression. In vitro study confirmed that
CSE could initiate oxidative stress in RLE-6TN cells and induce
apoptosis of alveolar epithelial cells by increasing p53
expression, while decreasing Bcl-2 expression, and NAC could
antagonize these changes.
An increase in oxidative stress of alveolar
epithelial cells can lead to decrease of intracellular GSH by
transversion of GSH to GSSG (33),
and the inhibitory effect of NAC suggests that oxidative stress
serves an important role in mediating the injury of alveolar
epithelial cells by CSE. NAC has been shown to reduce oxidative
stress by both direct antioxidant effects, as free sulfydryl groups
serve as a source of reducing equivalents, and by indirect
antioxidant effects through the replenishment of intracellular GSH
levels (18). Studies suggest that
NAC acts as a donor of sulfane sulfur when provided to cells
(14,34), which may form dependently or
independently of H2S, and is likely to exert a
cytoprotective effect independently of GSH replenishment. This may
occur by modulation of protein activity, protection of thiols from
irreversible modifications and/or increased oxidant scavenging
capacity (14).
Concomitant with the decrease in GSH, the presence
of cellular oxidative stress in CSE-treated cells was also
evidenced by the induction of HO-1, which supports the hypothesis
that CSE induction of HO-1 is regulated by changes in intracellular
GSH. As a precursor of GSH, NAC could significantly reduce the
expression of HO-1 stimulated by CSE. HO enzymes catalyze heme to
form equimolar amounts of ferrous iron, carbon monoxide and
biliverdin, all of which have cytoprotective properties, probably
by mitigating nitrosative/oxidative stress (35). Under basal conditions, HO-1 is
present at low to undetectable levels in most tissues, but its
expression is rapidly increased in response to environmental
stimuli producing oxidative stress and generating ROS, thus rapid
elevation in HO-1 expression as a response to oxidative stress is
usually considered to be a cellular defense mechanism (36). Although modest HO-1 expression is
cytoprotective, exacerbation of oxidative injury is often
associated with increased HO-1 expression, and increasing evidence
suggests that HO-1 induction can also lead to cell injury,
presumably through excessive ROS generation (37–39).
It is hypothesized that both excess HO activation and iron
accumulation generate ROS, and free iron-mediated Fenton reactions
have been implicated in lipid peroxidation during ferroptosis in
the pathogenesis of COPD (40).
In summary, CS could cause systemic (serum) and
local (lung tissue) oxidative stress and ultimately caused
apoptosis in lung tissues as well as ATII cells in vitro.
The protective effects of NAC on alveolar epithelial cells injured
by CS may be partially associated with the replenishment of
intracellular GSH. These results provide fundamental support for
the clinical application of NAC in the treatment of patients with
COPD.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81770044).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZJ designed the project. HC carried out the animal
experiments. YC, JC and LD carried out the in vitro
experiments, including mainly western blotting. SL carried out the
flow cytometry experiments. JC carried out statistical analysis and
drew the histograms. ZJ organized all data and wrote the
manuscript. All authors have read and approved the final
manuscript. ZJ and JC confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Animal procedures were approved by the Ethics
Committee of the Institutional Animal Care of Lanzhou University
(approval no. Lzujcyxy20240106; Lanzhou, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kahnert K, Jörres RA, Behr J and Welte T:
The diagnosis and treatment of COPD and its comorbidities. Dtsch
Arztebl Int. 120:434–444. 2023.PubMed/NCBI
|
|
2
|
Song Q, Chen P and Liu XM: The role of
cigarette smoke-induced pulmonary vascular endothelial cell
apoptosis in COPD. Respir Res. 22:392021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Devulder JV: Unveiling mechanisms of lung
aging in COPD: A promising target for therapeutics development.
Chin Med J Pulm Crit Care Med. 2:133–141. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kaur M, Chandel J, Malik J and Naura AS:
Particulate matter in COPD pathogenesis: An overview. Inflamm Res.
71:797–815. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen L, Zhu D, Huang J, Zhang H, Zhou G
and Zhong X: Identification of hub genes associated with COPD
through integrated bioinformatics analysis. Int J Chron Obstruct
Pulmon Dis. 17:439–456. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prieux R, Eeman M, Rothen-Rutishauser B
and Valacchi G: Mimicking cigarette smoke exposure to assess
cutaneous toxicity. Toxicol In Vitro. 62:1046642020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fischer BM, Voynow JA and Ghio AJ: COPD:
Balancing oxidants and antioxidants. Int J Chron Obstruct Pulmon
Dis. 10:261–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woźniak A, Górecki D, Szpinda M,
Mila-Kierzenkowska C and Woźniak B: Oxidant-antioxidant balance in
the blood of patients with chronic obstructive pulmonary disease
after smoking cessation. Oxid Med Cell Longev. 2013:8970752013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sies H and Jones DP: Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents. Nat
Rev Mol Cell Biol. 21:363–383. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barnes PJ: Oxidative stress in chronic
obstructive pulmonary disease. Antioxidants (Basel). 11:9652022.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cha SR, Jang J, Park SM, Ryu SM, Cho SJ
and Yang SR: Cigarette smoke-induced respiratory response: Insights
into cellular processes and biomarkers. Antioxidants (Basel).
12:12102023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Paintlia MK, Paintlia AS, Contreras MA,
Singh I and Singh AK: Lipopolysaccharide-induced peroxisomal
dysfunction exacerbates cerebral white matter injury: Attenuation
by N-acetyl cysteine. Exp Neurol. 210:560–576. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aldini G, Altomare A, Baron G, Vistoli G,
Carini M, Borsani L and Sergio F: N-Acetylcysteine as an
antioxidant and disulphide breaking agent: The reasons why. Free
Radic Res. 52:751–762. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pedre B, Barayeu U, Ezeriņa D and Dick TP:
The mechanism of action of N-acetylcysteine (NAC): The emerging
role of H(2)S and sulfane sulfur species. Pharmacol Ther.
228:1079162021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Millea PJ: N-acetylcysteine: Multiple
clinical applications. Am Fam Physician. 80:265–269.
2009.PubMed/NCBI
|
|
16
|
Sanguinetti CM: N-acetylcysteine in COPD:
Why, how, and when? Multidiscip Respir Med. 11:82016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Messier EM, Day BJ, Bahmed K, Kleeberger
SR, Tuder RM, Bowler RP, Chu HW, Mason RJ and Kosmider B:
N-Acetylcysteine protects murine alveolar type II cells from
cigarette smoke injury in a nuclear erythroid 2-related
factor-2-independent manner. Am J Respir Cell Mol Biol. 48:559–567.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cazzola M, Page CP, Wedzicha JA, Celli BR,
Anzueto A and Matera MG: Use of thiols and implications for the use
of inhaled corticosteroids in the presence of oxidative stress in
COPD. Respir Res. 24:1942023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Burkhardt BR, Lyle R, Qian K, Arnold AS,
Cheng H, Atkinson MA and Zhang YC: Efficient delivery of siRNA into
cytokine-stimulated insulinoma cells silences Fas expression and
inhibits Fas-mediated apoptosis. FEBS Lett. 580:553–560. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murphy MP, Bayir H, Belousov V, Chang CJ,
Davies KJA, Davies MJ, Dick TP, Finkel T, Forman HJ,
Janssen-Heininger Y, et al: Guidelines for measuring reactive
oxygen species and oxidative damage in cells and in vivo. Nat
Metab. 4:651–662. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dang X, He B, Ning Q, Liu Y, Guo J, Niu G
and Chen M: Alantolactone suppresses inflammation, apoptosis and
oxidative stress in cigarette smoke-induced human bronchial
epithelial cells through activation of Nrf2/HO-1 and inhibition of
the NF-κB pathways. Respir Res. 21:952020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vijayan VK: Chronic obstructive pulmonary
disease. Indian J Med Res. 137:251–269. 2013.PubMed/NCBI
|
|
23
|
Salvi SS and Barnes PJ: Chronic
obstructive pulmonary disease in non-smokers. Lancet. 374:733–743.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martinez FJ, Donohue JF and Rennard SI:
The future of chronic obstructive pulmonary disease
treatment-difficulties of and barriers to drug development. Lancet.
378:1027–1037. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Upadhyay P, Wu CW, Pham A, Zeki AA, Royer
CM, Kodavanti UP, Takeuchi M, Bayram H and Pinkerton KE: Animal
models and mechanisms of tobacco smoke-induced chronic obstructive
pulmonary disease (COPD). J Toxicol Environ Health B Crit Rev.
26:275–305. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Henrot P, Blervaque L, Dupin I, Zysman M,
Esteves P, Gouzi F, Hayot M, Pomiès P and Berger P: Cellular
interplay in skeletal muscle regeneration and wasting: Insights
from animal models. J Cachexia Sarcopenia Muscle. 14:745–757. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mineur YS, Abizaid A, Rao Y, Salas R,
DiLeone RJ, Gündisch D, Diano S, De Biasi M, Horvath TL, Gao XB and
Picciotto MR: Nicotine decreases food intake through activation of
POMC neurons. Science. 332:1330–1332. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen H, Hansen MJ, Jones JE, Vlahos R,
Bozinovski S, Anderson GP and Morris MJ: Cigarette smoke exposure
reprograms the hypothalamic neuropeptide Y axis to promote weight
loss. Am J Respir Crit Care Med. 173:1248–1254. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jomova K, Raptova R, Alomar SY, Alwasel
SH, Nepovimova E, Kuca K and Valko M: Reactive oxygen species,
toxicity, oxidative stress, and antioxidants: Chronic diseases and
aging. Arch Toxicol. 97:2499–2574. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Boatright KM and Salvesen GS: Caspase
activation. Biochem Soc Symp. 233–242. 2003.PubMed/NCBI
|
|
31
|
Ruaro B, Salton F, Braga L, Wade B,
Confalonieri P, Volpe MC, Baratella E, Maiocchi S and Confalonieri
M: The history and mystery of alveolar epithelial type II cells:
Focus on their physiologic and pathologic role in lung. Int J Mol
Sci. 22:25662021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Han S, Lee M, Shin Y, Giovanni R,
Chakrabarty RP, Herrerias MM, Dada LA, Flozak AS, Reyfman PA,
Khuder B, et al: Mitochondrial integrated stress response controls
lung epithelial cell fate. Nature. 620:890–897. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baglole CJ, Bushinsky SM, Garcia TM, Kode
A, Rahman I, Sime PJ and Phipps RP: Differential induction of
apoptosis by cigarette smoke extract in primary human lung
fibroblast strains: Implications for emphysema. Am J Physiol Lung
Cell Mol Physiol. 291:L19–L29. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ezeriņa D, Takano Y, Hanaoka K, Urano Y
and Dick TP: N-Acetyl cysteine functions as a fast-acting
antioxidant by triggering intracellular H(2)S and sulfane sulfur
production. Cell Chem Biol. 25:447–459.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wu CY, Cilic A, Pak O, Dartsch RC, Wilhelm
J, Wujak M, Lo K, Brosien M, Zhang R, Alkoudmani I, et al: CEACAM6
as a novel therapeutic target to boost HO-1-mediated antioxidant
defense in COPD. Am J Respir Crit Care Med. 207:1576–1590. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Consoli V, Sorrenti V, Grosso S and
Vanella L: Heme oxygenase-1 signaling and redox homeostasis in
physiopathological conditions. Biomolecules. 11:5892021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lee DW, Gelein RM and Opanashuk LA:
Heme-oxygenase-1 promotes polychlorinated biphenyl mixture aroclor
1254-induced oxidative stress and dopaminergic cell injury. Toxicol
Sci. 90:159–167. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin CC, Yang CC, Hsiao LD, Chen SY and
Yang CM: Heme oxygenase-1 induction by carbon monoxide releasing
molecule-3 suppresses interleukin-1β-mediated neuroinflammation.
Front Mol Neurosci. 10:3872017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Baglole CJ, Sime PJ and Phipps RP:
Cigarette smoke-induced expression of heme oxygenase-1 in human
lung fibroblasts is regulated by intracellular glutathione. Am J
Physiol Lung Cell Mol Physiol. 295:L624–L636. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|