Introduction
Drug-induced liver injury (DILI) is a common adverse
drug reaction, with an estimated worldwide incidence of 14–19 cases
per 100,000 people, 30% of which are associated with jaundice
(1). A 2019 retrospective study
involving 308 medical centers in the major cities of mainland China
noted that the incidence of DILI was 23.80 per 100,000 individuals,
which was higher than that in Western countries. It also reported
that DILI cases were rising every year, and that traditional
Chinese medicines and antituberculosis medicines, among others,
were the main drug classes that caused DILI (2). Notably, the incidence of DILI is
higher in hospitalized populations (3). DILI refers to an injury to the liver
or biliary system caused by the ingestion of hepatotoxic drugs.
Mechanistically, DILI can be attributed to either endogenous
toxicity (dose-related) or specific toxicity (dose-independent) and
indirect injury, a recently proposed type that refers to injury to
the liver or biliary system caused by a drug that alters a
patient's original liver disease or immune status (4). Endogenous toxicity is predictable and
can occur hours or days after an individual is exposed to a drug.
By contrast, specific toxicity is often unpredictable and is
determined by the interaction of environmental and host factors
with the drug (5). Sex is a
significant risk factor for DILI; young women are at a higher risk
of drug overdose events, which may be due to differences in
psychosocial factors, physiological differences and behavioral
patterns (6,7). According to the results of the Third
National Health and Nutrition Examination Survey in the United
States, it was found that women take acetaminophen (APAP) more
often than men (8). In addition,
it has been shown that the clearance of APAP from men's bodies is
22% higher than that from women's bodies, because men have higher
glucuronidation activity, which is a key enzyme in APAP metabolism
(9).
The primary cause of endogenous DILI is considered
to be the supratherapeutic use of APAP, which is one of the most
widely used and popular over-the-counter analgesic and antipyretic
medications across the world (10). APAP is included in the World Health
Organization's list of essential medicines recommended as a
first-line treatment for the majority of pain and fever cases, and
is considered safe at therapeutic doses; however, when overdosed,
it can cause life-threatening hepatotoxicity (11). The United States has the highest
proportion of DILI cases caused by APAP (~30% of cases), followed
by Pakistan (~16% of cases) (12).
The first reports of APAP poisoning in humans appeared in the 1960s
when Davidson and Eastham (13)
reported two patients who developed hepatotoxicity after overdose;
both patients succumbed on day 3 after overdosing, and in both
cases, the pathologic features of the liver sections were
suggestive of marked acute lobulocentric fulminant hepatic necrosis
(14). Notably, toxic effects are
highly likely with APAP at a dose of 150 mg/kg (15); this dose has remained virtually
unchanged over the past 20 years although N-acetylcysteine
(NAC) has been introduced as a clinical antidote to APAP poisoning.
Therefore, understanding the pathogenesis and risk factors of
APAP-induced liver injury (AILI), and proposing more effective
means of prevention and treatment are imperative.
Clinical manifestations and biomarkers
APAP toxicity is a time-dependent event, with the
liver being the first organ to come into contact with absorbed
APAP. This toxicity can occur from administration errors,
accidental ingestion and intentional self-administered overdose
(16). APAP hepatotoxicity can
generally be divided into four stages of presentation, with the
first stage characterized by nausea, vomiting, right upper
abdominal pain and other nonspecific symptoms that begin within
minutes to hours of ingestion. Some patients may initially be
asymptomatic, with hepatic function markers, such as serum
aspartate transferase (AST), prothrombin time (PT) and serum total
bilirubin (TBIL), at baseline levels (17). During the second stage,
abnormalities in liver function, such as an increase in AST
activity, PT and TBIL levels, occur and the patient may present
with nausea, pain in the right upper abdomen and jaundice (18). The third stage is often the peak of
hepatic injury, with marked necrosis of hepatocytes. AST levels can
peak at this stage, but some patients do not show typical symptoms.
In patients with more severe liver injury, fulminant liver failure,
including hepatic encephalopathy and hyperbilirubinemia, can occur
(19). The fourth stage is often
the recovery stage, when the indicators of various liver functions
may return to the baseline. Nevertheless, a small number of
patients experience liver and other organ failure, which is
life-threatening. Various factors can increase the risk for APAP
toxicity, which is mainly mediated by the metabolic pathway of APAP
(12). Plasma biomarkers detected
through liquid biopsies are currently considered the gold standard
for understanding the mechanisms of in vivo injury in
patients (20). Protein adducts in
plasma can serve as specific biomarkers of APAP toxicity. High
levels of APAP protein adducts (APAP-AD) in serum samples from
patients with acute liver failure (ALF) induced by APAP overdose
and lower levels of adducts in other types of liver injury, such as
pyrrole protein adducts, can be detected by high-sensitivity
high-pressure liquid chromatography combined with electrochemical
detection (21). A major advantage
of measuring these protein adducts is that their half-life in serum
is considerably long; therefore, they can be used to accurately
diagnose AILI long after APAP ingestion (22).
Pathophysiological mechanisms of AILI
Continuous improvements in research equipment and
methods in the fields of modern medicine and microbiology have
enabled a deeper understanding of the mechanisms underlying AILI.
Production of toxic metabolites, generation of protein adducts
leading to mitochondrial dysfunction and peroxynitrite formation,
and cell death due to DNA strand breaks are believed to be the
primary mechanisms of APAP-induced hepatotoxicity (Fig. 1). Recent studies have reported the
close involvement of several events in the onset of APAP toxicity,
which have been described in the following sub-sections.
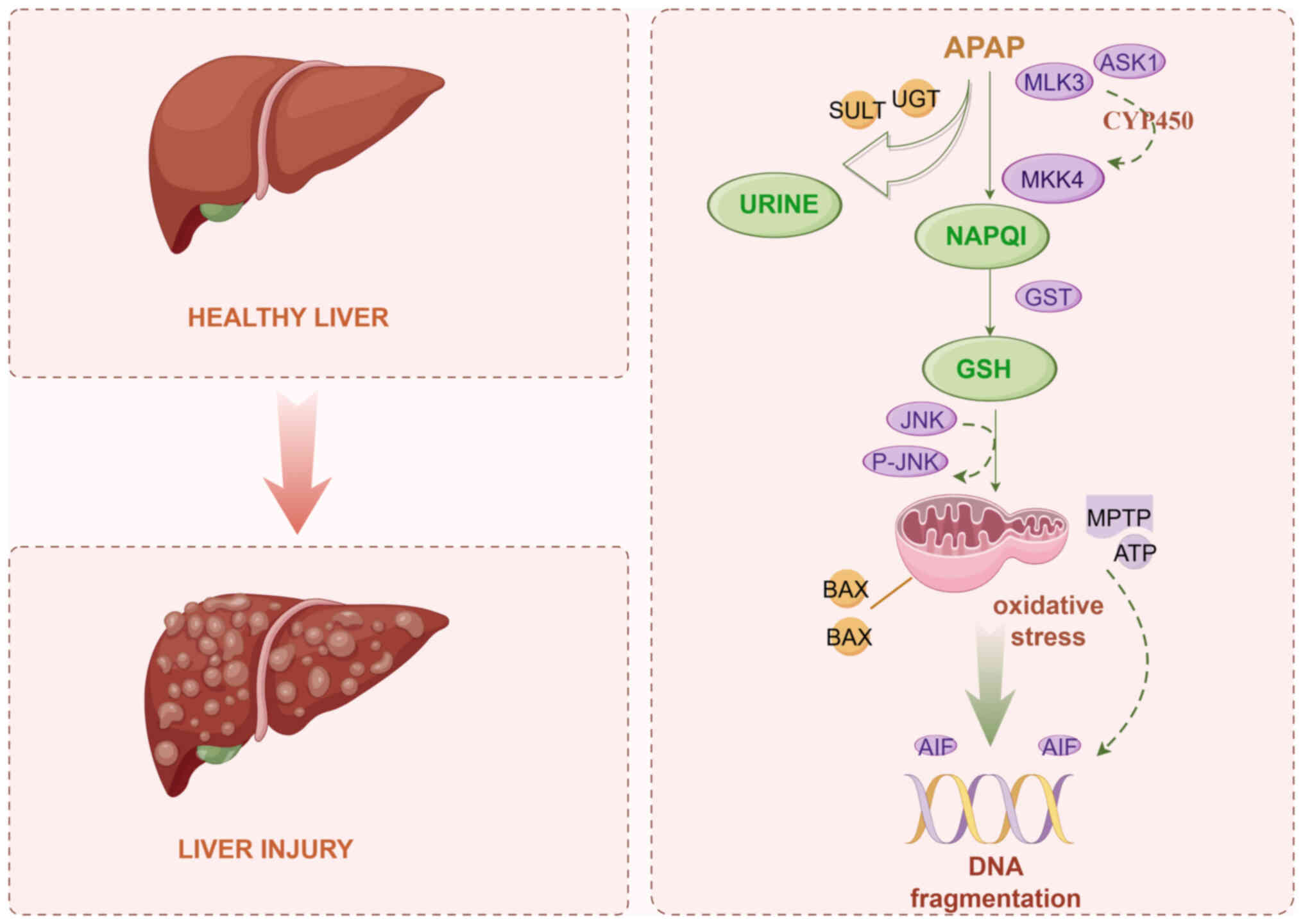 | Figure 1.Underlying mechanism of APAP-induced
liver injury. APAP, acetaminophen; UGT, glucuronosyltransferase;
SULT, sulfonyl transferase; ASK1, apoptosis signal-regulating
kinase 1; MLK3, mixed lineage kinase 3; MKK4, mitogen-activated
protein kinase kinase 4; NAPQI,
N-acetyl-p-benzoquinone imine; GST, glutathione
transferase; GSH, glutathione; MPTP, mitochondrial membrane
permeability transition pore; AIF, apoptosis-inducing factor. |
Toxic metabolites
The liver is a major target for drug toxicity
because it serves a crucial role in the metabolism of drugs, toxins
and other exogenous substances, and their removal from circulation.
The liver is not only responsible for the biotransformation of
drugs into more easily excretable forms, but it also protects the
body from the potentially harmful effects of drugs through its
detoxification system. Approximately 90% of APAP taken at
therapeutic doses is metabolized by phase II-coupled enzymes,
mainly glucuronosyltransferase (UGT) and sulfonyl transferases
(SULTs), which convert it into non-toxic compounds that are then
excreted in the urine via the biliary tract and renal system
(23). A small portion (~5%) is
excreted in the urine without biotransformation, whereas the
remaining portion (~5%) is bio-transformed by the cytochrome P450
(CYP450) system, specifically via CYP2E1 and CYP1A2 metabolism, to
produce the reactive intermediate molecule
N-acetyl-p-benzoquinone imine (NAPQI), which can be
readily detoxified to the water-soluble and innocuous mercapturic
acid by glutathione (GSH) (24).
Microsomal GSH transferase (GST) catalyzes the binding of NAPQI to
GSH, thus reducing its toxicity. Notably, the formation of NAPQI,
the active metabolite of APAP, is an important step in the
development of hepatotoxicity (25). Exceeding the therapeutic dose of
APAP leads to saturation of the phase II-coupled enzymes;
therefore, excess APAP is diverted to another metabolic pathway,
the CYP450 system, which results in the production of a large
amount of NAPQI in hepatocytes (26). This leads to GSH overconsumption,
which lowers GSH levels. Excess NAPQI binds to cellular
biomolecules, such as proteins, nucleic acids and lipids, a process
that occurs predominantly in the centrocytes and hepatocytes of
hepatic lobules. The degree of binding and the relative amount of
covalent binding have been reported to be associated with the
development of toxicity; the formation of protein adducts causes
oxidative stress and dysfunction, which further leads to hepatocyte
necrosis (27). Notably,
therapeutic doses of APAP do not cause liver injury if UGT and
SULTs are active and the hepatic GSH stores are adequately
maintained, because all of the NAPQI produced is efficiently
removed.
Oxidative stress in mitochondria
Mitochondria are vital organelles for cellular
energy production, which also have an important role in cell
signaling. Toxic doses of APAP can alter mitochondrial morphology
and function. Owing to the reactive nature of NAPQI, one of the
early events following its formation and the depletion of hepatic
GSH stores is the generation of NAPQI adducts on cellular proteins
(28). Mitochondria are a key
early target for NAPQI adduct formation. The levels of such adducts
markedly increase in mitochondria after APAP overdose into mice,
accounting for ~65% of the whole cytoplasm; mitochondrial protein
adducts, such as mitochondrial aldehyde dehydrogenase, α-subunit of
ATP synthetase and GSH peroxidase (GPX), have been found in mice
(29). Large amounts of NAPQI can
cause GSH depletion, which leads to increased release of
H2O2 in mitochondria, which is an important
oxidative stress signaling molecule that initiates the
auto-activation of apoptosis signal-regulating kinase 1 (ASK1),
further triggering the activation of mitogen-activated protein
kinase kinase 4. Ultimately, this leads to the cytoplasmic
activation of the MAPK, JNK, which belongs to a subgroup of MAPKs
that are mainly activated by cytokines and environmental stress
(30). JNK activation and
attenuation of liver injury have been observed in ASK1-deficient
mice, thus demonstrating that ASK1 is upstream of JNK; notably,
this activation is more pronounced at a late stage (i.e., ≥3 h
after APAP administration), with little effect at an earlier time
point (1.5 h) (31).
Another possible mechanism of JNK activation that
has recently been proposed is that it can be released from GSTπ
(32), an important detoxifying
enzyme that is a member of the GST family that binds to JNK.
Oxidative stress or direct binding of NAPQI can trigger the release
of JNK from GSTπ. Phosphorylated JNK can bind to the mitochondrial
outer membrane scaffolding protein and inhibit mitochondrial
electron transport through an Src-mediated process. Protein adducts
in mitochondria trigger initial electron leakage at the level of
complex III of the electron transport chain, causing the formation
of superoxide radicals outside the inner mitochondrial membrane
(IMM), which in turn triggers mild oxidative stress in the
cytoplasm (33). In addition,
activated JNK translocates from the cytoplasm to the mitochondria,
increasing the mitochondrial permeability transition pore (MPTP)
activity, which then elevates mitochondrial oxidative stress; this
strongly amplifies mitochondrial superoxide radical formation,
which can induce mitochondrial dysfunction and lead to
hepatocellular necrosis. NAPQI-induced formation of mitochondrial
adducts and the ensuing mitochondrial oxidative stress have been
well-studied in recent decades (34,35),
but the amplification of JNK is a more recent discovery (36).
Peroxynitrite formation
Although mitochondria exhibit strong antioxidant
defense activity, they can experience oxidative stress after
ectopic JNK expression, mainly due to the active disruption of
electron transfer by JNK-mediated signaling (37). Mitochondrial stress-generated
superoxide radicals can react rapidly with nitric oxide to form a
potent oxidant, peroxynitrite, which is a highly reactive and
nitrifying compound that causes APAP toxicity (38). During APAP hepatotoxicity,
peroxynitrite is mainly produced in hepatocytes and liver
sinusoidal endothelial cells (LSECs); the formation of
nitrotyrosine protein adducts, which are mainly found in
mitochondria, indirectly proves that peroxynitrite is formed in the
mitochondria of hepatocytes (39).
Time course studies of peroxynitrite formation have confirmed its
occurrence only in mitochondria after JNK translocation (40). This amplified oxidative and
nitrosative stress in the mitochondria, accompanied by inhibition
of the antioxidant system and nitrification of mitochondrial
components, can induce changes in mitochondrial membrane potential
and morphology, such as the opening of the MPTP, which leads to a
shift in MPT. This alteration, although reversible in the early
stages, eventually leads to an irreversible collapse of the IMM
potential and cessation of ATP synthesis. MPT induction may be
dependent on the magnitude of the upstream signal from APAP; mice
have been shown to exhibit transient MPT induction when the APAP
dose is low and irreversible induction at higher doses (41). The result is matrix swelling and
outer mitochondrial membrane rupture, which releases intermembrane
proteins such as endonuclease G and apoptosis-inducing factor (AIF)
into the cytoplasm; these proteins can later translocate into the
nucleus, leading to mitochondrial DNA breaks, as well as massive
mitochondrial dysfunction and swelling along with DNA damage, which
trigger programmed cellular necrosis (42). This event can directly cause the
cessation of ATP synthesis.
In addition to the unregulated release of
intermembrane proteins via the MPTP, they can be prematurely
released through the Bax pore. Bax, a member of the Bcl-2 family of
proteins, is located mainly in the cytoplasm; upon stimulation, it
can translocate to the outer mitochondrial membrane, form a pore,
and trigger the release of intermembrane proteins (43). In addition, JNK and phosphorylated
JNK are recruited to the mitochondria via the SH3
homology-associated BTK-binding protein, a mitochondrial outer
membrane protein that can further enhance mitochondrial damage and
necrosis (44). GSH supplements
not only provide excess cysteine as an energy substrate but also
serve an important role in scavenging free radicals and
peroxynitrite (45). Selective
clearance of mitochondrial peroxynitrite increases upon accelerated
restoration of mitochondrial GSH levels, further demonstrating the
critical role of peroxynitrite in pathophysiology (46).
Microcirculatory dysfunction
The hepatic microcirculatory milieu, which consists
mainly of LSECs, hepatic stellate cells and hepatic macrophages,
serves an important role in maintaining intrahepatic homeostasis
and hepatocyte function, regulating vascular tone and controlling
inflammation (47). Although
hepatocytes are the primary target of the cytotoxic effects of APAP
overdose, morphological alterations in LSECs are also associated
with APAP-induced liver disease. Drugs are transported through
blood sinusoids by various mechanisms, including organic anion and
cation transport proteins of the basolateral membrane, other
transport proteins such as Na+-taurine bile acid
co-transporting polypeptides or prostaglandin transport proteins,
and passive diffusion, before metabolism and clearance within
hepatocytes (48). APAP-induced
LSEC injury precedes liver injury; thus, LSEC injury is considered
an early event in liver injury (49). Intra-endothelial perturbations,
including swelling of LSECs, gap formation and estradiol
incorporation, occur as early as 2 h after APAP overdose,
suggesting that LSECs can metabolize APAP (49). In a previous study, gavage
administration of APAP to male C57Bl/6 mice revealed that a large
number of SECs swelled at 0.5 and 1 h, whereas the hepatocyte
morphology remained intact, and serum AST and alanine
amiotransferase (ALT) levels, which are indicators of hepatocyte
injury, were not significantly increased. These results suggested
that LSECs are damaged before hepatocytes (50). Furthermore, scanning electron
microscopy after APAP intoxication has revealed the induction of
large pores in LSECs, accompanied by the accumulation of
erythrocytes, enlargement of the Disse space and collapse of
sinusoidal lumen in a mouse model (51). Massive centrilobular congestion is
an important feature of APAP-induced hepatotoxicity in mice, which
occurs before the appearance of necrosis, and is induced by
hepatocytes and their relationship with LSECs. Vascular congestion,
interstitial congestion and sinusoidal collapse may impair hepatic
circulation, thereby exacerbating toxicity by causing additional
hypoxic injury (52). Furthermore,
serum hyaluronic acid (HA) levels have been shown to be elevated in
patients with APAP-induced acute hepatic injury (53); HA is mainly produced by mesenchymal
cells and is found at low levels in normal human plasma, as it is
rapidly cleared by LSECs. Notably, clearance of HA is impaired when
LSECs dysfunction, which may be related to the HA receptor and DNA
breaks on defective LSECs (54).
Hence, HA levels may be used to determine the extent of sinusoidal
vascular endothelial cell damage (55). Formaldehyde-treated serum albumin
(FSA) is a commonly used model ligand for the LSEC scavenger
receptor that has been widely used to study the scavenger receptor
function of LSECs, particularly when assessing the ability of LSECs
to scavenge blood-borne waste macromolecules. FSA expression in the
hemorrhagic centrilobular region has been reported to diminish 2 h
after APAP gavage in a mouse model, suggesting impaired LSEC
scavenger receptor function or a reduced number of cell surface
receptors (50).
Endoplasmic reticulum (ER) stress
The ER serves a crucial role in a number of cellular
processes, including protein folding, assembly, transport,
post-regulation, secretion and quality control of membrane
proteins, lipid synthesis and regulation of intracellular calcium
homeostasis. Disturbance of ER function by stimuli such as altered
cellular redox status, oxidative stress, calcium homeostasis
imbalance, hypoxia or energy deprivation can lead to the
accumulation of unfolded proteins in the ER, leading to ER stress
(56). The ER can respond to
stress through a complex series of events known as the unfolded
protein response (UPR). The UPR comprises a series of signaling
events, such as the activation of ER transmembrane proteins,
including activating transcription factor 6 (ATF6), which induces
various transcription factors such as pro-apoptotic proteins (e.g.,
CHOP) (57).
In contrast to other mechanisms of toxicity, such as
mitochondrial dysfunction and oxidative stress, ER stress has not
been comprehensively evaluated in the context of drug-induced side
effects. Some data from mouse experiments have suggested that ER
stress is a relatively late event in APAP toxicity, with
significant stress occurring only 12 h after APAP administration
(58). By contrast, hepatic
mitochondrial alterations, JNK activation and oxidative stress
occur much earlier in mice after the same dose of APAP (59). Sublethal doses of APAP can result
in a redox shift in ER compartments; notably, a shift of ER
oxidoreductase from the redox state to the oxidized state in
hepatic microsomes suggests that the ER is in a state of redox
imbalance. This state impairs ER oxidoreductase function and may
initiate ER-associated signaling pathways, including ATF6 and CHOP
activation, which inhibits the expression of anti-apoptotic genes
and activates pro-apoptotic genes (60). By contrast, CHOP deletion has been
reported to protect mice from liver injury after APAP poisoning
(61). Moreover, caspase-12, an
ER-resident caspase, is transiently activated by ER stress-related
factors such as calcium ion imbalance, misfolded protein
accumulation, and induction of ER stress inducers. It can be
considered that the activated state of caspase-12 does not last
long, but occurs for a short time and then disappears. By contrast,
the mode of cell death in APAP hepatotoxicity is
caspase-independent apoptosis, triggered instead by activation of
poly(ADP-ribose) polymerase 1 (62). The mode of cell death in APAP
hepatotoxicity is thought to be necrosis rather than apoptosis
because there is no caspase activation following APAP overdose,
despite mitochondrial rupture and release of intermembrane
proteins. Moreover, caspase inhibitors are ineffective in
protecting the liver from APAP toxicity (63).
NAPQI can covalently bind to a variety of microsomal
proteins, such as GST, protein disulfide isomerase (PDI) and
calreticulin. Since PDI and calreticulin have major roles in
protein folding and calcium chelation within the ER, covalent
binding of NAPQI to these proteins can induce ER stress (64). Furthermore, ER stress may be a
secondary consequence of mitochondrial dysfunction. The ER and
mitochondria are closely linked, and the physical binding of these
two organelles creates a structure called the
mitochondria-associated ER membrane; in a physiological context,
this connection allows for continuous calcium transfer. During ER
stress, calcium can be transferred to the mitochondria in large
amounts through these microstructural domains or as a result of
increased cytoplasmic calcium levels; thus, the induction of ER
stress can increase mitochondrial calcium overload, which in turn
favors mitochondrial membrane permeability and the release of
pro-apoptotic factors, such as cytochrome c and AIF
(65). Notably, ER stress can also
stimulate the expression of the mitochondrial stress factor heat
shock protein 60 (HSP60) in mouse hepatocytes, and may impair
mitochondrial respiration and decrease mitochondrial membrane
potential. This finding not only reveals the complex interaction
between the ER and mitochondria, but also demonstrates that ER
stress can induce mitochondrial stress (66).
Aseptic inflammation
Inflammatory liver diseases that are not caused by
pathogens, such as AILI, non-alcoholic steatohepatitis and
alcoholic liver disease, are considered ‘sterile’ (67). While hepatocellular necrosis is the
initial underlying mechanism of APAP-induced hepatotoxicity, the
second step in liver injury is a sterile inflammatory response to
necrotic hepatocytes. The total number of leukocytes in the livers
of mice has been shown to increase ~3-fold a total of 18 h after
APAP injection, suggesting a notable inflammatory response during
AILI (68). Hepatocytes release
intracellular components that can act as damage-associated
molecular patterns (DAMPs), such as high mobility group box 1
protein, DNA fragments and HSPs, which act as ligands for Toll-like
receptors on macrophages (69),
thus leading to transcriptional activation of cytokines and
chemokines. Most of these inflammatory factors can be released
directly into circulation, but a few cytokines require caspase-1
activation to trigger their activity. Activated caspase-1 induces
the production of the NLRP3 inflammasome complex, which comprises
NLRP3, caspase-activated deoxyribonuclease and procaspase-1
(70); this complex mediates a
pro-inflammatory response through neutrophil and monocyte
activation, and recruitment to the liver (71). When neutrophils are activated
following an APAP overdose, those in the peripheral circulation
tend to move toward the site of injury in the early stages, with
the majority migrating to areas of necrosis, after which the injury
begins to worsen, peaking at 24 h. Subsequently, the neutrophil
levels begin to decrease during the recovery phase of liver injury.
Monocytes participate in inflammation via phagocytosis, regulates
connective tissue remodeling and fibrosis processes and the
formation of extracellular traps (72). Monocytes also serve an important
role during the aseptic response. On the one hand, activated
monocytes can control neutrophil activation and recruitment through
chemokines; on the other hand, when monocytes reach the necrotic
area, they can be transformed into monocyte-derived macrophages,
which leads to a rapid increase in the number of hepatic
macrophages. CCL2 is a mediator of monocyte entry from bone marrow
into circulation (73). Although
the aseptic response removes necrotic cell debris and promotes
tissue repair, leukocytes contribute to tissue damage. Given the
dual role of inflammation in APAP hepatotoxicity, it could be
hypothesized that a moderate inflammatory response might contribute
to tissue repair and hepatocyte regeneration, whereas an excessive
response could exacerbate liver injury (74).
Treatments
The primary treatment for APAP poisoning is the use
of the specific detoxifying drug NAC, which can minimize APAP
damage to the liver. In addition, liver transplantation is an
option to consider for patients with ALF. However, NAC may cause
some adverse effects in clinical application and there is a problem
of drug resistance. Therefore, there is an urgent need to develop
new drugs to improve the therapeutic efficacy. The present study
explores the possible therapeutic targets for APAP poisoning and
reviews the current major therapeutic approaches to inform future
treatment strategies.
Inhibition of toxic metabolite
production
Historical progress of research on NAC is shown in
Table I. The first demonstration
of the detoxifying role of GSH in DILI was made by Mitchell et
al (75) at the National
Institutes of Health in 1973, and their first report on oral NAC
treatment for APAP overdose was published in May 1977. In 1985, NAC
treatment was found to be effective in detoxifying NAPQI in mice 1
h after APAP administration (76),
and it was suggested that this detoxification may be related to
enhancement in APAP-GSH metabolite formation and attenuation in
protein adduct synthesis. Notably, NAC is a precursor of GSH; NAC
is deacetylated to cysteine and conjugated to glutamate, which is
further converted to glutamylcysteine by glutamate-cysteine ligase
(GCL) in hepatocytes. Glutamylcysteine binds to glycine via GSH
synthetase to produce GSH, which effectively prevents
hepatotoxicity due to GSH depletion (77). NAC also reduces the covalent
bonding of APAP, thereby attenuating hepatic necrosis, and improves
mitochondrial energy metabolism through ATP maintenance in addition
to scavenging reactive oxygen species (ROS) and peroxynitrite
(78). This energy generation is
accomplished primarily through the Krebs cycle, also known as the
tricarboxylic acid cycle or aerobic respiration, which is part of
the cellular respiration process. The Krebs cycle maintains cell
survival and function by breaking down glucose into carbon dioxide
and water to produce energy (ATP) (77). Furthermore, NAC exhibits
anti-inflammatory and antioxidant properties (79). Approximately 40 years after its
introduction, NAC remains the only Food and Drug
Administration-approved antidote for APAP overdose. The standard
oral or intravenous dosing regimen of NAC is highly effective in
patients who develop a moderate overdose within 8 h of APAP
ingestion (80). NAC was initially
administered orally; however, intravenous infusion is currently
more common in clinical practice. Mainly due to the fact that the
patient is in a coma and cannot be administered orally, at the same
time, intravenous injection can ensure 100% of the drug enters the
blood circulation, avoiding uncertainty in the oral absorption
process.
 | Table I.Historical progression of NAC for the
treatment of APAP hepatotoxicity. |
Table I.
Historical progression of NAC for the
treatment of APAP hepatotoxicity.
| First author,
year | Research
progress | (Refs.) |
|---|
| Peterson, 1977 | The first report of
NAC treatment of APAP overdose was presented. | (120) |
| Akakpo, 1985 | Oral NAC was
approved by the FDA for APAP overdose. | (33) |
| Corcoran, 1985 | After 1 h of APAP
treatment, NAC effectively promoted detoxification of NAPQI in
mice, leading to attenuated protein adduct formation. | (121) |
| Smilkstein,
1988 | Administration of
NAC within the first 8–10 h after APAP ingestion was revealed to be
effective in preventing liver injury and liver failure. | (122) |
| Rumack, 2002 | The IV NAC dosing
regimen of 300 mg/kg infused over 21 h was approved by the
FDA. | (123) |
| Yarema, 2009 | When NAC was
initiated within 12 h, the relative risk of hepatotoxicity was
lower in the IV group vs. the oral group. | (124) |
| Yang, 2009 | Long-term NAC
treatment was revealed to impair APAP-induced liver regeneration in
patients with ALI, at least in part by inhibiting the NF-κB
activation pathway. | (125) |
| Blieden, 2010 | Common side
effects, such as nausea and vomiting, were reported to occur after
NAC treatment. | (126) |
| Saito, 2010 | The most effective
protective mechanism of NAC was revealed to be the enhancement of
hepatocyte clearance of NAPQI, which could prevent covalent
modification of cellular proteins, thereby blocking the initiation
of APAP toxicity. | (77) |
| Waring, 2012 | The side effects of
NAC were reported to be similar to the clinical manifestations of
allergic reactions, including rash, pruritus, angioedema and
bronchospasm. | (127) |
| de Andrade,
2015 | NAC was revealed to
attenuate hepatic injury inflammation during APAP-induced
hepatotoxicity and to exert antioxidant effects. | (128) |
| Khayyat, 2016 | After NAC
treatment, GSH was significantly elevated in an APAP-treated group;
the protective effect of NAC may occur by its promotion of GSH
biosynthesis. | (129) |
| Downs, 2021 | A standard dose of
NAC was sufficient to prevent 91% of hepatotoxicity in a large
number of patients with APAP overdose treated with NAC within 8
h. | (130) |
In the United States, the approved dose of oral NAC
is 140 mg/kg body weight followed by 70 mg/kg body weight every 4 h
for a total of 17 doses (81);
however, the efficacy of NAC reduces in patients with advanced
disease or after a large overdose because the drug has often been
metabolized in the body for some time before the patient is
admitted to the clinic (39). A
series of adverse reactions can occur during NAC treatment; some
examples are intravenous NAC side effects such as allergic
reactions, nausea, vomiting, diarrhea or constipation, fever,
headache, drowsiness and low blood pressure (82). These reactions mainly occur in the
first hour after NAC infusion, corresponding to the peak of NAC
concentration (83); therefore, a
drug that can prolong the therapeutic window period of NAC is
urgently awaited. Based on NAC trials, scientists have shown
through animal experiments that co-treatment of fomepizole (4MP)
with APAP can effectively prevent AILI (84,85);
4MP can strongly attenuate hepatic GSH depletion, and can almost
eliminate APAP-AD and all oxidative metabolites of APAP, suggesting
that it effectively inhibits CYP2E1 and largely prevents the
formation of NAPQI. 4MP may be considered a good candidate for
NAC-assisted treatment of patients with selective APAP overdose
(33).
Activation of autophagy
Autophagy is the conserved process by which
substrates in the cytoplasm are translocated to lysosomes through
intermediate double-membrane-bound vesicles called autophagosomes
(86). It is a tightly regulated
cellular process that is essential for the maintenance of cellular
homeostasis through lysosomal degradation to remove unwanted
cytoplasmic contents, including misfolded or aggregated proteins,
accumulated lipids, excessive or damaged organelles, and harmful
pathogens (87), in order to
achieve cell renewal (88). It has
been demonstrated that APAP treatment induces the formation of
autophagosomes in mouse livers and that autophagy defects enhance
hepatocyte apoptosis and necrotic cell death resulting from APAP
treatment, a process that relies on MPTP opening, depolarization of
mitochondrial membranes, swelling and loss of proteins such as
cytochrome c (89).
Mitochondrial damage is a key event in APAP
hepatotoxicity. Autophagy selectively removes damaged organelles,
especially mitochondria, thereby preventing oxidative stress and
necrotic cell death caused by mitochondrial damage (90). This process is largely dependent on
a mitochondrial serine/threonine kinase (PINK1). In healthy
mitochondria, the mitochondrial transmembrane potential drives
PINK1 into the IMM via outer mitochondrial membrane translocase;
mitochondrial damage induces the accumulation of PINK1, which
recruits Parkin to initiate mitosis. Activated Parkin mediates
ubiquitination of mitochondrial outer membrane proteins, which acts
as a signal to recruit autophagy aptamers, such as OPTN, NDP52 and
p62. As a result, the autophagy machinery is recruited to degrade
damaged mitochondria (91). Excess
APAP can activate autophagy in mouse livers and primary hepatocytes
for protective purposes, which can counteract the mitochondrial
oxidative stress induced by APAP and JNK activation, among others
(92). Mechanistically, this is
likely a compensatory response to excess ROS after APAP
intoxication, and in this regard, mitochondria are the main source
of ROS required for autophagy-inducing signaling; ROS accumulation
can induce autophagy by directly affecting core autophagy
mechanisms and indirectly affecting components of
autophagy-regulated signaling pathways (93). Considering that the accumulation of
APAP-AD is an early and critical step in mitochondrial damage, the
formation of such adducts poses a potential threat to mitochondria.
Therefore, timely and effective removal of APAP-AD may be an
important strategy to alleviate the toxic effects of APAP on
mitochondria, thus maintaining their normal function, ensuring
smooth intracellular energy metabolism, and providing sufficient
ATP for cells. Autophagy can selectively remove deleterious
APAP-AD, thereby protecting the liver from AILI to a certain extent
(89). Furthermore, activated
autophagy can remove damaged mitochondria, which are the sites of
ROS production; elevated ROS levels can induce autophagy. Removal
of damaged mitochondria can reduce ROS production and eliminate
inflammatory vesicles (e.g., NLRP3 inflammatory vesicles),
potentially inhibiting inflammatory responses and maintaining the
normal turnover and regulatory functions of the ER (94).
Activation of the nuclear factor
erythroid 2-related factor 2 (Nrf2) signaling pathway and
inhibition of ferroptosis
Nrf2 is involved in various aspects of cellular and
organismal detoxification against oxidative stress, regulation of
cellular metabolism and promotion of cellular proliferation, and
serves a crucial role in the pathological mechanisms of several
diseases as well as homeostasis (95). The Nrf2 pathway is considered to
have a key role in AILI, and activation of Nrf2 signaling is a
potential target for ameliorating APAP hepatotoxicity. NAPQI, the
intermediate metabolite produced by APAP, contributes to GSH
depletion and ROS formation; this triggers oxidative stress, which
leads to mitochondrial dysfunction, hepatocyte necrosis and liver
injury (70). To counteract
oxidative stress induced by ROS, the body maintains cellular redox
homeostasis through a series of antioxidant molecules and
detoxifying enzymes. The Nrf2/ARE pathway is one of the major
nuclear transcription factor-associated mechanisms that respond to
this process by activating phase II detoxification enzymes; Nrf2
can detoxify NAPQI by activating antioxidant enzymes (96). GCL, a key enzyme in GSH synthesis,
consists of catalytic (GCLC) and modifying subunits; GCLC is one of
the downstream targets of Nrf2 activation (97).
Iron-dependent cell death (ferroptosis) is a
regulated form of cell death that can be induced when intracellular
GPX4 is directly or indirectly inhibited by low GSH levels
(98). APAP has been extensively
studied and shown to induce hepatic lipid peroxidation and
ferroptosis, a cascading effect that ultimately leads to ALF in
mice (99). Notably, APAP-induced
ALF may be prevented by pharmacological and genetic inhibition of
ferroptosis, which acts as an initial event during the course of
APAP-induced hepatotoxicity and is not only capable of directly
damaging hepatocytes, but also triggers the release of
extracellular substances, such as DAMPs, that further exacerbate
inflammatory responses and intensify liver injury. Inhibition of
the Nrf2 pathway increases sensitivity to ferroptosis and GPX4 is a
downstream target of Nrf2 (100).
Several natural small-molecule drugs can activate the Nrf2 pathway,
and typically exhibit better biocompatibility and lower side
effects than conventional chemical drugs; they are of natural
origin, easily accessible and cost-effective. For example,
curcumin, a plant polyphenol extracted from turmeric, has been
shown to upregulate the expression of antioxidant enzymes, such as
GPX4, by promoting the release and activation of Nrf2, thereby
attenuating ferroptosis and alleviating APAP-induced
hepatotoxicity; this finding provides a new strategy and direction
for the treatment of APAP-induced ALF (101). Research progress on natural
small-molecule drugs for attenuating AILI has been summarized in
Table II, which lists the names
and mechanisms of action of various natural small-molecule drugs,
providing valuable references for researchers and clinicians.
| Table II.Advances in the study of natural
small molecule drug substances to ameliorate APAP liver injury. |
Table II.
Advances in the study of natural
small molecule drug substances to ameliorate APAP liver injury.
| First author,
year | Small molecule
drug | Mechanism | (Refs.) |
|---|
| Cai, 2022 | ASX | ASX ameliorated
AILI in vivo and in vitro by reducing inflammation,
inhibiting oxidative stress and ferroptosis via the NF-κB pathway,
and increasing autophagy via the Nrf2/HO-1 pathway. | (101) |
| Li, 2023 | KA | KA activated the
Nrf2 signaling pathway and inhibited ferroptosis. KA pretreatment
improved biochemical indices related to liver function, and
attenuated liver necrosis and inflammation in APAP-induced
mice. | (97) |
| Pang, 2016 | CA | CA prevented
APAP-induced hepatotoxicity by decreasing the expression of Keap1
and inhibiting the binding of Keap1 to Nrf2, which activated Nrf2,
leading to increased expression of antioxidant signaling. | (131) |
| Yang, 2020 | Limonin | Limonin attenuated
APAP-induced hepatotoxicity by activating the Nrf2 antioxidant
pathway and inhibiting the NF-κB inflammatory response through
upregulation of Sirt1. | (132) |
| An, 2023 | Abietic acid | Abietic acid
inhibited APAP-induced NF-κB activation, and increased Nrf2 and
HO-1 expression. In vitro, the inhibitory effects of Abietic
acid on APAP-induced inflammation and iron death were reversed when
Nrf2 was knocked down. | (133) |
| Lv, 2019 | Coriandrin | Coriandrin could
counteract APAP-induced ALF by inducing Nrf2 via the AMPK/GSK3β
pathway. | (134) |
| Wang, 2016 | EsA | EsA enhanced
Nrf2-regulated survival mechanisms through the AMPK/Akt/GSK3β
pathway and has been shown to possess protective potential against
APAP toxicity. | (135) |
| Wang, 2019 | Fariro | Activation of Nrf2
and induction of autophagy by Fariro through the AMPK/AKT pathway
contributed to its hepatoprotective activity in vitro, and
had protective potential against APAP-induced hepatotoxicity. | (96) |
| Zhu, 2023 | Xanthohumol | Xanthohumol
inhibited APAP-induced liver injury by suppressing oxidative stress
and mitochondrial dysfunction through activation of Nrf2 by
AMPK/Akt/GSK3β. | (136) |
| Yao, 2022 | Rosmarinic
acid | Rosmarinic acid was
shown to protect against AILI through Nrf2-mediated inhibition of
the NEK7-NLRP3 signaling pathway. | (137) |
| Jiang, 2022 | Sarmentosin | Sarmentosin
attenuated APAP-induced ALF and protected hepatocytes from APAP
injury in mice, and the important role of Nrf2-mediated oxidative
stress in the dynamics of this process was demonstrated. | (138) |
Liver repair and regeneration
Liver regeneration is a compensatory and beneficial
process in response to hepatocyte death and tissue damage;
stimulating this process may be a potential therapeutic strategy to
control APAP hepatotoxicity (102). The role played by innate immune
cells in APAP hepatotoxicity has been controversial, but evidence
has emphasized that innate immune cells are critical for liver
repair (103). Immune cells, such
as neutrophils and macrophages, stay mainly in necrotic areas after
APAP overdose; hepatic neutrophil infiltration is a key step of the
innate immune response to acute liver injury (104) and is critical for recovery after
APAP overdose (105). In
addition, platelets serve a key role in the course of AILI and
platelet depletion can attenuate hepatic injury to some extent.
Platelet-expressed c-type lectin-like receptor (CLEC)-2 is a key
mediator of platelet activation, and CLEC-2-mediated elimination of
platelet signaling may enhance recovery after acute liver injury
via TNF-α-dependent hepatic recruitment of neutrophils (103). In this context, neutrophil
infiltration represents an innate immune repair response that can
aid in rapid liver repair after acute injury.
Hepatocytes have a major role in the process of
liver regeneration. Liver recovery after APAP hepatotoxicity
involves a combination of numerous factors, such as hepatic
microvascular reconstruction and interactions between different
cell types. Among these factors, vascular endothelial growth factor
(VEGF), a major regulator of tumorigenesis, inflammation and wound
healing, can effectively promote liver regeneration (106). Cellular crosstalk between LSECs
and hepatocytes during liver regeneration serves an important role
in hepatic sinusoidal homeostasis and physiological angiogenesis
(107). A comparison of VEGF
receptor 1 (VEGFR1) tyrosine kinase-knockout (VEGFR1 TK2/2) and
wild-type (WT) mice revealed that the expression levels of
proliferating cell nuclear antigen and growth factors, such as
hepatocyte growth factor, CD31 and basic fibroblast growth factor,
were lower in the knockout mice than in WT mice. In addition, the
VEGFR1 TK2/2 mice showed impaired hepatic microvascular function,
which was reduced by ~40% compared with that in WT mice, a
phenomenon that was associated with enhanced hepatic MMP-9
expression (108). Selective
activation of VEGFR1 could serve as a target to promote tissue
repair by facilitating blood sinusoidal cell recovery after acute
liver injury; however, several risks are associated with VEGFR1
activation. Its activation on endothelial cells can promote
malignant angiogenesis, thereby enhancing the migration and
activity of endothelial cells, which may facilitate the invasion of
hepatocellular carcinoma cells (109).
Liver transplant
Hepatotoxicity can be completely prevented if NAC is
administered within 10 h of overdose. Patients who do not receive
NAC in time can develop severe liver injury, which may progress to
ALF. Therefore, in cases of severe liver injury caused by APAP,
liver transplantation may be the only treatment option to save the
life of the patient (110).
Moreover, liver transplantation is the only effective strategy for
the treatment of APAP-induced liver failure (111). The decision to undergo liver
transplantation for APAP overdose-induced ALF is challenging and
may be affected by the scarcity of donor liver grafts, acute graft
rejection, lifelong immunosuppression and unaffordable costs.
Psychosocial issues of patients with APAP overdose are of concern,
as >30% of those who meet the criteria for transplantation have
severe mental illness, or alcohol and/or drug abuse problems
(112).
Discussion
The prognosis of AILI is influenced by multiple
factors. APAP, a widely used analgesic and antipyretic, has become
a major cause of ALF in several countries and AILI is a major
public health problem worldwide. The causes of APAP intoxication
can be divided into two categories. For the first cause, i.e.,
self-administered overdose, patients are taken to the emergency
room for NAC treatment early and serious injury can be avoided. In
the other category, i.e., accidental overdoses, the optimal time
for treatment is often missed before an adverse reaction sets in
because the symptoms of poisoning are not obvious in the early
stages (24). The total ingested
dose of APAP is the most important determinant of the development
and severity of APAP-induced hepatotoxicity. The Rumack-Matthew
nomogram, developed in the 1970s as a tool for assessing the risk
of hepatotoxicity after ingestion of APAP, consists of two main
lines: The Treatment Line and the Rumack-Matthew Line, which
predict the risk of hepatotoxicity from 4 to 24 h after ingestion.
However, this chart does not apply to patients tested 24 h after
ingestion or with a history of multiple ingestions (113). In addition, patterns of use and
certain factors [e.g., chronic alcohol abuse (114), age, concomitant use of certain
medications, genetic factors, pre-existing liver disease and
nutritional status] can influence susceptibility to APAP
hepatotoxicity via multiple mechanisms (115). For example, a study of 1,039
children aged <7 years found no susceptibility to APAP-induced
hepatotoxicity after mild to moderate (up to 200 mg/kg) APAP
ingestion (116). This
observation could be explained by the fact that the sulfate
coupling of the drug is more pronounced in children, whereas the
glucuronide coupling is more predominant in adults, which means
that differences in the binding pathways may result in a reduced
risk of APAP-related toxicity in children. In addition, because
NAPQI formation is dependent on APAP metabolism via the CYP system
and coupling to mercapturic acid metabolites after therapeutic
doses, urinary concentrations of these metabolites are much lower
in children than in adults. This suggests that CYP serves a less
significant role in APAP metabolism in children than in adults
(117). Timely medical
intervention, such as the clinical application of NAC, can minimize
liver injury.
Research on the epidemiology, mechanism underlying
toxicity, diagnosis and treatment of AILI has progressed
considerably. However, some persistent challenges include the early
diagnosis of APAP-induced hepatotoxicity (when it is not clinically
apparent) to avoid severe injury later, and the identification of
safe doses for different populations in terms of their
sensitivities. Since LSEC damage often occurs in the early stages
of liver injury, it could be proposed that the detection of
LSEC-specific damage may provide a timely and accurate diagnosis of
patients when conventional markers, such as ALT and AST, do not
show notable changes. For example, changes in their morphology can
be observed by electron microscopy; in normal liver samples, the
thin and weak cytoplasm of LSECs contains a large number of pores,
which is its unique structure (118). However, when the liver is
diseased, the size of the pores will gradually change; for example,
in liver fibrosis, the pores will disappear progressively until a
continuous basement membrane is formed (119). To achieve this goal, future
studies are required to explore the specific relationship between
LSEC injury and APAP hepatotoxicity, and to develop more sensitive
and specific assays.
In conclusion, research on AILI is continuously
advancing, and researchers are exploring the mechanisms of
toxicity, biomarkers, therapeutic targets and new therapeutic
approaches from multiple perspectives. The results of these
research studies are expected to provide new ideas and methods for
the prevention and treatment of DILI, and to further reduce the
risk of liver injury caused by APAP and other drugs.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
RL and HL designed the subject of review. RL, HL,
HW, YuX, XX, YiX, HH, XL, CL and JY participated in writing and
reviewing the manuscript. All authors have read and approved the
final manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hoofnagle JH and Björnsson ES:
Drug-induced liver injury-types and phenotypes. N Engl J Med.
381:264–273. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shen T, Liu Y, Shang J, Xie Q, Li J, Yan
M, Xu J, Niu J, Liu J, Watkins PB, et al: Incidence and etiology of
drug-induced liver injury in mainland China. Gastroenterology.
156:2230–2241.e11. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Es B: The epidemiology of newly recognized
causes of drug-induced liver injury: An update. Pharmaceuticals
(Basel). 17:5202024. View Article : Google Scholar
|
|
4
|
Warnet JM, Bakar-Wesseling I, Thevenin M,
Serrano JJ, Jacqueson A, Boucard M and Claude JR: Effects of
subchronic low-protein diet on some tissue glutathione-related
enzyme activities in the rat. Arch Toxicol Suppl. 11:45–49.
1987.PubMed/NCBI
|
|
5
|
Andrade RJ, Chalasani N, Björnsson ES,
Suzuki A, Kullak-Ublick GA, Watkins PB, Devarbhavi H, Merz M,
Lucena MI, Kaplowitz N and Aithal GP: Drug-induced liver injury.
Nat Rev Dis Primers. 5:582019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kjartansdottir I, Bergmann OM and
Arnadottir RS: Paracetamol intoxications: A retrospective
population-based study in iceland. Scand J Gastroenterol.
47:1344–1352. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tan ST, Lo CH, Liao CH and Su YJ:
Sex-based differences in the predisposing factors of overdose: A
retrospective study. Biomed Rep. 16:492022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Paulose-Ram R, Hirsch R, Dillon C,
Losonczy K, Cooper M and Ostchega Y: Prescription and
non-prescription analgesic use among the US adult population:
Results from the third national health and nutrition examination
survey (NHANES III). Pharmacoepidemiol Drug Saf. 12:315–326. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rubin JB, Hameed B, Gottfried M, Lee WM
and Sarkar M; Acute Liver Failure Study Group, :
Acetaminophen-induced acute liver failure is more common and more
severe in women. Clin Gastroenterol Hepatol. 16:936–946. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Wu Q, Liu A, Anadón A, Rodríguez
JL, Martínez-Larrañaga MR, Yuan Z and Martínez MA: Paracetamol:
Overdose-induced oxidative stress toxicity, metabolism, and
protective effects of various compounds in vivo and in vitro. Drug
Metab Rev. 49:395–437. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brune K, Renner B and Tiegs G:
Acetaminophen/paracetamol: A history of errors, failures and false
decisions. Eur J Pain. 19:953–965. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chidiac AS, Buckley NA, Noghrehchi F and
Cairns R: Paracetamol (acetaminophen) overdose and hepatotoxicity:
Mechanism, treatment, prevention measures, and estimates of burden
of disease. Expert Opin Drug Metab Toxicol. 19:297–317. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davidson DG and Eastham WN: Acute liver
necrosis following overdose of paracetamol. Br Med J. 2:497–499.
1966. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
McGill MR and Hinson JA: The development
and hepatotoxicity of acetaminophen: Reviewing over a century of
progress. Drug Metab Rev. 52:472–500. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dart RC, Erdman AR, Olson KR, Christianson
G, Manoguerra AS, Chyka PA, Caravati EM, Wax PM, Keyes DC, Woolf
AD, et al: Acetaminophen poisoning: An evidence-based consensus
guideline for out-of-hospital management. Clin Toxicol (Phila).
44:1–18. 2006. View Article : Google Scholar
|
|
16
|
Chiew AL, Isbister GK, Stathakis P,
Isoardi KZ, Page C, Ress K, Chan BSH and Buckley NA: Acetaminophen
metabolites on presentation following an acute acetaminophen
overdose (ATOM-7). Clin Pharmacol Ther. 113:1304–1314. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rumack BH, Peterson RC, Koch GG and Amara
IA: Acetaminophen overdose. 662 cases with evaluation of oral
acetylcysteine treatment. Arch Intern Med. 141:380–385. 1981.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singer AJ, Carracio TR and Mofenson HC:
The temporal profile of increased transaminase levels in patients
with acetaminophen-induced liver dysfunction. Ann Emerg Med.
26:49–53. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Larson AM: Acetaminophen hepatotoxicity.
Clin Liver Dis. 11:525–548. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramachandran A and Jaeschke H:
Acetaminophen hepatotoxicity. Semin Liver Dis. 39:221–234. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ma J, Li M, Li N, Chan WY and Lin G:
Pyrrolizidine alkaloid-induced hepatotoxicity associated with the
formation of reactive metabolite-derived pyrrole-protein adducts.
Toxins. 13:7232021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McGill MR and Jaeschke H: Mechanistic
biomarkers in acetaminophen-induced hepatotoxicity and acute liver
failure: From preclinical models to patients. Expert Opin Drug
Metab Toxicol. 10:1005–1017. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nelson SD: Molecular mechanisms of the
hepatotoxicity caused by acetaminophen. Semin Liver Dis.
10:267–278. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee WM: Acetaminophen (APAP)
hepatotoxicity-isn't it time for APAP to go away? J Hepatology.
67:1324–1331. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang XP, Thiessen JJ, Spino M and
Templeton DM: Transport of iron chelators and chelates across MDCK
cell monolayers: Implications for iron excretion during chelation
therapy. Int J Hematol. 91:401–412. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Letelier ME, López-Valladares M,
Peredo-Silva L, Rojas-Sepúlveda D and Aracena P: Microsomal
oxidative damage promoted by acetaminophen metabolism. Toxicol In
Vitro. 25:1310–1313. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hinson JA, Pumford NR and Roberts DW:
Mechanisms of acetaminophen toxicity: Immunochemical detection of
drug-protein adducts. Drug Metab Rev. 27:73–92. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ramachandran A and Jaeschke H: A
mitochondrial journey through acetaminophen hepatotoxicity. Food
Chem Toxicol. 140:1112822020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
McGill MR, Sharpe MR, Williams CD, Taha M,
Curry SC and Jaeschke H: The mechanism underlying
acetaminophen-induced hepatotoxicity in humans and mice involves
mitochondrial damage and nuclear DNA fragmentation. J Clin Invest.
122:1574–1583. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nguyen NU and Stamper BD: Polyphenols
reported to shift APAP-induced changes in MAPK signaling and
toxicity outcomes. Chem Biol Interact. 277:129–136. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nakagawa H, Maeda S, Hikiba Y, Ohmae T,
Shibata W, Yanai A, Sakamoto K, Ogura K, Noguchi T, Karin M, et al:
Deletion of apoptosis signal-regulating kinase 1 attenuates
acetaminophen-induced liver injury by inhibiting c-jun N-terminal
kinase activation. Gastroenterology. 135:1311–1321. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thévenin AF, Zony CL, Bahnson BJ and
Colman RF: GST pi modulates JNK activity through a direct
interaction with JNK substrate, ATF2. Protein Sci. 20:834–848.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akakpo JY, Ramachandran A, Curry SC, Rumac
BH and Jaeschke H: Comparing N-acetylcysteine and 4-methylpyrazole
as antidotes for acetaminophen overdose. Arch Toxicol. 96:453–465.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moles A, Torres S, Baulies A, Garcia-Ruiz
C and Fernandez-Checa JC: Mitochondrial-lysosomal axis in
acetaminophen hepatotoxicity. Front Pharmacol. 9:4532018.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xu Y, Xia Y, Liu Q, Jing X, Tang Q, Zhang
J, Jia Q, Zhang Z, Li J, Chen J, et al: Glutaredoxin-1 alleviates
acetaminophen-induced liver injury by decreasing its toxic
metabolites. J Pharm Anal. 13:1548–1561. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chowdhury A, Lu J, Zhang R, Nabila J, Gao
H, Wan Z, Adelusi Temitope I, Yin X and Sun Y: Mangiferin
ameliorates acetaminophen-induced hepatotoxicity through APAP-cys
and JNK modulation. Biomed Pharmacother. 117:1090972019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jaeschke H, Adelusi OB, Akakpo JY, Nguyen
NT, Sanchez-Guerrero G, Umbaugh DS, Ding WX and Ramachandran A:
Recommendations for the use of the acetaminophen hepatotoxicity
model for mechanistic studies and how to avoid common pitfalls.
Acta Pharm Sin B. 11:3740–3755. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Radi R, Peluffo G, Alvarez MN, Naviliat M
and Cayota A: Unraveling peroxynitrite formation in biological
systems. Free Radic Biol Med. 30:463–488. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cover C, Mansouri A, Knight TR, Bajt ML,
Lemasters JJ, Pessayre D and Jaeschke H: Peroxynitrite-induced
mitochondrial and endonuclease-mediated nuclear DNA damage in
acetaminophen hepatotoxicity. J Pharmacol Exp Ther. 315:879–887.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saito C, Lemasters JJ and Jaeschke H:
c-jun N-terminal kinase modulates oxidant stress and peroxynitrite
formation independent of inducible nitric oxide synthase in
acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 246:8–17.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kon K, Kim JS, Uchiyama A, Jaeschke H and
Lemasters JJ: Lysosomal iron mobilization and induction of the
mitochondrial permeability transition in acetaminophen-induced
toxicity to mouse hepatocytes. Toxicol Sci. 117:101–108. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jaeschke H, Ramachandran A, Chao X and
Ding WX: Emerging and established modes of cell death during
acetaminophen-induced liver injury. Arch Toxicol. 93:3491–3502.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bajt ML, Farhood A, Lemasters JJ and
Jaeschke H: Mitochondrial bax translocation accelerates DNA
fragmentation and cell necrosis in a murine model of acetaminophen
hepatotoxicity. J Pharmacol Exp Ther. 324:8–14. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li GW, Liu J and Chen L: Continuous
ambulatory peritoneal dialysis treatment and blood glucose control
in diabetics with end-stage diabetic nephropathy. Zhonghua Nei Ke
Za Zhi. 28:360–363. 3821989.(In Chinese). PubMed/NCBI
|
|
45
|
Yoon E, Babar A, Choudhary M, Kutner M and
Pyrsopoulos N: Acetaminophen-induced hepatotoxicity: A
comprehensive update. J Clin Transl Hepatol. 4:131–142.
2016.PubMed/NCBI
|
|
46
|
Knight TR, Ho YS, Farhood A and Jaeschke
H: Peroxynitrite is a critical mediator of acetaminophen
hepatotoxicity in murine livers: Protection by glutathione. J
Pharmacol Exp Ther. 303:468–475. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Gracia-Sancho J, Marrone G and
Fernández-Iglesias A: Hepatic microcirculation and mechanisms of
portal hypertension. Nat Rev Gastroenterol. 16:221–234. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Gracia-Sancho J, Caparrós E,
Fernández-Iglesias A and Francés R: Role of liver sinusoidal
endothelial cells in liver diseases. Nat Rev Gastroenterol Hepatol.
18:411–431. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
McCuskey RS, Bethea NW, Wong J, McCuskey
MK, Abril ER, Wang X, Ito Y and DeLeve LD: Ethanol binging
exacerbates sinusoidal endothelial and parenchymal injury elicited
by acetaminophen. J Hepatol. 42:371–377. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ito Y, Bethea NW, Abril ER and McCuskey
RS: Early hepatic microvascular injury in response to acetaminophen
toxicity. Microcirculation. 10:391–400. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
McCuskey RS: Sinusoidal endothelial cells
as an early target for hepatic toxicants. Clin Hemorheol Microcirc.
34:5–10. 2006.PubMed/NCBI
|
|
52
|
Walker RM, Racz WJ and McElligott TF:
Acetaminophen-induced hepatotoxic congestion in mice. Hepatology.
5:233–240. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Damen L, Bruijn JKJ, Verhagen AP, Berger
MY, Passchier J and Koes BW: Symptomatic treatment of migraine in
children: A systematic review of medication trials. Pediatrics.
116:e295–e302. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Williams AM, Langley PG, Osei-Hwediah J,
Wendon JA and Hughes RD: Hyaluronic acid and endothelial damage due
to paracetamol-induced hepatotoxicity. Liver Int. 23:110–115. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
DeLeve LD, Wang X, Kaplowitz N, Shulman
HM, Bart JA and van der Hoek A: Sinusoidal endothelial cells as a
target for acetaminophen toxicity. Direct action versus requirement
for hepatocyte activation in different mouse strains. Biochem
Pharmacol. 53:1339–1345. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Walter P and Ron D: The unfolded protein
response: From stress pathway to homeostatic regulation. Science.
334:1081–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Maytin EV, Ubeda M, Lin JC and Habener JF:
Stress-inducible transcription factor CHOP/gadd153 induces
apoptosis in mammalian cells via p38 kinase-dependent and
-independent mechanisms. Exp Cell Res. 267:193–204. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Lee DH, Lee B, Park JS, Lee YS, Kim JH,
Cho Y, Jo Y, Kim HS, Lee YH, Nam KT and Bae SH: Inactivation of
Sirtuin2 protects mice from acetaminophen-induced liver injury:
Possible involvement of ER stress and S6K1 activation. BMB Rep.
52:190–195. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kalinec GM, Thein P, Parsa A, Yorgason J,
Luxford W, Urrutia R and Kalinec F: Acetaminophen and NAPQI are
toxic to auditory cells via oxidative and endoplasmic reticulum
stress-dependent pathways. Hear Res. 313:26–37. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang X, Xiong W, Chen LL, Huang JQ and
Lei XG: Selenoprotein V protects against endoplasmic reticulum
stress and oxidative injury induced by pro-oxidants. Free Radic
Biol Med. 160:670–679. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Uzi D, Barda L, Scaiewicz V, Mills M,
Mueller T, Gonzalez-Rodriguez A, Valverde AM, Iwawaki T, Nahmias Y,
Xavier R, et al: CHOP is a critical regulator of
acetaminophen-induced hepatotoxicity. J Hepatol. 59:495–503. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Nagy G, Kardon T, Wunderlich L, Szarka A,
Kiss A, Schaff Z, Bánhegyi G and Mandl J: Acetaminophen induces ER
dependent signaling in mouse liver. Arch Biochem Biophys.
459:273–279. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jaeschke H, Gujral JS and Bajt ML:
Apoptosis and necrosis in liver disease. Liver Int. 24:85–89. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Henderson CJ, Wolf CR, Kitteringham N,
Powell H, Otto D and Park BK: Increased resistance to acetaminophen
hepatotoxicity in mice lacking glutathione S-transferase pi. Proc
Natl Acad Sci USA. 97:12741–12745. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Foufelle F and Fromenty B: Role of
endoplasmic reticulum stress in drug-induced toxicity. Pharmacol
Res Perspect. 4:e002112016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Xiao T, Liang X, Liu H, Zhang F, Meng W
and Hu F: Mitochondrial stress protein HSP60 regulates ER
stress-induced hepatic lipogenesis. J Mol Endocrinol. 64:67–75.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Mihm S: Danger-associated molecular
patterns (DAMPs): Molecular triggers for sterile inflammation in
the liver. Int J Mol Sci. 19:31042018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sanz-Garcia C, Ferrer-Mayorga G,
González-Rodríguez Á, Valverde AM, Martín-Duce A, Velasco-Martín
JP, Regadera J, Fernández M and Alemany S: Sterile inflammation in
acetaminophen-induced liver injury is mediated by Cot/tpl2. J Biol
Chem. 288:15342–15351. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Jaeschke H, Williams CD, Ramachandran A
and Bajt ML: Acetaminophen hepatotoxicity and repair: The role of
sterile inflammation and innate immunity. Liver Int. 32:8–20. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Jaeschke H and Ramachandran A:
Acetaminophen hepatotoxicity: Paradigm for understanding mechanisms
of drug-induced liver injury. Ann Rev Pathol. 19:453–478. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Jaeschke H and Ramachandran A: Mechanisms
and pathophysiological significance of sterile inflammation during
acetaminophen hepatotoxicity. Food Chem Toxicol. 138:1112402020.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Guo H, Chen S, Xie M and Zheng M: The
complex roles of neutrophils in APAP-induced liver injury. Cell
Prolif. 54:e130402021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Krenkel O and Tacke F: Liver macrophages
in tissue homeostasis and disease. Nat Rev Immunol. 17:306–321.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Shan S, Shen Z and Song F: Autophagy and
acetaminophen-induced hepatotoxicity. Arch Toxicol. 92:2153–2161.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Mitchell JR, Jollow DJ, Potter WZ, Davis
DC, Gillette JR and Brodie BB: Acetaminophen-induced hepatic
necrosis. I. Role of drug metabolism. J Pharmacol Exp Ther.
187:185–194. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Renzi FP, Donovan JW, Martin TG, Morgan L
and Harrison EF: Concomitant use of activated charcoal and
N-acetylcysteine. Ann Emerg Med. 14:568–572. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Saito C, Zwingmann C and Jaeschke H: Novel
mechanisms of protection against acetaminophen hepatotoxicity in
mice by glutathione and N-acetylcysteine. Hepatology. 51:246–254.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lasram MM, Dhouib IB, Annabi A, El Fazaa S
and Gharbi N: A review on the possible molecular mechanism of
action of N-acetylcysteine against insulin resistance and type-2
diabetes development. Clin Biochem. 48:1200–1208. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Jones AL: Mechanism of action and value of
N-acetylcysteine in the treatment of early and late acetaminophen
poisoning: A critical review. J Toxicol Clin Toxicol. 36:277–285.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dell'Aglio DM, Sutter ME, Schwartz MD,
Koch DD, Algren DA and Morgan BW: Acute chloroform ingestion
successfully treated with intravenously administered
N-acetylcysteine. J Med Toxicol. 6:143–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fisher ES and Curry SC: Evaluation and
treatment of acetaminophen toxicity. Adv Pharmacol. 85:263–272.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Park BK, Dear JW and Antoine DJ:
Paracetamol (acetaminophen) poisoning. BMJ Clin Evid.
2015:21012015.PubMed/NCBI
|
|
83
|
Licata A, Minissale MG, Stankevičiūtė S,
Sanabria-Cabrera J, Lucena MI, Andrade RJ and Almasio PL:
N-acetylcysteine for preventing acetaminophen-induced liver injury:
A comprehensive review. Front Pharmacol. 13:8285652022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Akakpo JY, Jaeschke MW, Ramachandran A,
Curry SC, Rumack BH and Jaeschke H: Delayed administration of
N-acetylcysteine blunts recovery after an acetaminophen overdose
unlike 4-methylpyrazole. Arch Toxicol. 95:3377–3391. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Akakpo JY, Ramachandran A, Rumack BH,
Wallace DP and Jaeschke H: Lack of mitochondrial Cyp2E1 drives
acetaminophen-induced ER stress-mediated apoptosis in mouse and
human kidneys: Inhibition by 4-methylpyrazole but not
N-acetylcysteine. Toxicology. 500:1536922023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Matsuzawa-Ishimoto Y, Hwang S and Cadwell
K: Autophagy and inflammation. Annu Rev Immunol. 36:73–101. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Levine B and Klionsky DJ: Development by
self-digestion: Molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Moore MN: Autophagy as a second level
protective process in conferring resistance to
environmentally-induced oxidative stress. Autophagy. 4:254–256.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chao X, Wang H, Jaeschke H and Ding WX:
Role and mechanisms of autophagy in acetaminophen-induced liver
injury. Liver Int. 38:1363–1374. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: Cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Khaminets A, Heinrich T, Mari M, Grumati
P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N,
et al: Regulation of endoplasmic reticulum turnover by selective
autophagy. Nature. 522:354–358. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lin Z, Wu F, Lin S, Pan X, Jin L, Lu T,
Shi L, Wang Y, Xu A and Li X: Adiponectin protects against
acetaminophen-induced mitochondrial dysfunction and acute liver
injury by promoting autophagy in mice. J Hepatol. 61:825–831. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Rhodes DG, Sarmiento JG and Herbette LG:
Kinetics of binding of membrane-active drugs to receptor sites.
Diffusion-limited rates for a membrane bilayer approach of
1,4-dihydropyridine calcium channel antagonists to their active
site. Mol Pharmacol. 27:612–623. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Mochida K, Oikawa Y, Kimura Y, Kirisako H,
Hirano H, Ohsumi Y and Nakatogawa H: Receptor-mediated selective
autophagy degrades the endoplasmic reticulum and the nucleus.
Nature. 522:359–362. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhou J, Zheng Q and Chen Z: The Nrf2
pathway in liver diseases. Front Cell Dev. 10:8262042022.
View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wang L, Wei W, Xiao Q, Yang H and Ci X:
Farrerol ameliorates APAP-induced hepatotoxicity via activation of
Nrf2 and autophagy. Int J Biol Sci. 15:788–799. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Li H, Weng Q, Gong S, Zhang W, Wang J,
Huang Y, Li Y, Guo J and Lan T: Kaempferol prevents
acetaminophen-induced liver injury by suppressing hepatocyte
ferroptosis via Nrf2 pathway activation. Food Funct. 14:1884–1896.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Tao J, Xue C, Wang X, Chen H, Liu Q, Jiang
C and Zhang W: GAS1 promotes ferroptosis of liver cells in
acetaminophen-induced acute liver failure. Int J Med Sci.
20:1616–1630. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Wang C, Liu T, Tong Y, Cui R, Qu K, Liu C
and Zhang J: Ulinastatin protects against acetaminophen-induced
liver injury by alleviating ferroptosis via the SIRT1/NRF2/HO-1
pathway. Am J Transl Res. 13:6031–6042. 2021.PubMed/NCBI
|
|
101
|
Cai X, Hua S, Deng J, Du Z, Zhang D, Liu
Z, Khan NU, Zhou M and Chen Z: Astaxanthin activated the Nrf2/HO-1
pathway to enhance autophagy and inhibit ferroptosis, ameliorating
acetaminophen-induced liver injury. ACS Appl Mater Interfaces.
14:42887–42903. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yan M, Huo Y, Yin S and Hu H: Mechanisms
of acetaminophen-induced liver injury and its implications for
therapeutic interventions. Redox Biol. 17:274–283. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Chauhan A, Sheriff L, Hussain MT, Webb GJ,
Patten DA, Shepherd EL, Shaw R, Weston CJ, Haldar D, Bourke S, et
al: The platelet receptor CLEC-2 blocks neutrophil mediated hepatic
recovery in acetaminophen induced acute liver failure. Nat Commun.
11:19392020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
McDonald B and Kubes P: Innate immune cell
trafficking and function during sterile inflammation of the liver.
Gastroenterology. 151:1087–1095. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Calvente CJ, Tameda M, Johnson CD, Del
Pilar H, Lin YC, Adronikou N, De Mollerat Du Jeu X, Llorente C,
Boyer J and Feldstein AE: Neutrophils contribute to spontaneous
resolution of liver inflammation and fibrosis via microRNA-223. J
Clin Invest. 129:4091–4109. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shibuya M: Vascular endothelial growth
factor-dependent and -independent regulation of angiogenesis. BMB
Rep. 41:278–286. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Fernández M, Semela D, Bruix J, Colle I,
Pinzani M and Bosch J: Angiogenesis in liver disease. J Hepatol.
50:604–620. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kato T, Ito Y, Hosono K, Suzuki T, Tamaki
H, Minamino T, Kato S, Sakagami H, Shibuya M and Majima M: Vascular
endothelial growth factor receptor-1 signaling promotes liver
repair through restoration of liver microvasculature after
acetaminophen hepatotoxicity. Toxicol Sci. 120:218–229. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Li T, Zhu Y and Han L: VEGFR-1
activation-induced MMP-9-dependent invasion in hepatocellular
carcinoma. Future Oncol. 11:3143–3157. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Prescott LF, Illingworth RN, Critchley JA
and Proudfoot AT: Intravenous N-acetylcysteine: Still the treatment
of choice for paracetamol poisoning. Br Med J. 280:46–47. 1980.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Hu C, Zhao L, Wu Z and Li L:
Transplantation of mesenchymal stem cells and their derivatives
effectively promotes liver regeneration to attenuate
acetaminophen-induced liver injury. Stem Cell Res Ther. 11:882020.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Simpson KJ, Bates CM, Henderson NC,
Wigmore SJ, Garden OJ, Lee A, Pollok A, Masterton G and Hayes PC:
The utilization of liver transplantation in the management of acute
liver failure: Comparison between acetaminophen and
non-acetaminophen etiologies. Liver Transpl. 15:600–609. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Hodgman MJ and Garrard AR: A review of
acetaminophen poisoning. Crit Care Clin. 28:499–516. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Myers RP, Shaheen AAM, Li B, Dean S and
Quan H: Impact of liver disease, alcohol abuse, and unintentional
ingestions on the outcomes of acetaminophen overdose. Clin
Gastroenterol Hepatol. 6:918–925. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bunchorntavakul C and Reddy KR:
Acetaminophen (APAP or N-acetyl-p-aminophenol) and acute liver
failure. Clin Liver Dis. 22:325–346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mohler CR, Nordt SP, Williams SR,
Manoguerra AS and Clark RF: Prospective evaluation of mild to
moderate pediatric acetaminophen exposures. Ann Emerg Med.
35:239–244. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Kociancic T and Reed MD: Acetaminophen
intoxication and length of treatment: How long is long enough?
Pharmacotherapy. 23:1052–1059. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Liver sinusoidal endothelial cells, .
Physiology and role in liver diseases-PubMed[EB/OL]. [2024-12-29].
Available from:. https://pubmed.ncbi.nlm.nih.gov/27423426/
|
|
119
|
Pathological process of liver sinusoidal
endothelial cells in liver diseases-PubMed[EB/OL]. [2024-12-29].
Available from. https://pubmed.ncbi.nlm.nih.gov/29209108/
|
|
120
|
Peterson RG and Rumack BH: Treating acute
acetaminophen poisoning with acetylcysteine. JAMA. 237:2406–2407.
1977. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Corcoran GB and Wong BK: Role of
glutathione in prevention of acetaminophen-induced hepatotoxicity
by N-acetyl-L-cysteine in vivo: Studies with N-acetyl-D-cysteine in
mice. J Pharmacol Exp Ther. 238:54–61. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Smilkstein MJ, Knapp GL, Kulig KW and
Rumack BH: Efficacy of oral N-acetylcysteine in the treatment of
acetaminophen overdose. Analysis of the national multicenter study
(1976 to 1985). N Engl J Med. 319:1557–1562. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Rumack BH: Acetaminophen hepatotoxicity:
The first 35 years. J Toxicol Clin Toxicol. 40:3–20. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Yarema MC, Johnson DW, Berlin RJ,
Sivilotti ML, Nettel-Aguirre A, Brant RF, Spyker DA, Bailey B,
Chalut D, Lee JS, et al: Comparison of the 20-hour intravenous and
72-hour oral acetylcysteine protocols for the treatment of acute
acetaminophen poisoning. Ann Emerg Med. 54:606–614. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Yang R, Miki K, He X, Killeen ME and Fink
MP: Prolonged treatment with N-acetylcystine delays liver recovery
from acetaminophen hepatotoxicity. Crit Care. 13:R552009.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Blieden M, Paramore LC, Shah D and
Ben-Joseph R: A perspective on the epidemiology of acetaminophen
exposure and toxicity in the United States. Expert Rev Clin
Pharmacol. 7:341–348. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Waring WS: Novel acetylcysteine regimens
for treatment of paracetamol overdose. Ther Adv Drug Saf.
3:305–315. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
de Andrade KQ, Moura FA, dos Santos JM, de
Araújo OR, de Farias Santos JC and Goulart MO: Oxidative stress and
inflammation in hepatic diseases: Therapeutic possibilities of
N-acetylcysteine. Int J Mol Sci. 16:30269–30308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Khayyat A, Tobwala S, Hart M and Ercal N:
N-acetylcysteine amide, a promising antidote for acetaminophen
toxicity. Toxicol Lett. 241:133–142. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Downs JW, Cumpston KL, Kershner EK,
Troendle MM, Rose SR and Wills BK: Clinical outcome of massive
acetaminophen overdose treated with standard-dose N-acetylcysteine.
Clin Toxicol (Phila). 59:932–936. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Pang C, Zheng Z, Shi L, Sheng Y, Wei H,
Wang Z and Ji L: Caffeic acid prevents acetaminophen-induced liver
injury by activating the Keap1-Nrf2 antioxidative defense system.
Free Rad Biol Med. 91:236–246. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Yang R, Song C, Chen J, Zhou L, Jiang X,
Cao X, Sun Y and Zhang Q: Limonin ameliorates acetaminophen-induced
hepatotoxicity by activating Nrf2 antioxidative pathway and
inhibiting NF-κB inflammatory response via upregulating Sirt1.
Phytomedicine. 69:1532112020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
An Y, Luo Q, Han D and Guan L: Abietic
acid inhibits acetaminophen-induced liver injury by alleviating
inflammation and ferroptosis through regulating Nrf2/HO-1 axis. Int
Immunopharmacol. 118:1100292023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Lv H, Hong L, Tian Y, Yin C, Zhu C and
Feng H: Corilagin alleviates acetaminophen-induced hepatotoxicity
via enhancing the AMPK/GSK3β-Nrf2 signaling pathway. Cell Commun
Signal. 17:22019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Wang L, Zhang S, Cheng H, Lv H, Cheng G
and Ci X: Nrf2-mediated liver protection by esculentoside a against
acetaminophen toxicity through the AMPK/akt/GSK3β pathway. Free
Radic Biol Med. 101:401–412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Zhu L, Fan X, Cao C, Li K, Hou W and Ci X:
Xanthohumol protect against acetaminophen-induced hepatotoxicity
via Nrf2 activation through the AMPK/akt/GSK3β pathway. Biomed
Pharmacother. 165:1150972023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Yao Y, Li R, Liu D, Long L and He N:
Rosmarinic acid alleviates acetaminophen-induced hepatotoxicity by
targeting Nrf2 and NEK7-NLRP3 signaling pathway. Ecotoxicol Environ
Saf. 241:1137732022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Jiang Z, Yang X, Han Y, Li J, Hu C, Liu C
and Xiao W: Sarmentosin promotes USP17 and regulates Nrf2-mediated
mitophagy and cellular oxidative stress to alleviate APAP-induced
acute liver failure. Phytomedicine. 104:1543372022. View Article : Google Scholar : PubMed/NCBI
|














