Introduction
Coronary artery disease (CAD) is the most prevalent
form of cardiovascular disease, and the primary medical approach
for treating CAD involves timely reperfusion, including
percutaneous coronary intervention and coronary artery bypass
grafting (1). However, reperfusion
can exacerbate cardiac hypoxia/reoxygenation (H/R) injury, leading
to increased mortality. Mechanisms such as overproduction of
reactive oxygen species (ROS), apoptosis and imbalanced
mitochondrial dynamics can contribute to cardiac H/R injury
(2). Efforts have been made to
understand the cellular and molecular mechanisms of cardiac H/R
injury, including the interplay between autophagy and apoptosis
(3).
Autophagy is a cellular process that degrades and
recycles damaged organelles and proteins, which is critical in
regulating cardiomyocyte survival during H/R injury (4). Autophagy maintains cellular
homeostasis and promotes cell survival by clearing dysfunctional
cellular components (5); however,
the exact mechanisms by which autophagy is regulated during H/R
injury and how it interacts with other cellular stress responses
are not yet fully understood.
The maintenance of cardiac homeostasis depends on
the crucial process of eliminating dysfunctional mitochondria
through mitophagy (6). The primary
regulator of mitophagy is the PTEN-induced putative kinase
protein-1 (PINK1)/Parkin pathway, although the FUN14
domain-containing 1 (FUNDC1), Bcl2-interacting protein 3 (BNIP3)
and BNIP3-like (BNIP3L/NIX) pathways can also control mitophagy.
Dysregulated mitophagy is associated with cardiac dysfunction
related to aging, aortic stenosis, myocardial infarction or
diabetes (7). Notably,
PINK1-mediated mitophagy serves an important role in maintaining
mitochondrial quality control during ischemia/reperfusion injury
(7,8).
Semaglutide is a glucagon-like peptide-1 (GLP-1)
receptor agonist, and is a promising novel therapeutic agent that
has exhibited cardioprotective effects in previous studies
(9–12). As a member of the GLP-1 receptor
(GLP-1R) agonist class, semaglutide mimics the actions of
endogenous GLP-1, which is known to regulate glucose metabolism,
stimulate insulin secretion and inhibit glucagon release (13). In addition to its well-documented
benefits in managing blood glucose levels in type 2 diabetes,
semaglutide has shown potential in protecting the heart from damage
caused by ischemia and reperfusion (14). Despite these promising findings,
the specific molecular mechanisms underlying the cardioprotective
effects of semaglutide remain unclear. To address this, the present
study performed experiments using an in vitro model of H/R
in human AC16 cardiomyocytes. The aim of the current study was to
reveal the mechanisms underlying the protective effects of
semaglutide and to identify novel therapeutic targets for the
treatment of H/R injury, which may advance cardiovascular
medicine.
Materials and methods
Cell culture conditions and H/R
modeling
Numerous studies (15–17)
have adopted the AC16 cell line as an in vitro model to
investigate cardiac H/R injury. The selection of AC16 cells
(18) is attributed to their
shared characteristics with primary cardiomyocytes, such as the
expression of cardiac-specific markers (16,19,20)
and responsiveness to H/R conditions (21), making them a valuable tool for
exploring the mechanisms of heart diseases.
Human AC16 cardiomyocytes, sourced from the Cell
Resource Center, Peking Union Medical College of China (Beijing,
China), were maintained in DMEM (cat. no. D6429-500ML;
Sigma-Aldrich; Merck KGaA) supplemented with 10% FBS (cat. no.
FSP500; Shanghai Excell Biological Technology Co., Ltd.) and 1%
penicillin-streptomycin (cat. no. P1400; Beijing Solarbio Science
& Technology Co., Ltd.). The cells were incubated at 37°C in a
humidified environment of 95% air and 5% CO2
(CO2 incubator; CI-191C; Suzhou Jimei Electronic Co.,
Ltd.). AC16 cells were exposed to semaglutide (cat. no. HY-114118;
MedChemExpress) at concentrations ranging between 0 and 40 mmol/l
or DMSO (Beijing Solarbio Science & Technology Co., Ltd.) for
24, 48 and 72 h to identify the non-toxic concentration.
To stimulate H/R injury in myocardial cells, a H/R
model was established. Control cells were maintained in standard
DMEM conditions, while the experimental group underwent 8 h of
hypoxia in a specialized incubator (94% N2, 5%
CO2 and 1% O2 at 37°C), with nitrogen
replacing oxygen. Following this hypoxic phase, cells were
reoxygenated for 12 h at 37°C in a normoxic environment (95% air
and 5% CO2). To investigate the impact of semaglutide on
H/R injury, 5 mmol/l semaglutide was administered to the
experimental group during the reoxygenation phase. Additionally,
the autophagy activator rapamycin (0.1 µM; cat. no. HY-10219;
MedChemExpress) was used to stimulate autophagy and the autophagy
inhibitor 3-methyladenine (3-MA; cat. no. HY-19312; MedChemExpress)
was used to suppress autophagy for 48 h at 37°C in a normoxic
environment (95% air and 5% CO2) during reoxygenation;
cells were treated with these to evaluate the influence of
autophagy on H/R injury in cardiomyocytes.
Assessment of cell viability
According to the manufacturer's instructions, cell
viability was assessed using a Cell Counting Kit-8 (CCK-8) assay
(cat. no. CA1210; Beijing Solarbio Science & Technology Co.,
Ltd.). This assay was utilized to evaluate the cytotoxicity of
different concentrations of semaglutide on AC16 cell viability in a
dose- and time-dependent manner, both under normoxic conditions and
H/R conditions.
AC16 cells were incubated with 10 µl CCK-8 for 2 h
in the dark. The absorbance was measured at 450 nm using a
microplate reader (ReadMax 1200; Shanghai Flash Spectrum
Biotechnology Co., Ltd.).
ATP level determination
Intracellular ATP levels were measured using a
bioluminescent detection kit (ATP Assay Kit; cat. no. E-BC-F002;
Elabscience®; Elabscience Bionovation Inc.) according to
the manufacturer's instructions. A total of 2×106/ml
cells were used and luminescence intensities were recorded with a
multi-mode microplate reader (ReadMax 1200; Shanghai Shanpu Laser
Technology Co., Ltd.).
ROS measurement
ROS production was assessed using the ROS Assay Kit
(cat. no. S0033S; Beyotime Institute of Biotechnology), which
relies on measuring the fluorescence intensity of dihydroethidium
and 5-(and-6)-chloromethyl-2′,7′-dichlorodihydrofluorescein
diacetate (DCFH-DA). After treatment, the cells
(2×106/ml) were incubated with 10 µM/l DCFH-DA for 20
min at 37°C in the dark, washed three times with PBS and were then
observed under a fluorescence microscope (Nikon Eclipse ti2; Nikon
Corporation).
Transmission electron microscopy
AC16 cells (2×106/ml) were collected by
centrifugation at 1,000 × g for 3 min at 4°C the culture medium was
removed, and the cells were fixed with 2.5% glutaraldehyde electron
microscope fixative (cat. no. G1102; Wuhan Servicebio Technology
Co., Ltd.) at 4°C for 2–4 h. Subsequently, the samples were fixed
with 1% osmium tetroxide (cat. no. 18456; Ted Pella, Inc.),
dehydrated using a series of ethanol and propylene oxide solutions,
embedded and cut into 60–80-nm sections. The sections were then
stained with uranium-lead double staining (2% uranium acetate and
2.6% lead citrate) for 15 min at room temperature. After final
washes, the grids were blotted dry and left to dry overnight at
room temperature. The embedding molds were The cells were embedded
in acetone and SPI-Pon 812 Epoxy Resin, then the embedding molds
were placed in an oven at 60°C for 48 h for polymerization. Cells
were finally observed under a transmission electron microscope
(JEM-1200; Hitachi, Ltd.).
Monomeric red fluorescent protein
(mRFP)-green fluorescent protein (GFP)-LC3 assay and confocal
microscopy
AC16 cells were seeded at a density of
1×105 cells/well in 24-well plates and were then
incubated overnight at 37°C in a 5% CO2 environment. To
ensure optimal virus infection, the cell confluence rates were
maintained between 30 and 50%, taking into account cell size and
growth rate variations. The mRFP-GFP-LC3 adenoviral vectors used in
this assay were obtained from Hanbio Biotechnology Co., Ltd. This
assay relies on the differential pH stability of GFP and RFP.
Specifically, enhanced GFP fluorescence is quenched in acidic
lysosome conditions (pH <5), whereas mRFP fluorescence remains
unaffected. The merged green and red images reveal autophagosomes
as yellow puncta and autolysosomes as red puncta. An increase in
yellow and red puncta indicates enhanced autophagic flux, whereas
an increase solely in yellow puncta or a decrease in both suggests
autophagic flux blockage. Infection with the aforementioned
adenoviral vectors (1×109 TU/ml) was carried out
according to the manufacturer's guidelines, with an optimal
multiplicity of infection of 50 determined through preliminary
experiments, with the cells being infected for 36 h. LC3 puncta
were visualized using a confocal microscope [ICX41; Sunny Optical
Technology (Group) Company Limited] and were quantitatively
analyzed using ImageJ software (version 4.20; National Institutes
of Health) and GraphPad Prism software 19.0 (Dotmatics).
Western blot analysis
Protein lysates were extracted from cells using RIPA
buffer (cat. no. R0010; Beijing Solarbio Science & Technology
Co., Ltd.) and the protein concentration was determined using a BCA
Protein Assay Kit (cat. no. PC0020; Beijing Solarbio Science &
Technology Co., Ltd.). Proteins (30 µg/lane) were then separated by
SDS-PAGE on 8–10% gels (cat. no. ZS305; Beijing Zomanbio
International Biogene Technology Co., Ltd.) and were transferred
onto PVDF membranes. The membranes were blocked with 5% non-fat dry
milk (cat. no. D8340; Beijing Solarbio Science & Technology
Co., Ltd.) at room temperature for 1 h, then incubated with primary
antibodies at 4°C overnight. The following primary antibodies were
used: Sequestosome 1/p62 (1:1,000; cat. no. PM045; MBL
International Co.), Beclin1 (1:1,000; cat. no. PD017; MBL
International Co.), LC3B (1:1,000; cat. no. WL01506; Wanleibio Co.,
Ltd.), PINK1 (1:1,000; cat. no. DF7742; Affinity Biosciences),
Parkin (1:1,000; cat. no. AF0235; Affinity Biosciences) and β-actin
(1:5,000; cat. no. EM21002; HUABIO). The blots were then probed
with HRP-conjugated anti-rabbit and anti-mouse secondary antibodies
(1:10,000; cat. nos. ab97080 and ab97040; Abcam) for 60 min at
37°C. After secondary antibody incubation, the PVDF membrane was
immersed in TBS-0.05% Tween-20 and shaken for 45 min; this was
repeated three times. For visualization, ECL reagents A and B (cat.
no. P10300; Suzhou Xinsaimei Biotechnology Co., Ltd.) were mixed
equally and set aside. Subsequently, plastic wrap was placed on the
exposure platform, the membrane was blotted dry and placed on the
wrap, the ECL solution was applied and left to react for 1 min, and
the blots were visualized. The optical density values of the target
bands were analyzed using a gel image processing system (SH-523;
Shenhua Science Technology Co., Ltd.) to semi-quantify the blots.
All experiments were performed in triplicate.
Statistical analysis
Data were analyzed using SPSS 22.0 statistical
software (IBM Corp.). The measurement data are presented as the
mean ± SEM. One-way ANOVA was conducted to compare multiple groups,
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
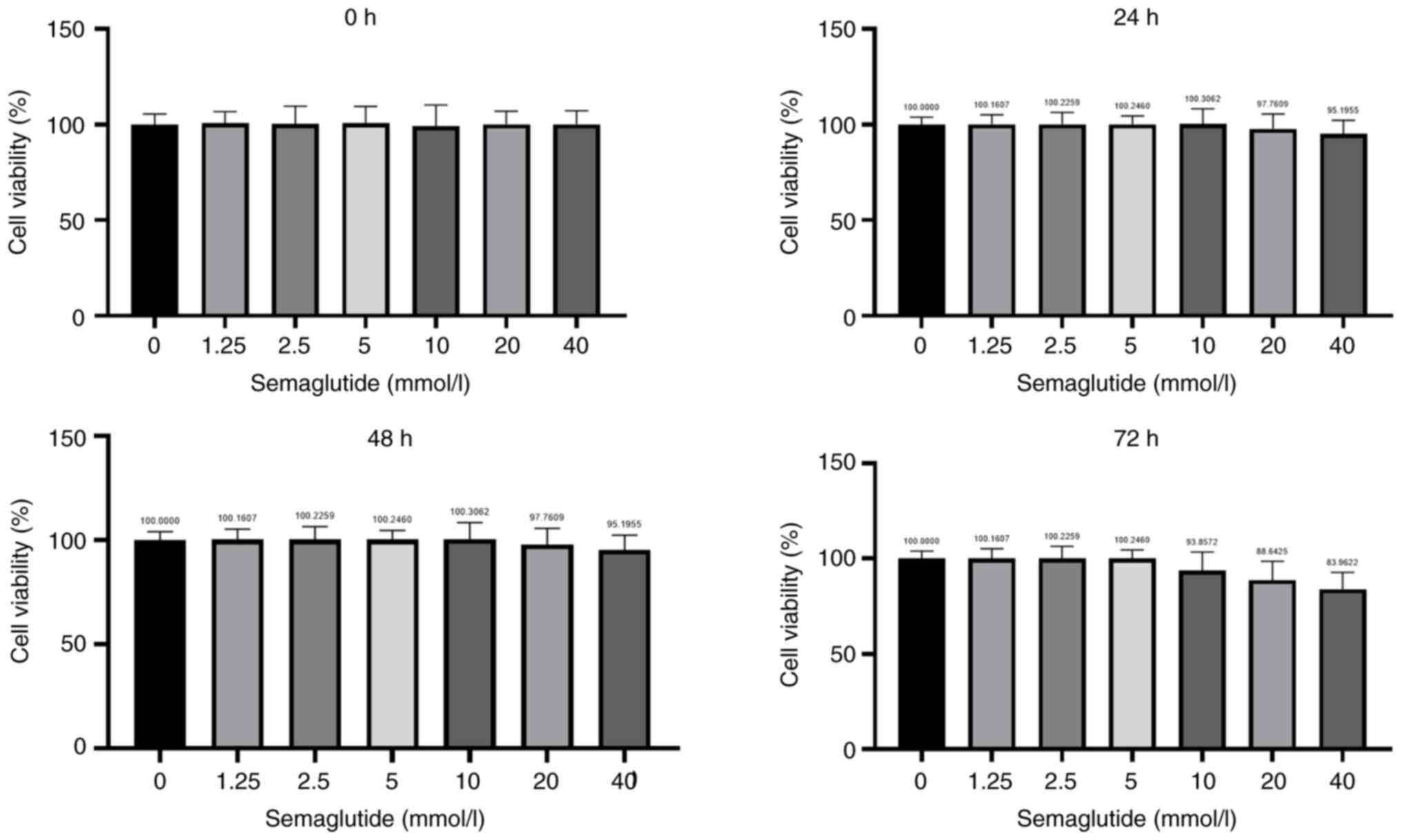
Effect of various concentrations of
semaglutide on AC16 cell viability
The optimal concentration of semaglutide varies
according to cell line and experimental setup. Although previous
studies have evaluated its concentration in other cell lines, such
as H9C2 cells (14,22), the concentration needed for
treating AC16 cells remains to be further explored. In the initial
stage, a CCK-8 assay was performed to assess the dose-dependent
cytotoxicity of different concentrations of semaglutide on AC16
cell viability under normoxic conditions (Fig. 1). AC16 cells were exposed to DMSO
or to semaglutide at concentrations ranging between 0 and 40 µmol/l
for 0 (Fig. 1A), 24 (Fig. 1B), 48 (Fig. 1C) and 72 h (Fig. 1D). As the concentration of
semaglutide increased, the proliferation rate of AC16 cells
decreased. For subsequent experiments, non-toxic concentrations of
semaglutide (1.25, 2.5, 5, and 10 mmol/l) were selected for cell
treatment.
The concentration of semaglutide used in the present
study was carefully selected after extensive experimentation and a
review of the literature (14,23).
Subsequently, a series of dose-response experiments were conducted
to determine the optimal concentration range that would allow the
effective investigation of the biological effects of semaglutide in
the H/R model. The CCK-8 assay was used to examine the effects of
different concentrations of semaglutide on AC16 cell viability
after H/R (Fig. 2). The CCK-8
assay demonstrated that, in AC16 cells treated with semaglutide +
H/R, cell viability increased with the increasing concentration of
semaglutide. The highest cell viability was observed when the
concentration of semaglutide was 5 mmol/l. However, when the
concentration of semaglutide reached 10 mmol/l, the cell viability
was reduced. Additionally, based on the present observations, the
association between semaglutide treatment and cell viability was
most significant at the 48-h time point, indicating a robust
response to treatment at this duration. Therefore, 5 mmol/l
semaglutide was chosen, and cells were treated for 48 h for all
subsequent experiments. In the current study, the H/R model was
established using AC16 cells, which is consistent with previous
studies (21,24). The concentration of semaglutide
administered and the duration of its application were aligned with
the parameters reported by Wu et al (23).
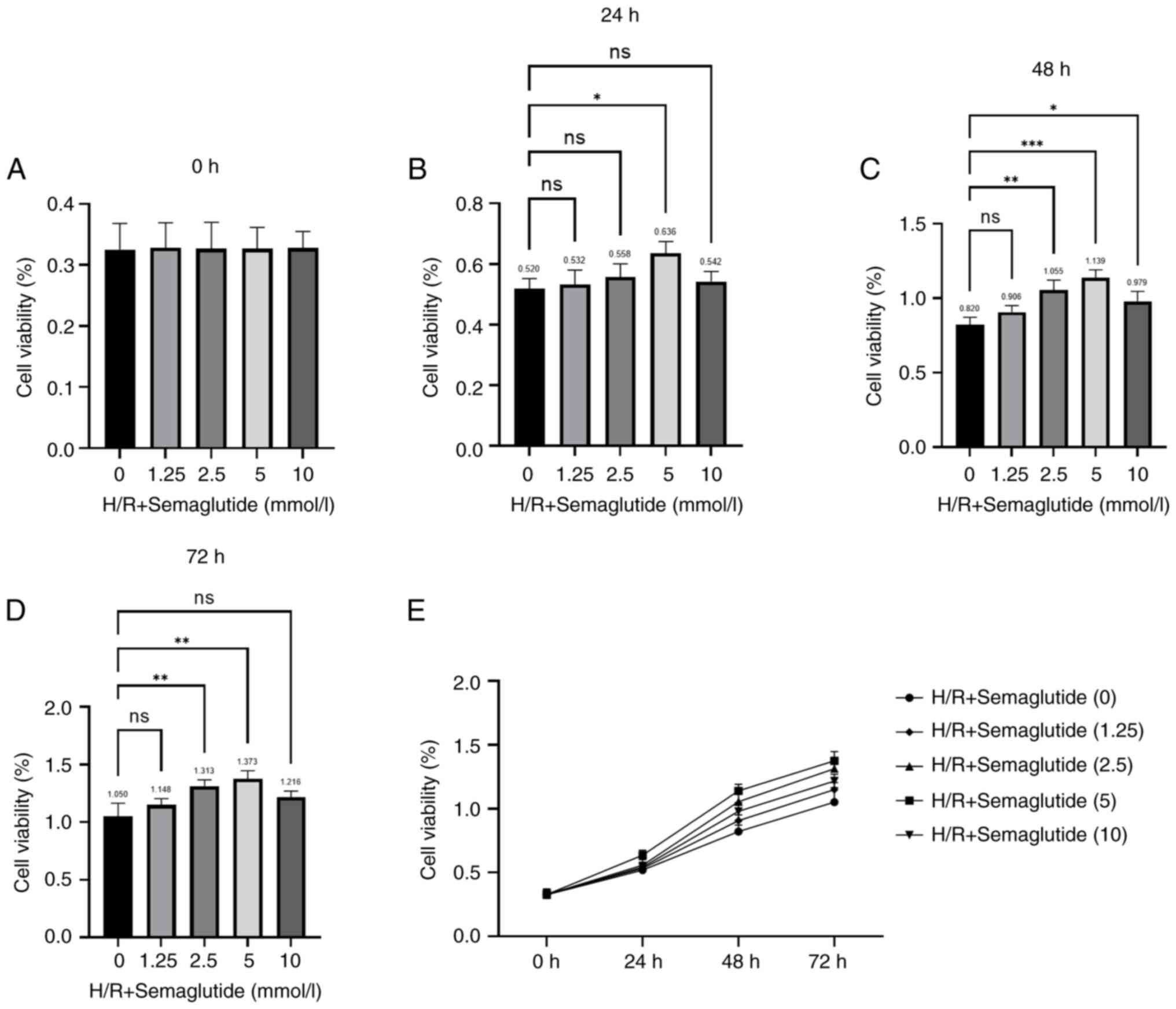 | Figure 2.Effects of different concentrations
of semaglutide on cell viability under H/R conditions. The AC16
cells were divided into the following groups: Normoxia group, H/R
group and H/R + semaglutide groups (1.25, 2.5, 5, or 10 mmol/l),
and cell viability was examined using a Cell Counting Kit-8 assay
after (A) 0, (B) 24, (C) 48 and (D) 72 h. Semaglutide was safe up
to a concentration of 5 mmol/l. (E) Treatment with 5 mmol/l
semaglutide for 48 h was the optimal treatment used for subsequent
experiments. The experiments were repeated three times. *P<0.05,
**P<0.01, ***P<0.001. H/R, hypoxia/reoxygenation; ns, not
significant. |
The cells were divided into the following five
groups: Normoxia, H/R, H/R + semaglutide (5 mmol/l), H/R +
semaglutide (5 mmol/l) + rapamycin (autophagy inducer), and H/R +
semaglutide (5 mmol/l) + 3-MA (autophagy inhibitor).
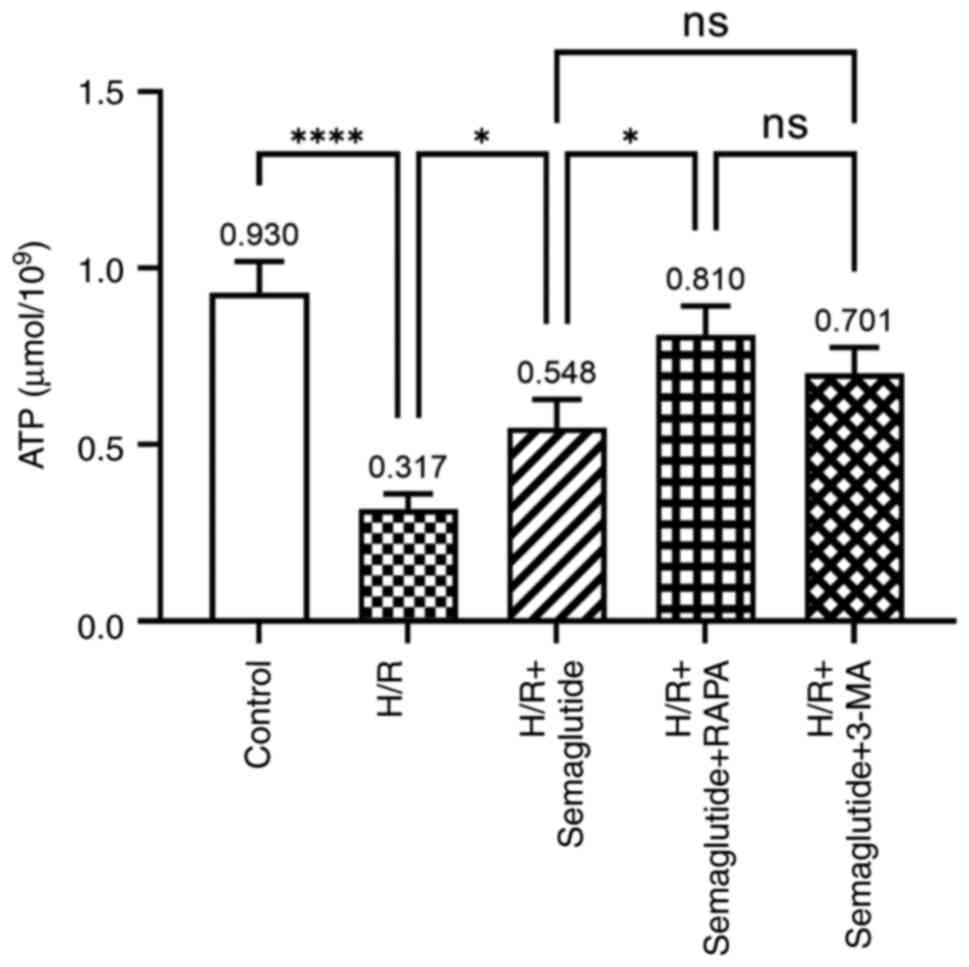
ATP production
The exposure of AC16 cardiomyocytes to H/R led to a
significant decrease in ATP levels compared with those in the
normoxia control group; however, compared with the H/R group,
treatment with semaglutide + H/R reversed this effect, resulting in
a substantial increase in ATP levels (Fig. 3). Additionally, compared with the
H/R + semaglutide group, further enhancement of ATP content was
observed when rapamycin was combined with semaglutide. Conversely,
the addition of 3-MA, an autophagy inhibitor, resulted in a marked
reduction in ATP levels.
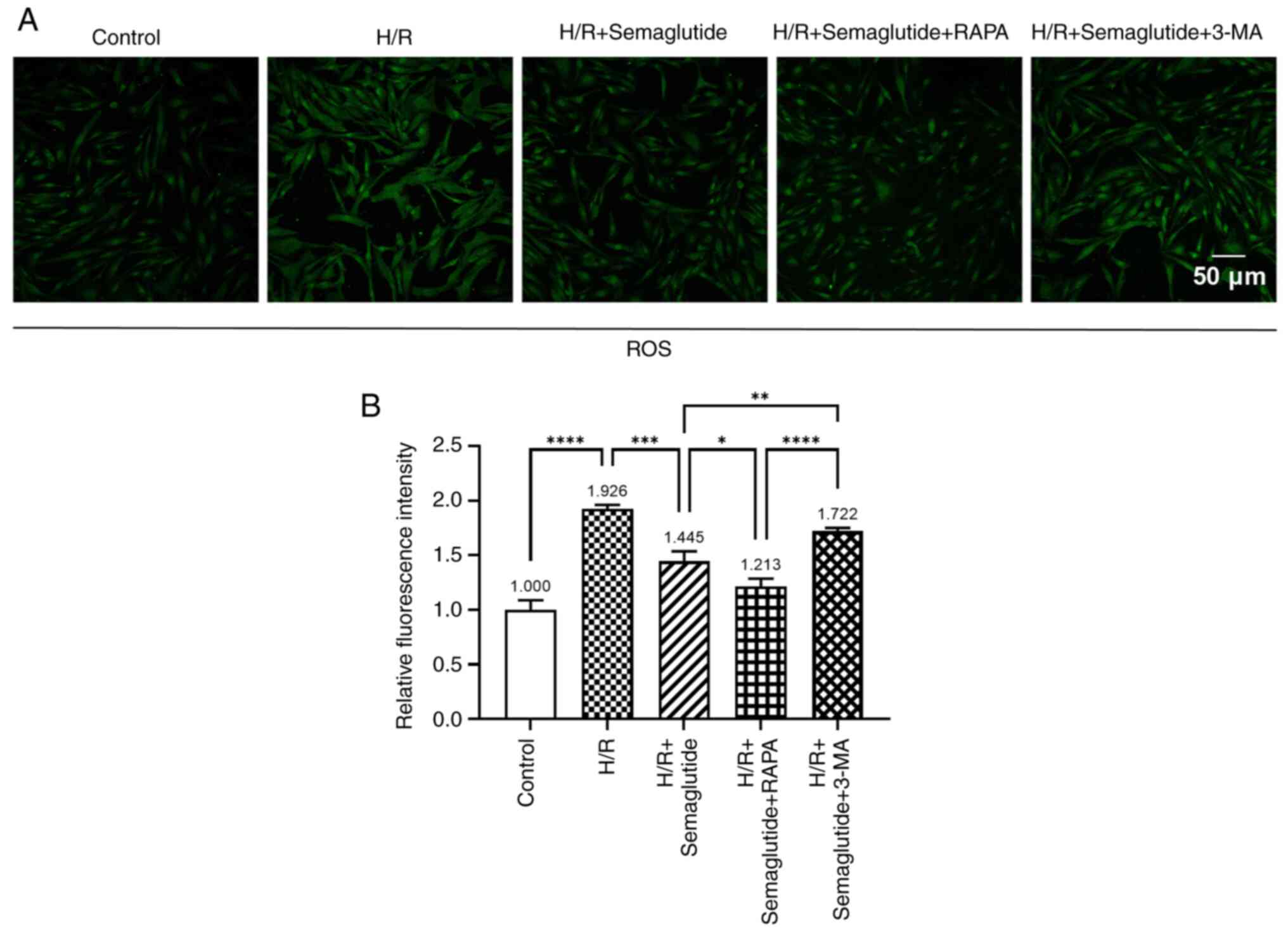
ROS levels
Subsequently, the present study explored the impact
of semaglutide on the regulation of oxidative stress. A notable
increase in ROS levels was detected in cells exposed to H/R
compared with those in the control group (Fig. 4). By contrast, treatment with
semaglutide resulted in a significant reduction in ROS fluorescence
intensity, indicating its ability to protect against
hypoxia-induced oxidative stress. Additionally, compared with in
the H/R + semaglutide group, the introduction of rapamycin further
decreased ROS levels, while treatment with 3-MA led to an increase
in ROS fluorescence intensity.
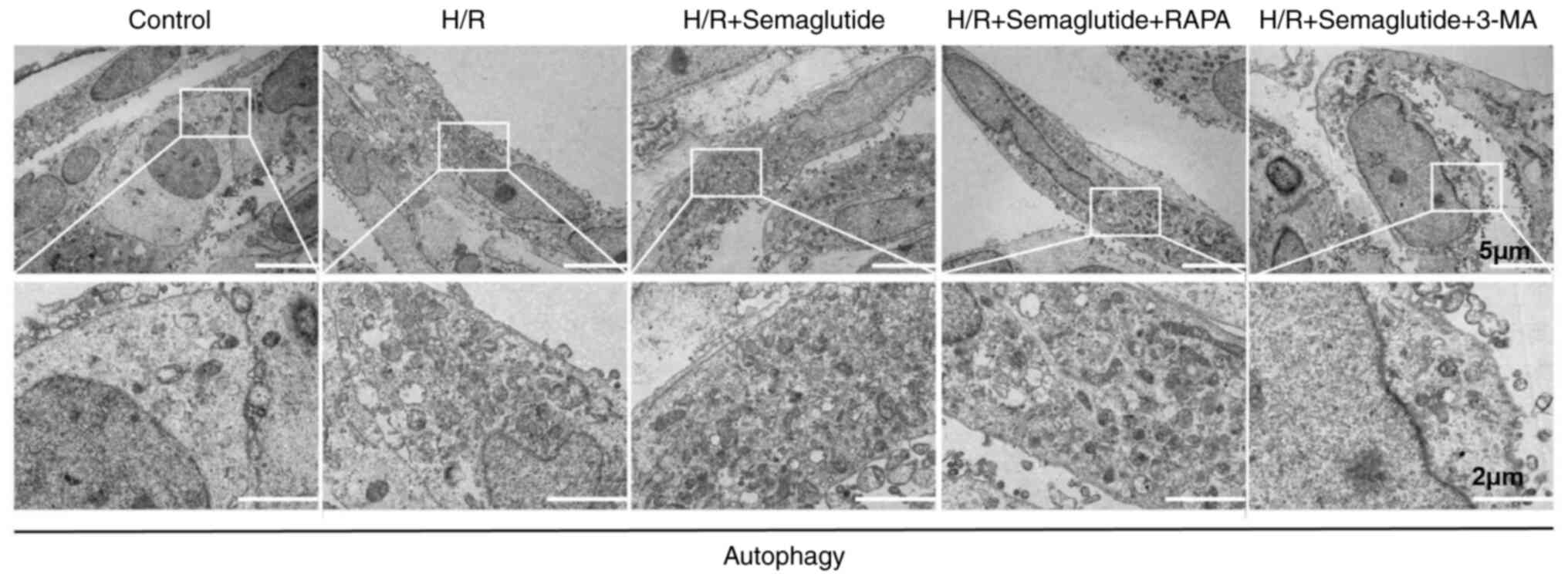
Autophagosome accumulation analyzed by
transmission electron microscopy
The present study investigated the impact of
semaglutide on autophagosome formation using transmission electron
microscopy. The observations using transmission electron microscopy
revealed an increase in the presence of autophagosomes in AC16
cells exposed to H/R compared with those in control cells (Fig. 5). Treatment with semaglutide
further amplified the number of autophagosomes in H/R-exposed
cells, suggesting that semaglutide may provide protective effects
against hypoxic damage by enhancing autophagy. Co-administration of
semaglutide and rapamycin led to a substantial augmentation in
autophagosome formation, as characterized by more autophagosomes,
compared with semaglutide alone, further supporting the stimulatory
effect of these compounds on autophagy. Conversely, the addition of
3-MA to semaglutide-treated H/R cells resulted in a partial
reduction in autophagosome accumulation, underscoring the
inhibitory effect of 3-MA on autophagy.
Analysis of autophagy by confocal
fluorescence microscopy
To further validate the impact of semaglutide on
autophagy in cardiomyocytes, tandem GFP-mRFP-LC3 adenovirus
microscopy, a method commonly used to study autophagic flux, was
performed to observe the progression of autophagy. The quenching of
GFP fluorescence in a lysosomal acid environment, while mRFP
fluorescence remains stable, allows for the identification of the
formation of autophagosomes and autolysosomes through the presence
of yellow puncta (GFP/mRFP) and red puncta (mRFP). Using the
mRFP-GFP-LC3 adenoviral assay to monitor autophagosome dynamics, it
was observed that semaglutide exerted a protective effect on cells
against hypoxia-induced damage by activating autophagy. Compared
with in the H/R group, the semaglutide + H/R group exhibited a
significant increase in the number of mRFP-GFP-LC3-positive
autophagosomes. Furthermore, when compared with the H/R +
semaglutide group, the rapamycin-treated group showed a marked
elevation in autophagosome count, whereas the 3-MA-treated group
demonstrated a significant reduction in autophagosome number
(Fig. 6).
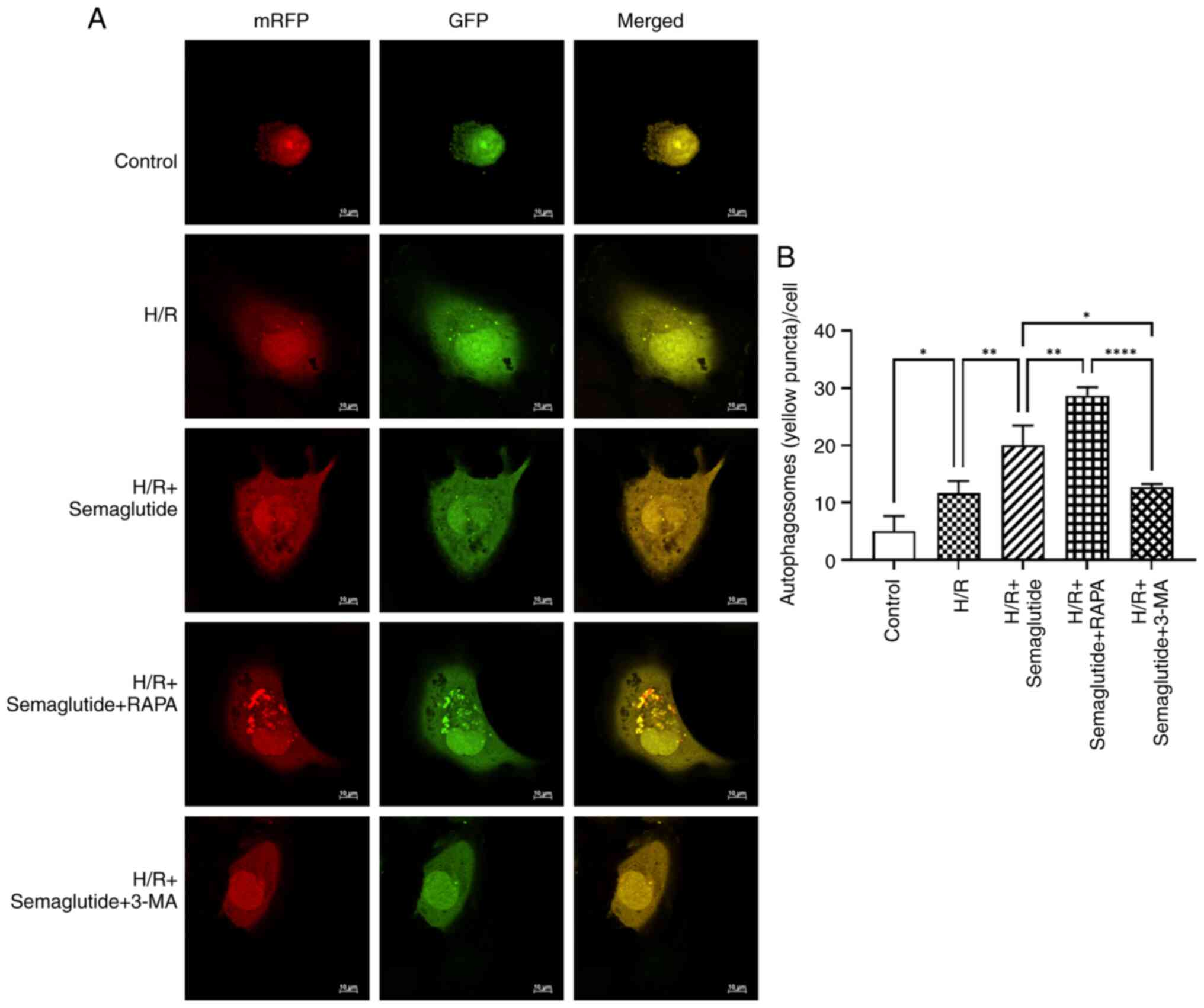 | Figure 6.Confocal microscopy analysis of
mRFP-GFP-LC3-labeled autophagosomes in AC16 cardiomyocytes under
H/R conditions in different groups. (A) AC16 cells were infected
with GFP-mRFP-LC3 adenoviral vectors for 36 h and were then
subjected to different treatments during reoxygenation. Both green
and red fluorescence changes were observed using a confocal
microscope. In the images, yellow dots indicate the presence of
unfused autophagosomes, while red dots represent fusion events,
signifying the formation of autolysosomes. (B) Number of GFP-LC3
and mRFP-LC3 puncta per cell in different groups was quantified.
The experiments were repeated three times. *P<0.05, **P<0.01,
****P<0.00. RAPA, rapamycin; 3-MA, 3-methyladenine; H/R,
hypoxia/reoxygenation; mRFP, monomeric red fluorescent protein;
GFP, green fluorescent protein. |
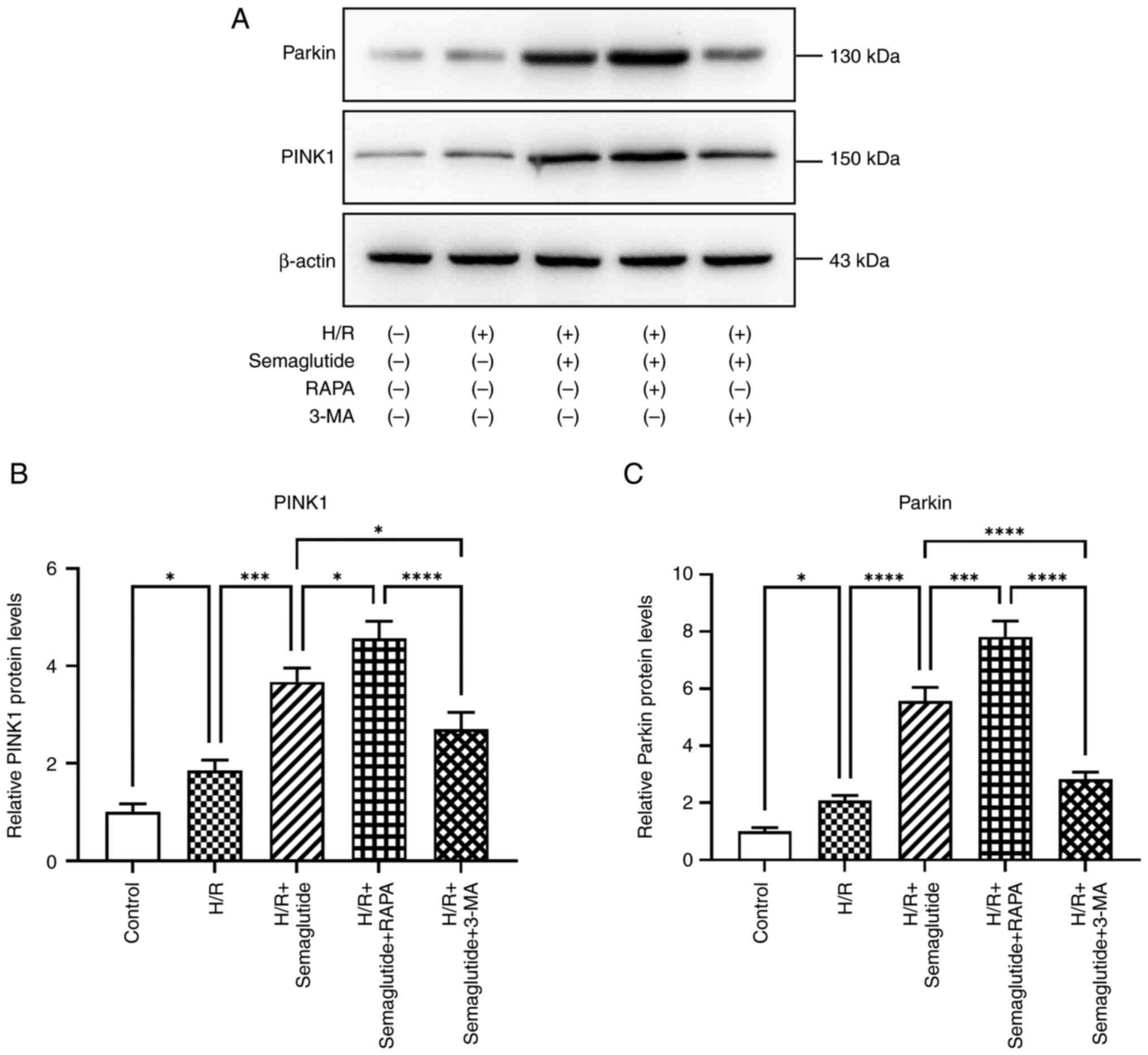
PINK1/Parkin pathway activation
The protein levels of Parkin and PINK1 were
significantly increased in cells exposed to H/R compared with those
in the control group (Fig. 7).
Additionally, treatment with semaglutide resulted in further
enhancement of the expression levels of Parkin and PINK1,
indicating its potential role in activating the PINK1/Parkin
pathway. These findings offer valuable insights into the mechanisms
responsible for the cardioprotective effects of semaglutide.
Compared with in the H/R + semaglutide group, the protein
expression levels of Parkin and PINK1 were significantly higher in
the rapamycin group, whereas they were significantly reduced in the
3-MA group.
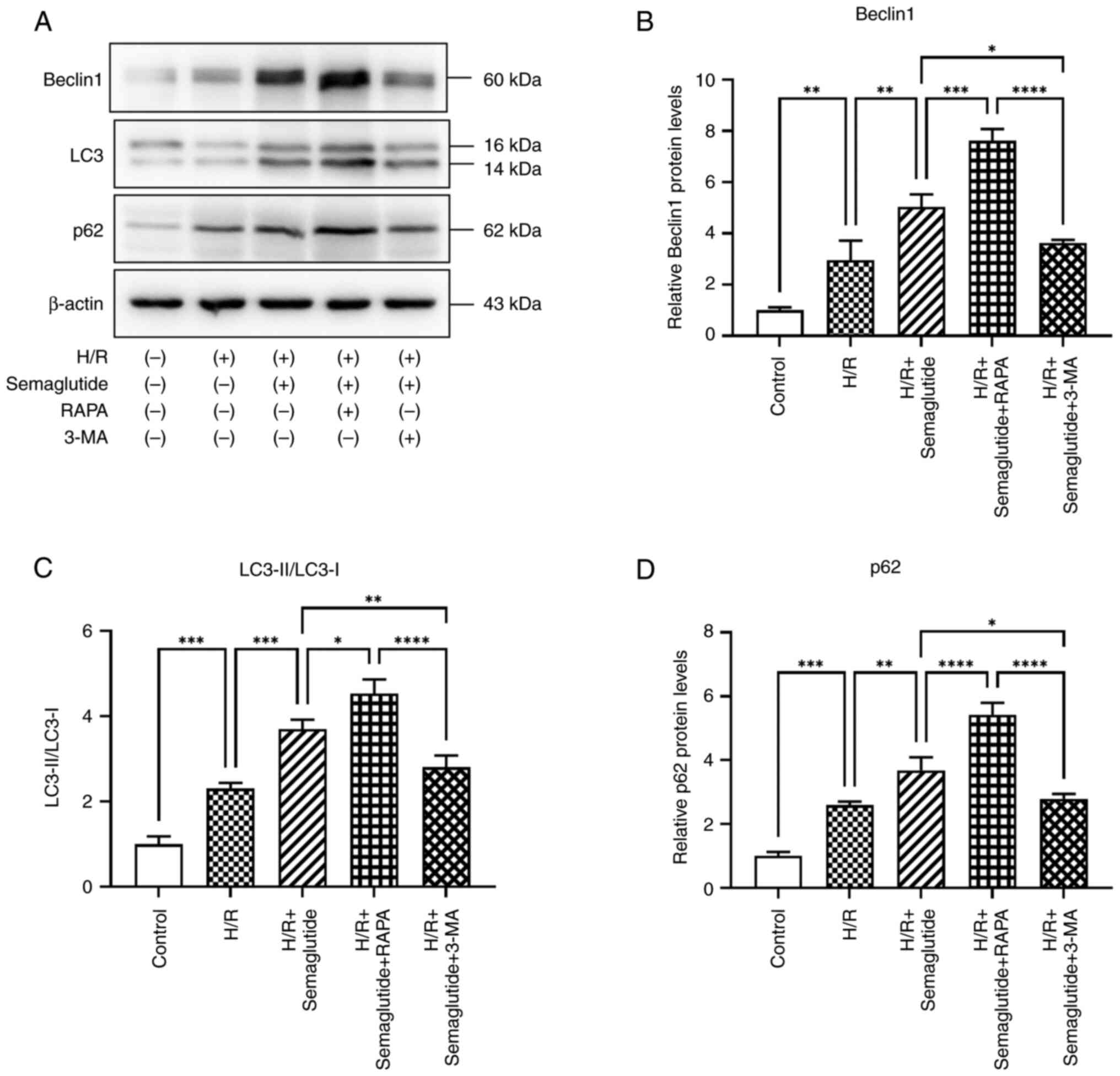
Autophagy markers
The present study investigated whether H/R triggered
autophagy in cardiomyocytes. Under normoxic conditions, western
blot analysis revealed minimal signaling for autophagy activation;
however, following H/R, autophagy markers were significantly
increased in cardiomyocytes, as indicated by the elevated
LC3B-II/LC3B-I ratio, along with increased expression levels of the
autophagy markers Beclin1 and p62 (Fig. 8). Furthermore, treatment with
semaglutide significantly enhanced the expression levels of these
proteins, thereby underscoring its pivotal role in promoting
autophagy. Notably, compared with in the H/R + semaglutide group,
rapamycin treatment led to even higher levels of autophagy marker
expression, whereas 3-MA reduced their expression levels.
Discussion
Extensive prospective studies (25–27)
have indicated that GLP-1R agonists offer cardioprotective benefits
for individuals with type 2 diabetes; these benefits include
improved cardiac function and myocardial blood flow, and reduced
cardiovascular events, all-cause mortality and progression of renal
disease. Additionally, GLP-1 has positive effects on blood
pressure, cholesterol levels and blood glucose regulation. Notable
options in the category of GLP-1R agonists, include liraglutide,
semaglutide, albiglutide and dulaglutide (9,28–31).
Insights from the SUSTAIN 1–7 trials have indicated that
semaglutide effectively improves glycemic control and promotes
weight loss, reducing cardiovascular risk in high-risk individuals
(32).
The novelty of the present study lies in
investigating the protective effects of semaglutide on
cardiomyocytes undergoing H/R injury and its relationship with
autophagy. While previous research (33,34)
has primarily examined the anti-diabetic properties of semaglutide,
to the best of our knowledge, its potential role in
cardioprotection has not been thoroughly explored. Limited research
has investigated the mechanisms underlying the cardioprotective
effects of semaglutide. A previous study demonstrated that
semaglutide can improve cardiac function, and reduce inflammation
and oxidative stress in obese mice, preventing lipid peroxidation
and cardiac impairment (35). Li
et al (36) demonstrated
that semaglutide protected against exercise-induced myocardial
injury in rats by activating the AMP-activated protein kinase
(AMPK) pathway, enhancing autophagy, decreasing ROS production and
reducing inflammation-related proteins. However, there is limited
research (14) focusing on the
effects of semaglutide on H/R-induced myocardial damage.
The present study investigated the protective
effects of semaglutide in a model of cardiac H/R. Pathological
conditions can cause mitochondria to generate elevated amounts of
ROS, inflicting macromolecular damage, and affecting cellular
homeostasis and function in several organs, including the heart
(37). Superoxide is the major
type of ROS that regulates autophagy (38); specifically, by promoting
macroautophagy, superoxide facilitates the protection of cardiac
myocytes from ischemia/reperfusion injury (39). Firstly, the present study
demonstrated that H/R exposure led to a marked increase in
oxidative stress in AC16 cells, as evidenced by elevated ROS
production. This finding is consistent with the results of a
previous report that indicated that oxidative stress serves a
pivotal role in myocardial H/R injury (40). Notably, treatment with semaglutide
effectively reduced ROS levels, indicating its antioxidant
properties. Additionally, semaglutide increased ATP content, which
is crucial for sustaining cellular energy under hypoxic conditions,
suggesting improved mitochondrial function and energy
metabolism.
Autophagy has emerged as a crucial process for
maintaining cardiac homeostasis and response to various stresses,
such as H/R injury (41).
Autophagy, also known as ‘self-eating’, is a highly conserved
catabolic process responsible for degrading and recycling cellular
components, thereby ensuring cellular health and survival (42). The role of autophagy in cardiac
diseases is complex (43). On the
one hand, autophagy can protect cardiomyocytes from damage by
eliminating dysfunctional organelles and toxic protein aggregates
(44). On the other hand,
dysregulation of autophagy has been linked to the development of
various cardiac disorders (45).
Therefore, understanding the mechanisms that regulate autophagy in
cardiomyocytes is essential for the development of novel
therapeutic strategies.
Among the well-recognized autophagy pathways, the
PINK1/Parkin pathway, as well as other pathways such as the FUNDC1,
BNIP3 and BNIP3L/NIX pathways, are notable. The PINK1/Parkin
pathway is triggered in response to mitochondrial damage, resulting
in the recruitment of Parkin to impaired mitochondria and
subsequent mitophagy; the PINK1/Parkin pathway is the most clearly
defined pathway for mitochondrial autophagy (46). When the mitochondrial membrane
potential becomes depolarized, PINK1, which is anchored to the
mitochondrial outer membrane, will recruit the Parkin protein from
the cytoplasm to the mitochondrial outer membrane. Following this,
Parkin can enlist the autophagy receptor p62 to facilitate the
development of autophagy lysosomes (8,47).
The present study indicated an increase in autophagy
activity in H/R-exposed cells, characterized by an elevated
LC3-II/LC3-I ratio and increased number of autophagosomes. The
present study demonstrated that semaglutide treatment further
enhanced the H/R-induced changes in autophagy activity,
autophagosome levels and LC3-II/LC3-I ratio, suggesting a
regulatory role of semaglutide in autophagy. Additionally, this
effect was strengthened by rapamycin, an autophagy inducer, and
weakened by 3-MA, an autophagy inhibitor.
The present results demonstrated that H/R induced
oxidative stress, increased ROS generation and activated the
PINK1/Parkin pathway. Semaglutide treatment further upregulated
PINK1 and Parkin expression, suggesting that semaglutide may
promote mitophagy through this pathway. However, the lack of a
PINK1/Parkin knockout in the present study limited our ability to
conclusively attribute the observed effects to a single pathway.
Other pathways, including the AMPK pathway or alternative pathways,
may also serve roles in the biological processes studied. Without
genetic manipulation to confirm their involvement, the primary or
exclusive pathway remains unclear. Thus, the present findings
should be cautiously interpreted, recognizing the potential for
multiple interacting pathways and the need for further
research.
Some studies (48,49)
have shown that an increase in the LC3-II/LC3-I ratio is
accompanied by a decrease in p62 levels, whereas the present study
observed an increase in p62 levels in the semaglutide group
undergoing H/R, despite the elevated LC3B-II/LC3B-I ratio, which is
indicative of impaired autophagic flux. The interplay between p62,
a recognized autophagy substrate protein, and autophagic activity
is complex. Under typical physiological conditions (50), active autophagy results in the
degradation of p62 within lysosomes, leading to decreased p62
levels; however, an intriguing paradox emerges under conditions of
oxidative stress or toxin exposure, where autophagic activation
coexists with elevated p62 levels (51,52).
This suggests that p62 may serve as a stress-responsive protein
(53), which is upregulated in
response to oxidative challenges. Upon oxidative stress, nuclear
factor erythroid 2-related factor binds directly to an antioxidant
response element in the p62 promoter to induce its expression.
Given the complexity of the relationship between p62 levels,
autophagic activity and cellular stress responses, a comprehensive
analysis of autophagic flux is essential for accurately assessing
cellular autophagic activity. This analysis should include detailed
examinations of the various stages of autophagosome formation,
lysosome fusion and substrate degradation. By assessing these
stages, a more thorough understanding of the autophagic processes
within the cell may be gained. Moreover, a detailed investigation
into the degradation of p62, which primarily occurs through
autophagy, is crucial.
Semaglutide might influence autophagic activity by
interacting with key regulators of autophagy, including mTOR, AMPK
or unc-51-like autophagy activating kinase 1. Studying the impact
of semaglutide on these autophagy-related proteins and their
downstream effects on p62 degradation would further elucidate its
role in autophagic flux. This approach will provide deeper insights
into the mechanisms by which semaglutide modulates autophagic
activity and its potential therapeutic implications.
The findings of the present study indicated that
semaglutide may confer protective effects against H/R-induced
cardiomyocyte injury by modulating autophagy via the
ROS/PINK1/Parkin pathway. Given that both excessive and deficient
autophagy can precipitate pathological states, precise regulation
of this process is paramount. Consequently, elucidating the optimal
timing and methodologies for inhibiting or activating these
pathways is of utmost importance, particularly in striking a
balance between autophagy-induced cell death and cell survival.
Notably, the present study has some limitations.
Firstly, conclusions obtained from investigations in only one cell
line are limited and we fully acknowledge the limitations of
relying solely on the AC16 cell line to study cardiac H/R. Heart
tissue in vivo exhibits a high degree of complexity and
heterogeneity, which the AC16 cell line may not fully capture as a
single cell type. To more comprehensively understand the biological
mechanisms involved in the cardiac H/R process, it is necessary to
consider using different cell lines or combining animal experiments
with in vitro studies. Furthermore, the cell experiments
should include H/R with RAPA or 3-MA alone, as these groups could
provide further insights into the autophagy process. However, given
the constraints of the current study, including resource
limitations and the need to balance experimental complexity with
feasibility, the current grouping strategy was used. Notably,
previous studies (14,54), such as Zhu et al (14) have demonstrated the expression of
GLP-1Rs in cardiomyocytes in control, H/R, H/R + saline and H/R +
semaglutide groups, and have investigated the mechanisms by which
GLP-1R agonists such as semaglutide exert their cardioprotective
effects. In the present study, GLP-1R protein levels were not
measured in the control, H/R or H/R + semaglutide groups; we aim to
address this limitation by measuring GLP-1R levels in future
studies.
In conclusion, the present study demonstrated that
semaglutide may protect AC16 cardiomyocytes against H/R-induced
injury by reducing oxidative stress and modulating autophagy,
particularly through the ROS/PINK1/Parkin/p62 pathway. These
findings offer novel insights into the mechanisms of the
cardioprotective effects of semaglutide and suggest its potential
therapeutic application in myocardial ischemia/reperfusion injury.
Future studies are required to further explore the precise
mechanisms of action of semaglutide and its clinical implications
in the treatment of myocardial ischemia/reperfusion injury.
Acknowledgements
Not applicable.
Funding
This work was supported by the Medical Science Research Project
of Hebei (grant no. 20232033).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
LQL, LLJ and JW contributed to the research design,
manuscript drafting and overall manuscript revision process. YPT
performed the data analysis. LQL took the lead in writing the
manuscript, while JW and LLJ provided substantial revisions. LQL,
LLJ, YPT and JW all participated in data acquisition, analysis and
interpretation. LQL and LLJ oversaw the research program, reviewed
the manuscript, and confirm the authenticity of all the raw data.
All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yetgin T, Manintveld OC, Boersma E,
Kappetein AP, van Geuns RJ, Zijlstra F, Duncker DJ and van der
Giessen WJ: Remote ischemic conditioning in percutaneous coronary
intervention and coronary artery bypass grafting. Circ J.
76:2392–2404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu Y, Tang C, Tan S, Duan J, Tian H and
Yang Y: Cardioprotective effect of isorhamnetin against myocardial
ischemia reperfusion (I/R) injury in isolated rat heart through
attenuation of apoptosis. J Cell Mol Med. 24:6253–6262. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang A, Zhang H, Liang Z, Xu K, Qiu W,
Tian Y, Guo H, Jia J, Xing E, Chen R, et al: U0126 attenuates
ischemia/reperfusion-induced apoptosis and autophagy in myocardium
through MEK/ERK/EGR-1 pathway. Eur J Pharmacol. 788:280–285. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dong Y, Undyala VV, Gottlieb RA, Mentzer
RM Jr and Przyklenk K: Autophagy: Definition, molecular machinery,
and potential role in myocardial ischemia-reperfusion injury. J
Cardiovasc Pharmacol Ther. 15:220–230. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mokhtari B and Badalzadeh R: Protective
and deleterious effects of autophagy in the setting of myocardial
ischemia/reperfusion injury: An overview. Mol Biol Rep.
49:11081–11099. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Onishi M, Yamano K, Sato M, Matsuda N and
Okamoto K: Molecular mechanisms and physiological functions of
mitophagy. EMBO J. 40:e1047052021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Turkieh A, El Masri Y, Pinet F and
Dubois-Deruy E: Mitophagy regulation following myocardial
infarction. Cells. 11:1992022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gu J, Zhang T, Guo J, Chen K, Li H and
Wang J: PINK1 Activation and translocation to
mitochondria-associated membranes mediates mitophagy and protects
against hepatic ischemia/reperfusion injury. Shock. 54:783–793.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leiter LA, Bain SC, Bhatt DL, Buse JB,
Mazer CD, Pratley RE, Rasmussen S, Ripa MS, Vrazic H and Verma S:
The effect of glucagon-like peptide-1 receptor agonists liraglutide
and semaglutide on cardiovascular and renal outcomes across
baseline blood pressure categories: Analysis of the LEADER and
SUSTAIN 6 trials. Diabetes Obes Metab. 22:1690–1695. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nauck MA and Quast DR: Cardiovascular
safety and benefits of semaglutide in patients with type 2
Diabetes: Findings from SUSTAIN 6 and PIONEER 6. Front Endocrinol
(Lausanne). 12:6455662021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ryan DH, Lingvay I, Colhoun HM, Deanfield
J, Emerson SS, Kahn SE, Kushner RF, Marso S, Plutzky J,
Brown-Frandsen K, et al: Semaglutide effects on cardiovascular
outcomes in people with overweight or obesity (SELECT) rationale
and design. Am Heart J. 229:61–69. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lin K, Wang A, Zhai C, Zhao Y, Hu H, Huang
D, Zhai Q, Yan Y and Ge J: Semaglutide protects against
diabetes-associated cardiac inflammation via Sirt3-dependent RKIP
pathway. Br J Pharmacol. Dec 22–2024.doi: 10.1111/bph.17327 (Epub
ahead of print). View Article : Google Scholar
|
|
13
|
Blundell J, Finlayson G, Axelsen M, Flint
A, Gibbons C, Kvist T and Hjerpsted JB: Effects of once-weekly
semaglutide on appetite, energy intake, control of eating, food
preference and body weight in subjects with obesity. Diabetes Obes
Metab. 19:1242–1251. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu Q, Luo Y, Wen Y, Wang D, Li J and Fan
Z: Semaglutide inhibits ischemia/reperfusion-induced cardiomyocyte
apoptosis through activating PKG/PKCε/ERK1/2 pathway. Biochem
Biophys Res Commun. 647:1–8. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ouyang M, Lu J, Ding Q, Qin T, Peng C and
Guo Q: Knockdown of long non-coding RNA PVT1 protects human AC16
cardiomyocytes from hypoxia/reoxygenation-induced apoptosis and
autophagy by regulating miR-186/Beclin-1 axis. Gene.
754:1447752020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yan X and Hou J: miR-22 host gene enhances
nuclear factor-kappa B activation to aggravate hypoxia-induced
injury in AC16 Cardiomyocytes. Cell Transplant.
30:9636897219903232021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu CJ, Xia F, Ruan L, Hu SP, Zhu WJ and
Yang K: Circ_0004771 promotes Hypoxia/Reoxygenation induced
Cardiomyocyte injury via activation of mitogen-activated protein
kinase signaling pathway. Int Heart J. 64:1125–1132. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Davidson MM, Nesti C, Palenzuela L, Walker
WF, Hernandez E, Protas L, Hirano M and Isaac ND: Novel cell lines
derived from adult human ventricular cardiomyocytes. J Mol Cell
Cardiol. 39:133–147. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sun K, Chen M, Kong X, Hou W, Xu Z and Liu
L: Cardiac-specific Suv39h1 knockout ameliorates high-fat diet
induced diabetic cardiomyopathy via regulating Hmox1 transcription.
Life Sci. 360:1232582025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li Z, Zhao J, Li H, Li Y and Lin C:
Catalpol protects AC16 cells from hypoxia/reoxygenation injury by
regulating the miR-22-3p/DPP4 axis. J Biochem Mol Toxicol.
36:e230342022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng CL, Jiang N, Zhao JF, Liu K, Jiang W
and Cao PG: Metformin relieves H/R-induced cardiomyocyte injury
through miR-19a/ACSL axis-possible therapeutic target for
myocardial I/R injury. Toxicol Appl Pharmacol. 414:1154082021.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang YF, Zhang D, Hu WM, Liu DX and Li L:
Semaglutide-mediated protection against Aβ correlated with
enhancement of autophagy and inhibition of apotosis. J Clin
Neurosci. 81:234–239. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu L, Zhan Y and Wang Y: Semaglutide may
ameliorate fibrosis and inhibit epithelial-mesenchymal transition
in intrauterine adhesion models. Int J Mol Sci. 25:61962024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li Y, Zhou Y, Pei H and Li D: Disruption
of BACH1 protects AC16 Cardiomyocytes against
hypoxia/reoxygenation-evoked injury by diminishing CDKN3
transcription. Cardiovasc Toxicol. 24:1105–1115. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bajaj HS, Al-Jabri B and Verma S:
Glucagon-like peptide-1 receptor agonists and cardiovascular
protection in type 2 diabetes: A pathophysiology-based review of
clinical implications. Curr Opin Cardiol. 33:665–675. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yao H, Zhang A, Li D, Wu Y, Wang CZ, Wan
JY and Yuan CS: Comparative effectiveness of GLP-1 receptor
agonists on glycaemic control, body weight, and lipid profile for
type 2 diabetes: Systematic review and network meta-analysis. BMJ.
384:e0764102024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marx N, Husain M, Lehrke M, Verma S and
Sattar N: GLP-1 receptor agonists for the reduction of
atherosclerotic cardiovascular risk in patients with type 2
diabetes. Circulation. 146:1882–1894. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kristensen SL, Rørth R, Jhund PS, Docherty
KF, Sattar N, Preiss D, Køber L, Petrie MC and McMurray JJV:
Cardiovascular, mortality, and kidney outcomes with GLP-1 receptor
agonists in patients with type 2 diabetes: A systematic review and
meta-analysis of cardiovascular outcome trials. Lancet Diabetes
Endocrinol. 7:776–785. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Marso SP, Daniels GH, Brown-Frandsen K,
Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR,
Ravn LS, et al: Liraglutide and cardiovascular outcomes in type 2
diabetes. N Engl J Med. 375:311–322. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gerstein HC, Colhoun HM, Dagenais GR, Diaz
R, Lakshmanan M, Pais P, Probstfield J, Riesmeyer JS, Riddle MC,
Rydén L, et al: Dulaglutide and cardiovascular outcomes in type 2
diabetes (REWIND): A double-blind, randomized placebo-controlled
trial. Lancet. 394:121–130. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Williams TC and Stewart E: Semaglutide and
cardiovascular outcomes in patients with type 2 diabetes. N Engl J
Med. 376:8912017.PubMed/NCBI
|
|
32
|
Aroda VR, Ahmann A, Cariou B, Chow F,
Davies MJ, Jódar E, Mehta R, Woo V and Lingvay I: Comparative
efficacy, safety, and cardiovascular outcomes with once-weekly
subcutaneous semaglutide in the treatment of type 2 diabetes:
Insights from the SUS TAIN 1–7 trials. Diabetes Metab. 45:409–418.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Davies M, Færch L, Jeppesen OK, Pakseresht
A, Pedersen SD, Perreault L, Rosenstock J, Shimomura I, Viljoen A,
Wadden TA, et al: Semaglutide 2·4 mg once a week in adults with
overweight or obesity, and type 2 diabetes (STEP 2): A randomised,
double-blind, double-dummy, placebo-controlled, phase 3 trial.
Lancet. 397:971–984. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li A, Su X, Hu S and Wang Y: Efficacy and
safety of oral semaglutide in type 2 diabetes mellitus: A
systematic review and meta-analysis. Diabetes Res Clin Pract.
198:1106052023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pan X, Yue L, Ban J, Ren L and Chen S:
Effects of semaglutide on cardiac protein expression and cardiac
function of obese mice. J Inflamm Res. 15:6409–6425. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Q, Tuo X, Li B, Deng Z, Qiu Y and Xie
H: Semaglutide attenuates excessive exercise-induced myocardial
injury by inhibiting oxidative stress and inflammation in rats.
Life Sci. 250:1175312020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan P, Xie XH, Chen CH, Peng X, Zhang P,
Yang C and Wang YT: Molecular regulation mechanisms and
interactions between reactive oxygen species and mitophagy. DNA
Cell Biol. 38:10–22. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hamacher-Brady A, Brady NR and Gottlieb
RA: Enhancing macroautophagy protects against ischemia/reperfusion
injury in cardiac myocytes. J Biol Chem. 281:29776–29787. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen Y, Azad MB and Gibson SB: Superoxide
is the major reactive oxygen species regulating autophagy. Cell
Death Differ. 16:1040–1052. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lei S, Feng Y, Huang P, Chen B, Bao K, Wu
Q, Zhang H and Huang X: Ophiopogonin D'-induced mitophagy and
mitochondrial damage are associated with dysregulation of the
PINK1/Parkin signaling pathway in AC16 cells. Toxicology.
477:1532752022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sciarretta S, Maejima Y, Zablocki D and
Sadoshima J: The role of autophagy in the heart. Annu Rev Physiol.
80:1–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ashrafizadeh M, Ahmadi Z, Farkhondeh T and
Samarghandian S: Autophagy as a molecular target of quercetin
underlying its protective effects in human diseases. Arch Physiol
Biochem. 128:200–208. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Przyklenk K, Dong Y, Undyala VV and
Whittaker P: Autophagy as a therapeutic target for
ischaemia/reperfusion injury? Concepts, controversies, and
challenges. Cardiovasc Res. 94:197–205. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sciarretta S, Yee D, Shenoy V, Nagarajan N
and Sadoshima J: The importance of autophagy in cardioprotection.
High Blood Press Cardiovasc Prev. 21:21–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nishida K, Taneike M and Otsu K: The role
of autophagic degradation in the heart. J Mol Cell Cardiol.
78:73–79. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Carmichael PL and Peng S:
Doxorubicin-induced mitophagy and mitochondrial damage is
associated with dysregulation of the PINK1/parkin pathway. Toxicol
In Vitro. 51:1–10. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Quinn PMJ, Moreira PI, Ambrósio AF and
Alves CH: PINK1/PARKIN signalling in neurodegeneration and
neuroinflammation. Acta Neuropathol Commun. 8:1892020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhou P, Xie W, Meng X, Zhai Y, Dong X,
Zhang X, Sun G and Sun X: Notoginsenoside R1 ameliorates diabetic
retinopathy through PINK1-dependent activation of mitophagy. Cells.
8:2132019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Jin C, Cao Y and Li Y: Bone mesenchymal
stem cells origin exosomes are effective against sepsis-induced
acute kidney injury in rat model. Int J Nanomedicine. 18:7745–7758.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bjørkøy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang Y, Mun SR, Linares JF, Ahn J, Towers
CG, Ji CH, Fitzwalter BE, Holden MR, Mi W, Shi X, et al:
ZZ-dependent regulation of p62/SQSTM1 in autophagy. Nat Commun.
9:43732018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yang X, Zhang R, Nakahira K and Gu Z:
Mitochondrial DNA mutation, diseases, and nutrient-regulated
mitophagy. Annu Rev Nutr. 39:201–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
McLean BA, Wong CK, Kabir MG and Drucker
DJ: Glucagon-like Peptide-1 receptor Tie2+ cells are essential for
the cardioprotective actions of liraglutide in mice with
experimental myocardial infarction. Mol Metab. 66:1016412022.
View Article : Google Scholar : PubMed/NCBI
|