The central dogma of molecular biology is that
genetic information is transferred from deoxyribonucleic acid (DNA)
to ribonucleic acid (RNA) and then from RNA to proteins (1). In this process, messenger RNA (mRNA)
plays a crucial role as a template for protein synthesis, carrying
genetic information that is central to the dogma of molecular
biology (2). Of the RNA found in
mammalian genomes, ~50% is unable to produce long transcripts
because of the absence of large open reading frames; such RNA is
known as long noncoding RNA (lncRNA). However, advances in genome
sequencing have revealed that, in addition to mRNAs, lncRNAs can
also contain short open reading frames (sORFs) that encode fewer
than 100 codons. These sORFs within lncRNAs encode functionally
stable peptides or microproteins (3–5).
Mitochondria are indispensable for cellular aerobic
respiration, facilitating ATP synthesis and sustaining the
oxidative respiratory chain (6,7).
Beyond their role in energy production, mitochondria regulate redox
signaling, cellular senescence, calcium homeostasis, apoptosis and
inflammation. Thus, maintaining mitochondrial homeostasis is vital
for proper cellular function (7,8).
Processes such as pyroptosis, ferroptosis and necroptosis may
result from a reduction in mitochondrial numbers. Furthermore,
mitochondrial dysfunction can lead to autophagic degradation, loss
of membrane potential, excessive reactive oxygen species (ROS)
production and structural damage (9). Mitochondrial autophagy impairment has
been linked to myocardial fibrosis (10), atherosclerosis (11) and myocardial ischemia/reperfusion
injury (12). Additionally,
dysregulation of mitochondrial dynamics contributes to the onset
and progression of heart failure (13), myocardial infarction (14) and hypertension (15). These findings underscore the
critical role of mitochondrial dysfunction in the pathogenesis of
cardiovascular diseases (CADs). Therefore, investigating the
mechanisms of mitochondrial involvement in CVDs and exploring
potential therapeutic strategies could provide new insights and
targets for CVD treatments (16).
Unlike the single genome found in prokaryotic cells,
eukaryotic cells possess multiple genomes. The nuclear genome and
the mitochondrial genome of eukaryotic cells have coevolved and
continuously adapted to each other, ultimately forming a unified
dual-genome system within animal cells that stores genetic
information and performs various functions (17). The mitochondrial genome contains
only 13 protein-coding genes; therefore, ~98% of mitochondrial
function-related proteins are encoded by the nuclear genome.
Notably, the mitochondrial genome lacks introns and contains only a
few noncoding nucleotides between adjacent genes, as well as sORFs
that encode functional mitochondria-derived peptides (MDPs). This
ability of the mitochondrial genome to encode functionally
significant sORFs through short gene sequences not only highlights
the complexity of the mitochondrial transcriptome but also provides
new avenues for exploring gene expression and functions within the
mitochondria (18–22).
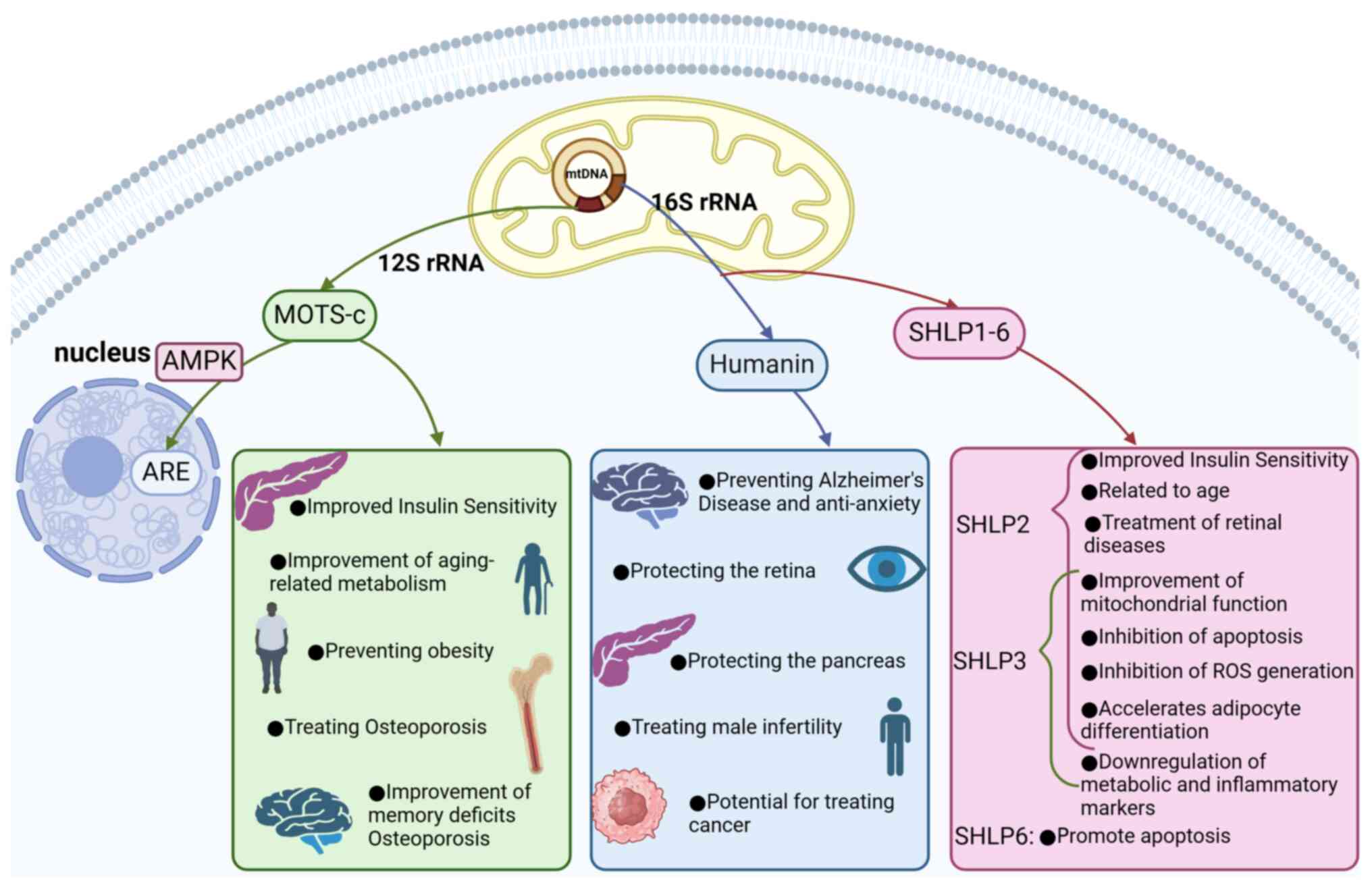
Since the discovery of the first
mitochondria-derived peptide, humanin (HN), in 2001 (23), eight additional MDPs have been
identified, including small HN-like peptides 1–6 (SHLP1-6)
(24), the mitochondrial open
reading frame of the 12S ribosomal RNA type-c (MOTS-c) (25) and SHMOOSE (26) (Fig.
1). According to the 2024 report by the American Heart
Association, CVDs, including coronary heart disease, acute heart
failure and stroke, account for ~30% of deaths worldwide, which
underscores their significant morbidity and mortality (27). These conditions are characterized
by high mortality rates, significant morbidity and increasing
prevalence, making them major contributors to population health
issues. In recent years, the incidence and mortality rates of CVDs
have been increasing among both elderly and younger populations
(28–30). MDPs have been shown to have
regulatory effects on various CVDs and to have considerable
therapeutic potential. The present review discussed the advances in
research regarding MDPs in CVDs, aiming to provide new insights for
future studies and developments.
As a cytoprotective peptide, HN can interact with
heat shock protein 90, thereby exerting a protective effect on
cardiomyoblasts, dopaminergic neuronal cells and fibroblasts
(37). Studies have shown that HN
and its analogs can protect retinal pigment epithelium (RPE) cells
from oxidative stress by improving mitochondrial function in RPE
cells, thereby providing a protective effect on the retina
(38,39). Subsequent research revealed that HN
regulates mitochondrial function by increasing intracellular ATP
levels and respiratory rates, thereby preventing oxidative stress
within cells and ultimately providing cytoprotective effects
(37–41).
A single injection of HN and its analogs has been
shown to improve systemic insulin sensitivity and markedly decrease
blood glucose levels in diabetic rats. Therefore, HN is considered
to have potential as a therapeutic agent for diabetes (42). In addition, HN can induce the
phosphorylation of signal transducer and activator of transcription
3 (STAT3) and extracellular signal-regulated kinase (ERK), thereby
reducing cytokine-induced apoptosis in β-cells. These findings
suggest that HN has protective effects on pancreatic islets and
could aid in the treatment of diabetes (43).
In addition to the aforementioned effects, HN can
also participate in the IL-12/IL-27 cytokine network to exert
immunoregulatory functions within the testicular environment,
suggesting potential therapeutic effects on male infertility and
reproductive health (44). Studies
have shown that HN is highly expressed in patients with gastric and
bladder cancers (45,46), suggesting its potential as a novel
therapeutic target for overcoming chemotherapy resistance in cancer
treatment (47). HN, as the first
identified MDP, has been studied more extensively than other MDPs.
However, a number of aspects of HN functions remain unclear,
warranting further in-depth exploration.
In 2015, through a computational search for
potential sORFs in human 12S rRNA, Kim et al identified an
sORF consisting of 51 base pairs; this sORF can be translated into
a 16-amino acid peptide, which they named MOTS-c (48). MOTS-c is a bioactive peptide that
can regulate gene expression and cellular metabolism. Although
MOTS-c originates from mtDNA, it can translocate from the
mitochondria to the nucleus in response to metabolic stress
triggers (48). The nuclear
translocation of MOTS-c is dependent on 5′-adenosine
monophosphate-activated protein kinase (AMPK). Once MOTS-c enters
the nucleus, it can bind to nuclear DNA and interact with
transcription factors associated with antioxidant response elements
(AREs), such as nuclear factor erythroid 2-related factor 2 (Nrf2),
thereby regulating gene expression and enhancing cellular
resistance to metabolic stress (25,48).
MOTS-c can also increase the levels of
5-aminoimidazole-4-carboxamide ribonucleoside (AICAR), an AMPK
activator, by inhibiting the folate cycle and de novo purine
biosynthesis, thereby activating AMPK. The mechanism of action of
AICAR involves the phosphorylation-induced inactivation of
acetyl-CoA carboxylase, which activates AMPK and stimulates fatty
acid oxidation. This action alleviates the allosteric inhibition of
carnitine palmitoyl transferase 1 and enhances glucose uptake in
muscle cells (21,25,49).
MOTS-c targets skeletal muscle, enhancing systemic insulin
sensitivity and increasing glucose processing rates by promoting
AMPK activation and GLUT4 expression in muscle tissue. This
evidence supports the notion that MOTS-c can prevent insulin
resistance and may have therapeutic effects against diabetes
(25,50).
MOTS-c can increase NAD+ levels and NAD+ serves as
an effective activator of sirtuins, playing a crucial role in the
aging process (25,51,52).
Additionally, MOTS-c levels decline with age, which implies that
MOTS-c may play a role in age-related metabolic abnormalities such
as reduced insulin sensitivity, impaired fatty acid oxidation,
mitochondrial dysfunction, increased obesity, chronic inflammation
and decreased metabolic flexibility (25). Treatment with MOTS-c could prevent
obesity in mice that were fed a high-fat diet by regulating lipid
metabolism and inflammation, as well as by improving mitochondrial
function and insulin sensitivity (25). Moreover, MOTS-c treatment has been
shown to prevent bone loss by promoting type I collagen production
and inhibiting osteoclastogenesis (53), which suggests that MOTS-c acts as a
promising therapeutic approach for osteoporosis (54–56).
Peripheral treatment with MOTS-c has been shown to enhance the
formation and consolidation of objects and spatial recognition
memory while also ameliorating Aβ-induced memory deficits.
Therefore, MOTS-c is considered a novel potential target for the
treatment of cognitive decline in AD (57). As the second MDP discovered after
HN, MOTS-c has become a focal point of research in recent years
because of its unique association with nuclear DNA. With the
continuous advance of MOTS-c research, its multifunctional roles in
various diseases have been discovered and confirmed. For example,
MOTS-c exerts anti-inflammatory effects by inhibiting the MAPK
pathway and stimulating the aryl hydrocarbon receptor (AhR)/STAT3
signaling pathway, which suggests its therapeutic potential for
treating sepsis (58). In a murine
model of colitis, treatment with MOTS-c increased the levels of
phosphorylated AMPK and inhibited the activation of the ERK/JNK
pathway, which led to a reduced inflammatory response, increased
antiapoptotic capacity and ultimately contributed to the protection
against colitis (59). MOTS-c also
protects rotenone-treated PC12 neuronal cells from oxidative stress
by activating the Nrf2/heme oxygenase 1/NAD(P)H quinone
dehydrogenase 1 pathway, which suggests its therapeutic benefits
for Parkinson's disease (60).
Additionally, MOTS-c improves mitochondrial membrane potential
stability by activating the AMPK pathway and reducing inflammation,
oxidative stress and cellular damage, thus alleviating
cancer-induced bone pain (61). In
osteoblasts, MOTS-c promotes the synthesis of type I collagen by
activating the TGF-β/SMAD signaling pathway and leading to
improvements in osteoporosis (53). With respect to its role in cancer,
MOTS-c inhibited the pathological progression of ovarian cancer by
attenuating USP7-mediated deubiquitination of LARS1 (62). In addition to its described
mechanisms and functions in various diseases, MOTS-c exerts
beneficial effects on CADs by regulating inflammation, oxidative
stress and apoptosis (63). This
aspect is further detailed in the following section. The
multifunctionality of MOTS-c in these diseases underscores its
immense potential as a therapeutic target.
SHLP2 is an insulin sensitizer that markedly
improves insulin sensitivity by increasing the glucose infusion
rate, inhibiting hepatic glucose production and enhancing
peripheral glucose uptake (24).
As age increases, the level of SHLP2 in the bloodstream decreases,
which may be indicative of an association with age-related diseases
(24). SHLP2 treatment of
senescent RPE cells revealed that SHLP2 can inhibit cell apoptosis,
primarily through the downregulation of caspase family gene
expression (64). SHLP3 can
downregulate the expression of metabolic and inflammatory markers,
thereby inhibiting the generation of ROS; additionally, it mediates
ERK signaling pathways, promoting adipocyte differentiation and
ultimately enhancing mitochondrial function and cell survival
(44,65). SHLP2 and SHLP3 can both enhance
mitochondrial function, reduce cell apoptosis, decrease the
production of ROS and accelerate adipocyte differentiation
(24). SHLP2 and SHLP3 can prevent
staurosporine-induced damage to the mitochondrial membrane and the
activation of caspase-3, thereby exerting protective effects on
cells. Additionally, SHLP2 can induce the phosphorylation of STAT3
and ERK, further contributing to its protective effects on cellular
health (24,43,66).
In experiments assessing the effects of SHLP1-6 on cell viability,
SHLP2 and SHLP3 increased cell viability and reduced apoptosis in
NIT-1 and 22Rv1 cells; by contrast, SHLP6 markedly increased
apoptosis in NIT-1 and 22Rv1 cells, demonstrating effects opposite
to those of SHLP2 and SHLP3 (24).
In addition to identifying the roles of SHLPs in human diseases,
researchers sequenced mtDNA from five rodent species and found
highly homologous fragments when the sequences of MOTS-c, SHLP4 and
SHLP6 were compared. This discovery has significant research
implications for exploring the reasons behind hibernation behaviors
in these animals as adaptations to cold climates (67).
Currently, research on the functions of SHLP1-6 has
not received widespread attention. However, the protective effects
and insulin-sensitizing properties of SHLP2 and SHLP3, which are
similar to those of HN, highlight their significant potential for
treating certain related diseases. There remains a substantial gap
in our understanding of the roles and mechanisms of SHLP1, SHLP4,
SHLP5 and SHLP6. Nevertheless, existing studies suggest that these
peptides can influence cell function and survival, indicating their
potential role in regulating cellular homeostasis (Table I). In summary, the discovery and
investigation of SHLP1-6 may provide new insights and approaches
for future research on various diseases.
The endothelium is a monolayer of endothelial cells
that forms a barrier throughout the entire vascular system,
separating the vascular wall from the bloodstream; it plays a
crucial role in regulating vascular development and maintaining
homeostasis (68,69). The core function of the endothelium
is to maintain the balance between vascular dilation and
constriction (70,71). When the endothelium is stimulated
and damaged, the balance between vascular dilation and constriction
is disrupted, ultimately leading to endothelial dysfunction
(72). Endothelial dysfunction is
considered an early manifestation of atherosclerosis (69,73).
In the initial stages of atherosclerosis, a dysfunctional
endothelium produces proinflammatory cytokines and decreases the
activity of nitric oxide (NO), which has anti-inflammatory
properties. This process promotes the recruitment and migration of
monocytes from the circulation into the intima, ultimately leading
to their differentiation into macrophages (74,75).
Macrophages engulf oxidized lipoproteins, forming foam cells, which
serve as a hallmark of early atherosclerosis (76). Moreover, when the endothelial layer
of arterial blood vessels is damaged, low-density lipoprotein (LDL)
permeates the subendothelial space. ROS oxidatively modify LDL,
resulting in the formation of oxidized LDL (Ox-LDL). Ox-LDL further
increases ROS production, ultimately leading to increased oxidative
stress, inflammation and atherosclerotic plaque formation (77–79).
In addition to endothelial dysfunction, factors such as
inflammation, dyslipidemia, plaque rupture and smoking also
contribute to the development of atherosclerosis (80,81).
Studies have shown that serum levels of HN are
markedly lower in patients with endothelial dysfunction than in
healthy individuals (82,83). This decrease may be associated with
coronary endothelial dysfunction, including impaired vascular
dilatation, which is typically associated with increased oxidative
stress and inflammation. These factors disrupt mitochondrial energy
production and structural integrity, leading to mitochondrial
dysfunction, which, in turn, contributes to reduced levels of HN
(84–86). These findings suggest that serum HN
levels could serve as potential indicators for monitoring
endothelial dysfunction. ROS can contribute to endothelial
dysfunction by reducing the activity of NO in blood vessels and
promoting cellular damage (87).
Therefore, oxidative stress is considered a crucial mechanism
involved in the pathogenesis of endothelial dysfunction and plays a
significant role in the occurrence and progression of
atherosclerosis (77). Research
has shown that HN is expressed in the endothelial cells of both
human arteries and veins. Additionally, HN can reduce the levels of
ROS and ceramides induced by Ox-LDL in human aortic endothelial
cells in a dose-dependent manner, thereby preventing cell apoptosis
(88,89). HN exerts its antioxidant effects by
inhibiting the production of ROS through the suppression of reduced
nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2)
(90). These findings suggest that
HN may serve as a potential therapeutic target for atherosclerosis
by mitigating oxidative stress.
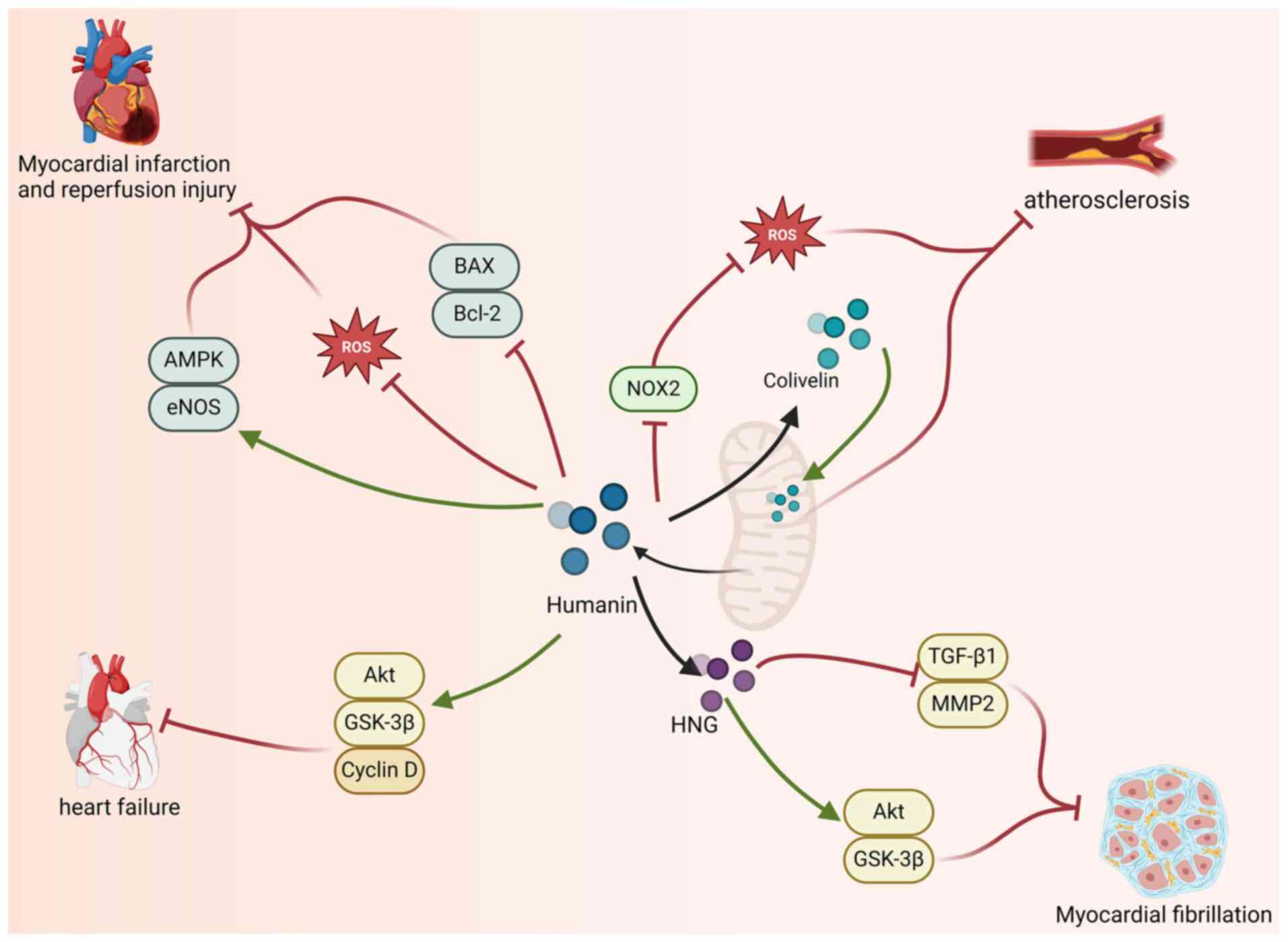
Colivelin is a hybrid peptide composed of the
C-terminus of activity-dependent neurotrophic factor (ADNF) fused
with the HN derivative AGA-(C8R) humanin-G (HNG)17 (91). Research has shown that Colivelin
can alleviate mitochondrial dysfunction in damaged endothelial
cells, playing a critical role in maintaining the structural
integrity of the vascular wall (92,93).
As Colivelin can help maintain normal vascular structure, it has
the potential to serve as a therapeutic agent for treating
endothelial dysfunction, thereby exerting beneficial effects on
atherosclerosis.
Acute myocardial infarction results from the acute
blockage of blood flow to the myocardium (94). This sudden blockage can lead to a
reduction or complete loss of blood flow to a specific part of the
heart, ultimately resulting in cardiac damage or necrosis (95,96).
Percutaneous coronary intervention can rapidly and effectively
restore blood flow to previously ischemic areas. However, the
restoration of blood flow to these ischemic regions results in the
production of large amounts of ROS, which can induce cardiomyocyte
death and lead to myocardial damage. This phenomenon is referred to
as myocardial ischemia-reperfusion injury (96–98).
HNG, in which the 14th amino acid (serine) is
replaced by glutamic acid, is a more potent analog of HN.
Thummasorn et al (97)
demonstrated that pretreatment with HNG in a mouse model of
myocardial ischemia-reperfusion injury conferred beneficial effects
against ischemia-reperfusion damage. Pretreatment with HNG can
reduce the generation of ROS, thereby alleviating mitochondrial
dysfunction in the heart (97).
Additionally, HNG pretreatment decreases the infarct area and the
protein expression of B-cell lymphoma-2-associated X protein (Bax),
resulting in cardioprotective effects (97). Researchers have shown that in
vitro treatment with HNG in a mouse model of established
ischemia can reduce myocardial infarct size in a dose-dependent
manner, resulting in cardioprotective effects (99); after HNG treatment, there was a
significant increase in the phosphorylation levels of AMP-activated
protein kinase (AMPK) and endothelial nitric oxide synthase (eNOS)
in the hearts of the mice, as well as a notable decrease in the
levels of Bax and B-cell lymphoma-2 (Bcl-2). These findings suggest
that HNG may provide cardioprotection in myocardial reperfusion
injury by activating AMPK-eNOS-mediated signaling pathways and
regulating apoptotic factors (99). Moreover, administering a high dose
of HNG (252 µg/kg) during the ischemic phase increases the level of
HN in the damaged myocardium, thereby enhancing the
cardioprotective effects of HN against myocardial
ischemia-reperfusion injury (94).
HNG not only increases the level of HN but also reduces the
myocardial infarct size and alleviates cardiac mitochondrial
dysfunction (94). The results of
the aforementioned studies demonstrate that HN and its derivatives
play a beneficial role in the prevention and treatment of
ischemia-reperfusion injury. This beneficial effect is attributed
primarily to the ability of HN and its derivatives to mitigate
mitochondrial dysfunction in damaged myocardial cells, thereby
protecting the heart. These findings highlight the potential of HN
and its derivatives as novel therapeutic agents for
ischemia-reperfusion injury.
The endonuclease G (EndoG) gene has been identified
as a pressure-independent determinant of cardiac hypertrophy.
Specifically, the absence of EndoG induces cardiomyocyte
hypertrophy and increases the production of ROS in vitro
(100). Cardiomyocyte hypertrophy
can be induced through the activation of AKT/ERK phosphorylation
and the activation of the mTOR signaling pathway, as well as the
inhibition of glycogen synthase kinase-3β (GSK-3β) phosphorylation.
These processes collectively induce the transcription of multiple
genes, including myocyte enhancer factor 2. During the progression
of cardiomyocyte hypertrophy, mitochondrial ROS serve as
stimulators that affect these signaling pathways. If such
stimulation persists, it may ultimately lead to heart failure
(101–104).
The addition of low concentrations of HN to
cardiomyocytes from EndoG gene-deficient mice can prevent the
accumulation of mitochondrial ROS in these EndoG-deficient
cardiomyocytes. Moreover, HN can also inhibit the abnormal growth
of EndoG-deficient cardiomyocytes in vitro, thereby exerting
an antihypertrophic effect on these cells (105). Experiments have demonstrated that
HN inhibits cardiomyocyte hypertrophy by restoring ROS levels, cell
size and normal proliferation capabilities in EndoG-deficient cells
(105). In addition, HN restores
the proliferation capacity of EndoG-deficient cells while
simultaneously normalizing the phosphorylation ratio of
phosphorylated Akt/Akt and the expression of cyclin D (106). This occurs because HN can
mitigate the impact of ROS on Akt signaling, thereby restoring Akt
phosphorylation and cell proliferation in EndoG-deficient cells;
that is, HN can overcome the effects of ROS and restore normal
proliferation rates in the absence of EndoG (106). The results from these studies
demonstrate that HN can beneficially maintain normal cellular
function by reducing intracellular ROS levels, particularly by
markedly inhibiting cardiomyocyte hypertrophy. Therefore, HN may
exert a beneficial effect on heart failure by suppressing
cardiomyocyte hypertrophy, indicating its potential as a
therapeutic agent for heart failure.
Cardiac fibroblasts are the most abundant stromal
cell type in the heart and serve as the primary producers of the
extracellular matrix (ECM) within the myocardium. These fibroblasts
play crucial roles in maintaining the integrity of the ECM network.
The main component of the ECM is collagen, which is deposited by
cardiac fibroblasts, provides structural support to cardiac tissue,
maintains structural integrity and regulates cell communication and
function (107,108). In response to pathological
stimuli such as myocardial infarction, cardiac fibroblasts are
activated and differentiate into myofibroblasts, disrupting
homeostasis within the cardiac tissue. This process initiates and
promotes the occurrence and progression of cardiac fibrosis
(109–112). Cardiac fibrosis increases with
age and ultimately leads to cardiac dysfunction, which is
characterized by the deposition of ECM in the myocardium and the
production of myofibroblasts and is often accompanied by diastolic
or systolic heart failure. The activation of fibroblasts and their
differentiation into myofibroblasts are primarily mediated by
TGF-β, which contributes to a profibrotic cardiac microenvironment
(113–116).
In a study involving the long-term administration of
exogenous HNG to aged mice, HNG increased the percentage of
cardiomyocytes in aging hearts, reduced collagen deposition in the
cardiac stroma and decreased the proliferation of fibroblasts in
the aging myocardium; additionally, HNG downregulated the
expression of TGF-β1 and MMP2 in the aging myocardium (117). MMPs are enzymes responsible for
the degradation of the ECM and the expression of MMP2 plays a
profibrotic role in the heart (113,118). Additionally, HNG can attenuate
cardiac fibrosis by activating the Akt/GSK-3β pathway. HNG
activates the Akt pathway both in vitro and in vivo.
Activated Akt directly phosphorylates GSK-3β at Ser9, negatively
regulating GSK-3β kinase activity, inhibiting the opening of the
mitochondrial permeability transition pore and ultimately
suppressing myocardial failure and fibrosis (117,119,120). The results from the
aforementioned studies indicate that treatment with HN and its
derivatives can mitigate myocardial fibrosis in aging hearts
(117). Whether HN and its
derivatives can serve as potential therapeutic approaches for
myocardial fibrosis and related diseases in aging hearts requires
further investigation. (Fig.
2).
In the treatment of patients with ST-segment
elevation myocardial infarction (STEMI), performing percutaneous
coronary intervention (PCI) after diagnosis leads to more favorable
outcomes than do conventional therapies (121). However, PCI does not always yield
positive outcomes. The ‘no-reflow phenomenon’ is a potential
complication associated with PCI (122). The pathogenesis of the no-reflow
phenomenon is complex and involves multiple factors, such as
atherosclerosis, ischemic injury, myocardial reperfusion injury and
coronary microvascular dysfunction, all of which can contribute to
its occurrence (123).
A comparison of serum MOTS-c levels in patients with
STEMI undergoing PCI with those in healthy individuals revealed
that MOTS-c levels are markedly lower in patients with STEMI
(124). This decrease is more
pronounced as the thrombolysis in myocardial infarction (TIMI)
blood flow worsens. Therefore, low MOTS-c levels could be an
important predictor of STEMI and reduced MOTS-c levels may play a
role in the onset and progression of STEMI (124). Furthermore, studies indicate that
as MOTS-c levels decline, TIMI flow worsens, ultimately leading to
the no-reflow phenomenon. In patients with STEMI undergoing PCI,
MOTS-c levels are markedly elevated, indicating that increased
MOTS-c has high sensitivity and specificity for predicting the
no-reflow phenomenon. Therefore, MOTS-c is considered a robust and
independent predictor of no-reflow occurrence (124). Although an association between
MOTS-c levels and STEMI has been established, the underlying
physiological mechanisms remain unclear and warrant further
investigation.
Vascular calcification refers to the abnormal
deposition of calcium phosphate crystals in the arterial wall
(125); it commonly occurs in
vascular lesions associated with diabetes, chronic kidney disease,
hypertension and aging, leading to medial sclerosis and the
calcification of atherosclerotic plaques (125,126). The prevalence of vascular
calcification increases with age (126,127). Vascular calcification typically
occurs in both the intima and media layers of the arterial wall.
The intimal layer consists of endothelial cells, which are
surrounded by a thick layer of elastic fibers. Intimal
calcification is associated with dyslipidemia and inflammation,
with inflammation contributing to thickening of the intimal layer,
ultimately leading to atherosclerosis (128–130). Studies have shown that metformin
(131), growth hormone-releasing
peptides (132) and
death-associated protein kinase 3 (133) can alleviate vascular
calcification to varying degrees. These substances achieve this
effect by influencing AMPK signaling pathways, which play a key
role in reducing vascular calcification. These findings suggest
that the AMPK signaling pathway plays a crucial role in regulating
the progression of vascular calcification.
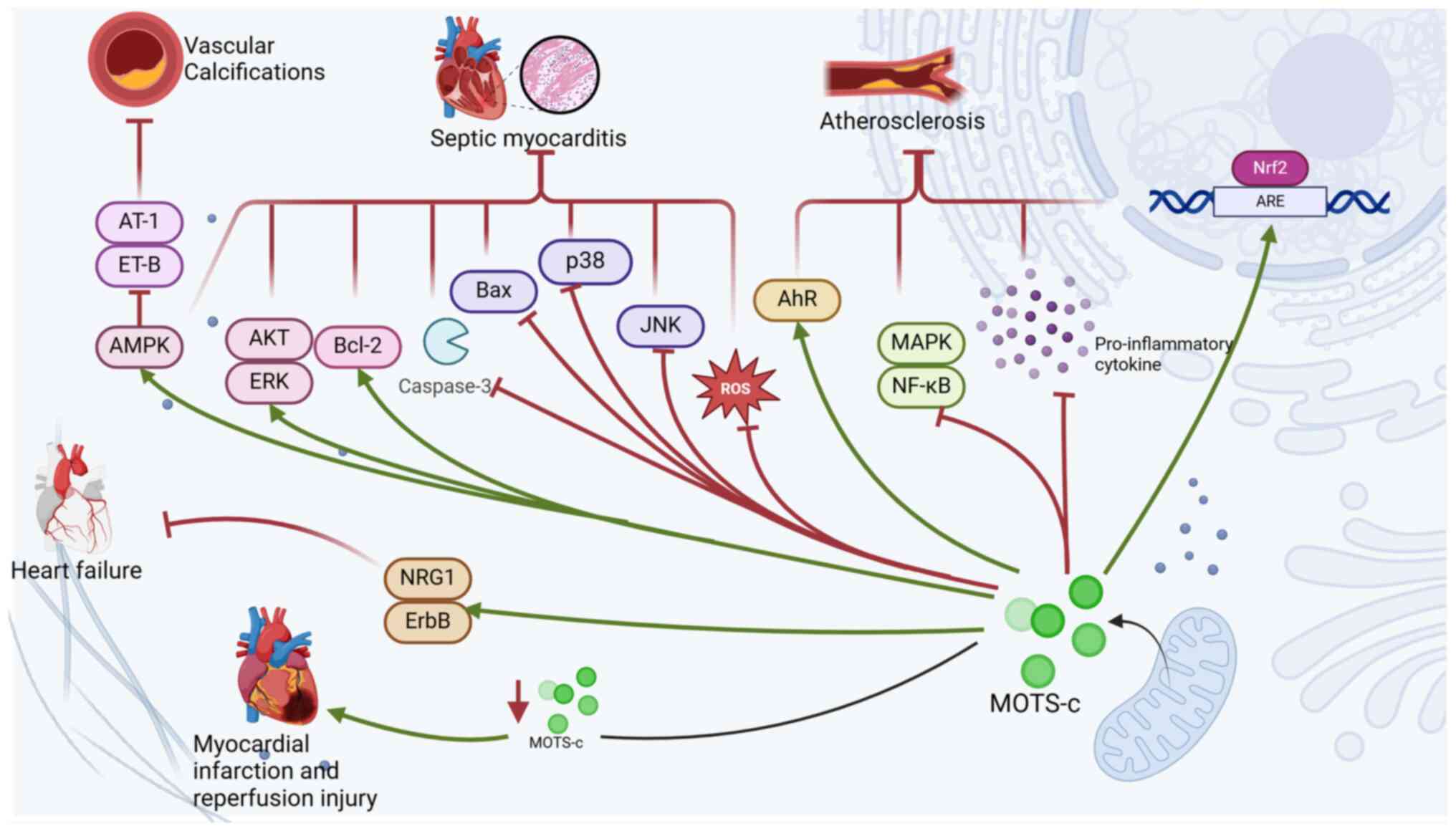
In a study in which vascular calcification was
induced in rats through treatment with vitamin D3 and nicotine,
subsequent treatment with MOTS-c markedly reduced blood pressure,
preserved normal heart structure and decreased vascular stiffness
(134). These findings
demonstrate that MOTS-c can improve cardiovascular and vascular
abnormalities caused by vascular calcification (134). Further research revealed that
MOTS-c can reverse the downregulation of AMPK expression caused by
vascular calcification. Additionally, MOTS-c reduces the levels of
angiotensin II type 1 (AT-1) and endothelin B (ET-B), contributing
to its protective effects on the cardiovascular system (134). AT-1 and ET-B can participate in
the AMPK signaling pathway by binding to their respective receptors
(134). A decrease in AT-1
receptor levels plays a crucial role in reducing oxidative stress
and preventing the progression of myocardial contractile
dysfunction, whereas elevated AT-1 receptor levels can induce
myocardial fibrosis and heart failure (135,136). These findings suggest that MOTS-c
may alleviate vascular calcification and improve associated cardiac
abnormalities by activating AMPK signaling and suppressing the
expression of AT-1 and ET-B receptors. Therefore, it is proposed
that MOTS-c could serve as a potential anti-calcifying agent for
intervention in vascular calcification and may have therapeutic
effects in this context.
A study revealed that patients with endothelial
dysfunction have markedly lower plasma levels of MOTS-c than
patients with normal endothelial function (137). Furthermore, the plasma levels of
MOTS-c are positively associated with microvascular and epicardial
coronary endothelial function, indicating that lower levels of
MOTS-c in plasma are associated with endothelial dysfunction
(137). Additionally,
preconditioning with MOTS-c in rats or aortic arteries from mice
with renal artery stenosis can enhance acetylcholine (ACh)-induced
vasodilation, indicating that MOTS-c can improve vascular
endothelial function in vitro (137).
In the initial stage of atherosclerosis, a
dysfunctional endothelium produces proinflammatory factors and
reduces the activity of NO, which has anti-inflammatory properties.
This leads to the recruitment and transport of circulating
monocytes to the intima, resulting in their differentiation into
macrophages (74,75). Research has demonstrated that
MOTS-c can inhibit the expression of proinflammatory cytokines,
such as TNF-α, IL-6 and IL-1β, while simultaneously increasing the
levels of the anti-inflammatory cytokine IL-10 (58). Additionally, MOTS-c exerts its
anti-inflammatory effects by inhibiting the phosphorylation of
mitogen-activated protein kinase (MAPK)-related proinflammatory
pathways and activating AhR-associated anti-inflammatory pathways
(58). Furthermore, MOTS-c can
inhibit oxidative stress and inflammatory states induced by
H2O2 in H2c2 cells by activating the Nrf2/ARE
pathway and suppressing the nuclear factor
kappa-light-chain-enhancer of activated B cells (NF-κB) pathway
(138). Additionally, MOTS-c
alleviates endothelial dysfunction by inhibiting the MAPK/NF-κB
pathway in activated B cells (139).
In summary, MOTS-c can improve vascular endothelial
function through anti-inflammatory and antioxidative stress
pathways, thereby inhibiting the occurrence and progression of
endothelial dysfunction. Given the critical role of endothelial
dysfunction in the development of atherosclerosis, MOTS-c is
hypothesized to exert therapeutic effects on atherosclerosis by
ameliorating endothelial dysfunction (77–79).
The effect of MOTS-c on atherosclerosis primarily involves
addressing inflammation and endothelial dysfunction; however, the
underlying mechanisms warrant further investigation. Additionally,
it remains unclear whether MOTS-c has similar therapeutic potential
against atherosclerosis induced by other factors, which requires
further exploration.
Heart failure is a syndrome that occurs when the
heart is unable to pump sufficiently to meet the energy demands of
the body (140). The pathogenesis
of heart failure includes inflammation, increased oxidative stress,
abnormalities in energy metabolism, cardiac cell apoptosis,
mitochondrial dysfunction and interstitial fibrosis (140). In a study where heart failure was
induced in mice through transverse aortic constriction surgery, the
administration of MOTS-c markedly attenuated the progression of
cardiac dysfunction and structural deterioration in the mice;
additionally, MOTS-c treatment resulted in a notable reduction in
inflammatory responses and an increase in antioxidant capacity
(141). Given that inflammation
and oxidative stress play crucial roles in the development of heart
failure, MOTS-c may hold potential for the prevention and treatment
of heart failure progression (141).
The protein levels of neuregulin 1β (NRG1-β) in the
serum of heart failure patients are markedly lower than those in
healthy individuals (142). NRG1
is a membrane-bound vasoactive peptide that belongs to the
epidermal growth factor family; it is released through proteolytic
cleavage in response to stimuli such as inflammation, ischemia and
oxidative stress in various tissue types, including the heart
(143). NRG1 influences
cardiomyocytes by activating ErbB tyrosine kinase receptors,
leading to downstream signaling through the phosphoinositide
3-kinase (PI3K) and MAPK pathways. This activation ultimately
inhibits cell apoptosis and promotes cardiomyocyte proliferation
(143,144). In mice with heart failure induced
by diabetes, the expression levels of NRG1 and ErbB mRNA decreased;
however, following treatment with MOTS-c, the levels of NRG1 and
ErbB increased (145). These
studies suggest that MOTS-c may achieve therapeutic effects on
heart failure by restoring the NRG1/ErbB signaling pathway.
Although research on MOTS-c for treating heart failure is limited,
the current findings indicate that MOTS-c may hold therapeutic
potential. However, further investigations are needed to elucidate
the broader mechanisms and physiological roles of MOTS-c in heart
failure treatment.
Sepsis is defined as life-threatening organ
dysfunction resulting from a dysregulated host response to
infection (146). Septic
cardiomyopathy is one of the most severe complications of sepsis,
with an incidence of 30–60% among septic patients and a mortality
rate of 70–90% (147–149). Its primary features include left
ventricular dilation and a reduced ejection fraction (EF), which
severely impair cardiac function, leading to circulatory failure
and multi-organ dysfunction. These complications markedly
exacerbate the progression of sepsis (147,148,150,151). Timely intervention in septic
cardiomyopathy has the potential to reverse the onset and
progression of sepsis, underscoring the critical importance of
early detection and targeted therapeutic strategies (152,153). The pathogenesis of septic
cardiomyopathy involves dysregulated inflammatory responses,
imbalances in calcium homeostasis, mitochondrial dysfunction,
oxidative stress and endothelial dysfunction (147,154,155).
Research has shown that in a mouse model of septic
cardiomyopathy induced by lipopolysaccharide (LPS), treatment with
MOTS-c can reverse the increased transcription levels of
inflammatory factors such as IL-1β, IL-4, IL-6 and TNF-α caused by
LPS administration. These findings indicate that MOTS-c can
effectively decrease the inflammatory response associated with
LPS-induced septic cardiomyopathy (155). Additionally, MOTS-c has been
shown to reverse the LPS-induced decrease in the expression of the
antiapoptotic protein BCL-2 as well as the increase in the levels
of the proapoptotic proteins BAX and cleaved caspase-3. Thus,
MOTS-c is capable of alleviating cardiomyocyte apoptosis in
LPS-induced septic cardiomyopathy (155). In the MOTS-c treatment group, the
levels of cellular antioxidants were greater than those in the
untreated LPS group; furthermore, MOTS-c was able to eliminate
excessive ROS production in cardiomyocytes, indicating that MOTS-c
treatment can alleviate the mitochondrial dysfunction and oxidative
stress induced by LPS in cardiomyocytes (155). MOTS-c also activates various
cardioprotective signaling pathways, including the AMPK, AKT and
ERK pathways, while inhibiting multiple proinflammatory signaling
pathways, such as the c-Jun N-terminal kinase (JNK) and p38
mitogen-activated protein kinase (p38) pathways (155). In summary, MOTS-c may protect
normal myocardial function by reducing inflammatory responses in
cardiomyocytes, inhibiting cardiomyocyte apoptosis and maintaining
mitochondrial homeostasis. Research on the therapeutic effects of
MOTS-c in septic cardiomyopathy is still in its early stages.
Whether MOTS-c can provide effective clinical treatment for human
septic cardiomyopathy and whether it can become a successful
therapeutic target are questions that warrant further exploration
(Fig. 3).
Hyperlipidemia (HLP) is characterized by lipid
metabolism abnormalities and issues related to fat transport,
manifesting as elevated cholesterol levels or dysregulated
lipoproteins; It is a significant risk factor for conditions such
as coronary atherosclerosis, heart failure, hypertension,
myocardial infarction and stroke, which are related to CADs
(156–159). Increasing evidence suggests that
sphingolipids may serve as primary regulators of lipid metabolism
(160). A high-fat diet has been
shown to increase sphingolipid levels in the liver, adipose tissue
and plasma (161). Sphingolipids
are synthesized from ceramides through enzymatic transfer mediated
by phosphocholine transferase; thus, preventing the de novo
synthesis of ceramides is crucial for alleviating obesity-related
conditions such as hyperlipidemia and atherosclerosis (162,163). The injection of SHLP2 into
diet-induced obese mice markedly reduced the plasma levels of
CID3126 after treatment (163),
with ceramide (CID3126) serving as a precursor for sphingolipid
synthesis (163). Concurrently,
two glycosylated ceramides, glycosyl-N-palmitoyl-sphingosine and
glycosyl-N-steryl-sphingosine, decreased substantially, along with
a marked reduction in various types of sphingolipids (163). Additionally, the levels of
diacylglycerol, a byproduct generated during sphingolipid
synthesis, also tended to decrease in the plasma (163). Collectively, these findings
suggest that ceramide synthesis for sphingolipid production is
impaired, indicating that SHLP2 may exert beneficial effects on
hyperlipidemia by modulating sphingolipid metabolic pathways and
altering the concentrations of lipid metabolites in the plasma
(163). Therefore, it is proposed
that SHLP2 could serve as a potential therapeutic target for
conditions associated with hyperlipidemia (Table II).
The present review offered an extensive exploration
of the roles, mechanisms and therapeutic potential of MDPs in CVDs.
Distinct from prior reviews (63,96,164), the present analysis introduced
several innovative elements that enhanced the comprehension of the
role of MDPs in cardiovascular biology and its therapeutic
implications.
In summary, the innovations of this review lie in
its comprehensive exploration of the molecular mechanisms of MDPs
in CVDs. By systematically summarizing the roles of different MDPs
across various types of CVDs and incorporating the most recent
research developments, this review offered a thorough and intuitive
perspective on the topic. The innovative structural design of this
manuscript presented the functions and mechanisms of MDPs in a
cohesive manner, enhancing the understanding of their potential
therapeutic applications. Additionally, it highlighted the
limitations of current research and clinical trials, while
proposing future directions to address existing challenges, thereby
advancing the scientific knowledge of MDPs in CVDs.
Currently, the confirmed microproteins identified
within the sORFs of mtDNA include HN (23), SHLP1-6 (24), MOTS-c (25) and the recently discovered SHMOOSE
(26). Owing to their antioxidant,
anti-inflammatory and antiapoptotic properties, MDPs have
significant effects on various conditions and diseases, including
aging, chronic inflammatory diseases, cancer, neurodegenerative
diseases and CVDs (36,47,58,145). The recent discovery of SHMOOSE
underlines that our exploration of mitochondrial-derived peptides
is just beginning; there may still be a number of unknowns within
the mitochondria awaiting discovery, along with numerous questions
that remain to be answered.
HN, the first identified MDP, is widely recognized
for its therapeutic effects on neurodegenerative diseases (34–36).
Research has revealed its significant roles in CADs, diabetes and
cancer, particularly in relation to cardiovascular health (43,45,88,94,117). Owing to the antioxidant,
antiapoptotic and cytoprotective properties of HN and its analogs,
these peptides may have therapeutic potential for cardiovascular
conditions such as atherosclerosis (88,90,92),
ischemia-reperfusion injury (94,97,99)
and myocardial fibrosis (117),
suggesting the possibility of developing new therapeutic targets
for various CADs. While research on HN and its analogs in the
context of cardiovascular health is more advanced than research on
other MDPs, further investigations are necessary to explore their
clinical applications in the future.
MOTS-c is a bioactive peptide that regulates gene
expression and cellular metabolism (25,48).
Although it originates from mtDNA, it can reach the nucleus in
response to metabolic stress, thereby modulating gene expression to
cope with such stress (48). The
unique properties of MOTS-c have made it a focal point of research
in recent years. While studies have established associations
between MOTS-c and various cardiovascular-related diseases,
including atherosclerosis (58,137–139), vascular calcification (134), heart failure (141,145) and reperfusion injury (124), most studies have focused
primarily on observational phenomena rather than elucidating the
underlying mechanisms of action. Additionally, the potential
relationships of MOTS-c with other CVDs remain to be validated.
Research on SHLP1-6 has focused primarily on SHLP2
and SHLP3, which have been shown to enhance mitochondrial function,
reduce apoptosis and ROS production and accelerate adipocyte
differentiation (24,43,64).
By contrast, SHLP6 has opposite effects (24). The study of SHLP1-6 is still in its
early stages and current evidence linking these peptides to CVDs is
limited (163). However, the
relationship between SHLP2 and hyperlipidemia raises the question
of whether SHLP1-6 might also play a role in cardiovascular health,
which remains uncertain.
At present, investigations into the role of MDPs in
CVDs have focused mainly on HN and MOTS-c, while the effects of
other MDPs on cardiovascular health have yet to be thoroughly
studied. Regarding HN and MOTS-c, numerous questions remain
unanswered. The recent discovery of SHMOOSE prompts the
consideration of whether more types of MDPs have yet to be
identified. These should be the main issues of concern: First, the
molecular mechanisms of MDPs remain incompletely understood,
suggesting that their actions may vary across different diseases.
Second, most research on MDPs has been conducted in cellular and
animal models, with limited clinical data, hindering their
application (63,165,166). Third, MDPs exhibit low
bioavailability and stability in vivo (167,168). Finally, due to technical
constraints, the synthesis and detection of MDPs remain costly,
presenting both technological and economic challenges to expanding
related research (169).
In summary, the associations between MDPs and CVDs
are undeniable; however, the underlying mechanisms require further
investigation. Thus, it is important to explore the interactions
between MDPs and relevant signaling pathways in the regulation of
mitochondrial function, inflammation, oxidative stress and
apoptosis. Additionally, developing more efficient and stable
delivery systems for MDPs could enhance their bioavailability.
Furthermore, research on MDPs should focus on bridging the gap
between basic studies and clinical trials to verify their safety
and therapeutic efficacy in CVD patients. As modern science
continues to explore the mitochondrial genome and advances in
medical research progress, the therapeutic potential of MDPs for
CVD treatment will gradually be revealed.
Not applicable.
The present study was supported by the Weifang City Youth Talent
Support Program and Weifang Science and Technology Development
Projects (grant no. 2023YX092).
Not applicable.
YR and ZG wrote and drafted the manuscript and
figures. LZ, HL and XZ revised the manuscript. XG, XC and HC
assisted in manuscript structuring and revisions. MC reviewed and
edited the manuscript before submission. Data authentication is not
applicable. All the authors read and approved the final
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Crick F: Central dogma of molecular
biology. Nature. 227:561–563. 1970. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
van den Akker GGH, Caron MMJ, Peffers MJ
and Welting TJM: Ribosome dysfunction in osteoarthritis. Curr Opin
Rheumatol. 34:61–67. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Carninci P, Kasukawa T, Katayama S, Gough
J, Frith MC, Maeda N, Oyama R, Ravasi T, Lenhard B, Wells C, et al:
The transcriptional landscape of the mammalian genome. Science.
309:1559–1563. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Plaza S, Menschaert G and Payre F: In
search of lost small peptides. Annu Rev Cell Dev Biol. 33:391–416.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Leong AZX, Lee PY, Mohtar MA, Syafruddin
SE, Pung YF and Low TY: Short open reading frames (sORFs) and
microproteins: An update on their identification and validation
measures. J Biomed Sci. 29:192022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Huang J, Hao J, Wang P and Xu Y: The role
of mitochondrial dysfunction in CKD-related vascular calcification:
From mechanisms to therapeutics. Kidney Int Rep. 9:2596–2607. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pang B, Dong G, Pang T, Sun X, Liu X, Nie
Y and Chang X: Advances in pathogenesis and treatment of vascular
endothelial injury-related diseases mediated by mitochondrial
abnormality. Front Pharmacol. 15:14226862024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chang X, Lochner A, Wang HH, Wang S, Zhu
H, Ren J and Zhou H: Coronary microvascular injury in myocardial
infarction: Perception and knowledge for mitochondrial quality
control. Theranostics. 11:6766–6785. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang B, Wu H, Zhang J, Cong C and Zhang
L: The study of the mechanism of non-coding RNA regulation of
programmed cell death in diabetic cardiomyopathy. Mol Cell Biochem.
479:1673–1696. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao J, Yang T, Yi J, Hu H, Lai Q, Nie L,
Liu M, Chu C and Yang J: AP39 through AMPK-ULK1-FUNDC1 pathway
regulates mitophagy, inhibits pyroptosis, and improves
doxorubicin-induced myocardial fibrosis. iScience. 27:1093212024.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y,
Ding K, Zheng S, Yang N, Zhang Z, et al: Novel role for caspase 1
inhibitor VX765 in suppressing NLRP3 inflammasome assembly and
atherosclerosis via promoting mitophagy and efferocytosis. Cell
Death Dis. 13:5122022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zang GY, Yin Q, Shao C, Sun Z, Zhang LL,
Xu Y, Li LH and Wang ZQ: CD137 signaling aggravates myocardial
ischemia-reperfusion injury by inhibiting mitophagy mediated NLRP3
inflammasome activation. J Geriatr Cardiol. 20:223–237. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Forte M, Schirone L, Ameri P, Basso C,
Catalucci D, Modica J, Chimenti C, Crotti L, Frati G, Rubattu S, et
al: The role of mitochondrial dynamics in cardiovascular diseases.
Br J Pharmacol. 178:2060–2076. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ramachandra CJA, Hernandez-Resendiz S,
Crespo-Avilan GE, Lin YH and Hausenloy DJ: Mitochondria in acute
myocardial infarction and cardioprotection. EBioMedicine.
57:1028842020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu X, Tan H, Liu X and Wu Q: Correlation
between the expression of Drp1 in vascular endothelial cells and
inflammatory factors in hypertension rats. Exp Ther Med.
15:3892–3898. 2018.PubMed/NCBI
|
|
16
|
Brown DA, Perry JB, Allen ME, Sabbah HN,
Stauffer BL, Shaikh SR, Cleland JGF, Colucci WS, Butler J, Voors
AA, et al: Expert consensus document: Mitochondrial function as a
therapeutic target in heart failure. Nat Rev Cardiol. 14:238–250.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bensasson D, Zhang D, Hartl DL and Hewitt
GM: Mitochondrial pseudogenes: Evolution's misplaced witnesses.
Trends Ecol Evol. 16:314–321. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Popov LD: Mitochondrial
peptides-appropriate options for therapeutic exploitation. Cell
Tissue Res. 377:161–165. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Benayoun BA and Lee C: MOTS-c: A
mitochondrial-encoded regulator of the nucleus. Bioessays.
41:e19000462019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mercer TR, Neph S, Dinger ME, Crawford J,
Smith MA, Shearwood AM, Haugen E, Bracken CP, Rackham O,
Stamatoyannopoulos JA, et al: The human mitochondrial
transcriptome. Cell. 146:645–658. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim SJ, Xiao J, Wan J, Cohen P and Yen K:
Mitochondrially derived peptides as novel regulators of metabolism.
J Physiol. 595:6613–6621. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Son JM and Lee C: Mitochondria:
Multifaceted regulators of aging. BMB Rep. 52:13–23. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashimoto Y, Niikura T, Tajima H, Yasukawa
T, Sudo H, Ito Y, Kita Y, Kawasumi M, Kouyama K, Doyu M, et al: A
rescue factor abolishing neuronal cell death by a wide spectrum of
familial Alzheimer's disease genes and Abeta. Proc Natl Acad Sci
USA. 98:6336–6341. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cobb LJ, Lee C, Xiao J, Yen K, Wong RG,
Nakamura HK, Mehta HH, Gao Q, Ashur C, Huffman DM, et al: Naturally
occurring mitochondrial-derived peptides are age-dependent
regulators of apoptosis, insulin sensitivity, and inflammatory
markers. Aging (Albany NY). 8:796–809. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lee C, Zeng J, Drew BG, Sallam T,
Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R,
et al: The mitochondrial-derived peptide MOTS-c promotes metabolic
homeostasis and reduces obesity and insulin resistance. Cell Metab.
21:443–454. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Miller B, Kim SJ, Mehta HH, Cao K, Kumagai
H, Thumaty N, Leelaprachakul N, Braniff RG, Jiao H, Vaughan J, et
al: Mitochondrial DNA variation in Alzheimer's disease reveals a
unique microprotein called SHMOOSE. Mol Psychiatry. 28:1813–1826.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Martin SS, Aday AW, Almarzooq ZI, Anderson
CAM, Arora P, Avery CL, Baker-Smith CM, Barone Gibbs B, Beaton AZ,
Boehme AK, et al: 2024 Heart disease and stroke statistics: A
report of US and global data from the american heart association.
Circulation. 149:e347–e913. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Virani SS, Alonso A, Aparicio HJ, Benjamin
EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng
S, Delling FN, et al: Heart disease and stroke statistics-2021
update: A report from the american heart association. Circulation.
143:e254–e743. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Westman PC, Lipinski MJ, Luger D, Waksman
R, Bonow RO, Wu E and Epstein SE: Inflammation as a driver of
adverse left ventricular remodeling after acute myocardial
infarction. J Am Coll Cardiol. 67:2050–2060. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roy P, Orecchioni M and Ley K: How the
immune system shapes atherosclerosis: Roles of innate and adaptive
immunity. Nat Rev Immunol. 22:251–265. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamagishi Y, Hashimoto Y, Niikura T and
Nishimoto I: Identification of essential amino acids in humanin, a
neuroprotective factor against Alzheimer's disease-relevant
insults. Peptides. 24:585–595. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thiankhaw K, Chattipakorn K, Chattipakorn
SC and Chattipakorn N: Roles of humanin and derivatives on the
pathology of neurodegenerative diseases and cognition. Biochim
Biophys Acta Gen Subj. 1866:1300972022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Niikura T: Humanin and Alzheimer's
disease: The beginning of a new field. Biochim Biophys Acta Gen
Subj. 1866:1300242022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao H, Sonada S, Yoshikawa A, Ohinata K
and Yoshikawa M: Rubimetide, humanin, and MMK1 exert
anxiolytic-like activities via the formyl peptide receptor 2 in
mice followed by the successive activation of DP1, A2A, and GABAA
receptors. Peptides. 83:16–20. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Murakami M, Nagahama M, Maruyama T and
Niikura T: Humanin ameliorates diazepam-induced memory deficit in
mice. Neuropeptides. 62:65–70. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hashimoto Y, Ito Y, Niikura T, Shao Z,
Hata M, Oyama F and Nishimoto I: Mechanisms of neuroprotection by a
novel rescue factor humanin from Swedish mutant amyloid precursor
protein. Biochem Biophys Res Commun. 283:460–468. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gong Z, Tasset I, Diaz A, Anguiano J, Tas
E, Cui L, Kuliawat R, Liu H, Kühn B, Cuervo AM and Muzumdar R:
Humanin is an endogenous activator of chaperone-mediated autophagy.
J Cell Biol. 217:635–647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sreekumar PG, Ishikawa K, Spee C, Mehta
HH, Wan J, Yen K, Cohen P, Kannan R and Hinton DR: The
mitochondrial-derived peptide humanin protects RPE cells from
oxidative stress, senescence, and mitochondrial dysfunction. Invest
Ophthalmol Vis Sci. 57:1238–1253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gong Z and Tasset I: Humanin enhances the
cellular response to stress by activation of chaperone-mediated
autophagy. Oncotarget. 9:10832–10833. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qin Q, Jin J, He F, Zheng Y, Li T, Zhang Y
and He J: Humanin promotes mitochondrial biogenesis in pancreatic
MIN6 β-cells. Biochem Biophys Res Commun. 497:292–297. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sreekumar PG and Kannan R: Mechanisms of
protection of retinal pigment epithelial cells from oxidant injury
by humanin and other mitochondrial-derived peptides: Implications
for age-related macular degeneration. Redox Biol. 37:1016632020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Muzumdar RH, Huffman DM, Atzmon G,
Buettner C, Cobb LJ, Fishman S, Budagov T, Cui L, Einstein FH,
Poduval A, et al: Humanin: A novel central regulator of peripheral
insulin action. PLoS One. 4:e63342009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hoang PT, Park P, Cobb LJ,
Paharkova-Vatchkova V, Hakimi M, Cohen P and Lee KW: The
neurosurvival factor humanin inhibits beta-cell apoptosis via
signal transducer and activator of transcription 3 activation and
delays and ameliorates diabetes in nonobese diabetic mice.
Metabolism. 59:343–349. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lue Y, Swerdloff R, Jia Y and Wang C: The
emerging role of mitochondrial derived peptide humanin in the
testis. Biochim Biophys Acta Gen Subj. 1865:1300092021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mottaghi-Dastjerdi N, Soltany-Rezaee-Rad
M, Sepehrizadeh Z, Roshandel G, Ebrahimifard F and Setayesh N:
Genome expression analysis by suppression subtractive hybridization
identified overexpression of humanin, a target gene in gastric
cancer chemoresistance. Daru. 22:142014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Omar NN, Tash RF, Shoukry Y and ElSaeed
KO: Breaking the ritual metabolic cycle in order to save acetyl
CoA: A potential role for mitochondrial humanin in T2 bladder
cancer aggressiveness. J Egypt Natl Canc Inst. 29:69–76. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang SF, Chen S, Tseng LM and Lee HC: Role
of the mitochondrial stress response in human cancer progression.
Exp Biol Med (Maywood). 245:861–878. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Kim KH, Son JM, Benayoun BA and Lee C: The
mitochondrial-encoded peptide MOTS-c translocates to the nucleus to
regulate nuclear gene expression in response to metabolic stress.
Cell Metab. 28:516–524.e17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wu Y, Sun L, Zhuang Z, Hu X and Dong D:
Mitochondrial-derived peptides in diabetes and its complications.
Front Endocrinol (Lausanne). 12:8081202021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bonkowski MS and Sinclair DA: Slowing
ageing by design: The rise of NAD+ and
sirtuin-activating compounds. Nat Rev Mol Cell Biol. 17:679–690.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Imai S and Guarente L: NAD+ and sirtuins
in aging and disease. Trends Cell Biol. 24:464–471. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Che N, Qiu W, Wang JK, Sun XX, Xu LX, Liu
R and Gu L: MOTS-c improves osteoporosis by promoting the synthesis
of type I collagen in osteoblasts via TGF-β/SMAD signaling pathway.
Eur Rev Med Pharmacol Sci. 23:3183–3189. 2019.PubMed/NCBI
|
|
54
|
Yan Z, Zhu S, Wang H, Wang L, Du T, Ye Z,
Zhai D, Zhu Z, Tian X, Lu Z and Cao X: MOTS-c inhibits osteolysis
in the mouse calvaria by affecting osteocyte-osteoclast crosstalk
and inhibiting inflammation. Pharmacol Res. 147:1043812019.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sartori M, Vincenzi F, Ravani A, Cepollaro
S, Martini L, Varani K, Fini M and Tschon M: RAW 264.7 co-cultured
with ultra-high molecular weight polyethylene particles
spontaneously differentiate into osteoclasts: An in vitro model of
periprosthetic osteolysis. J Biomed Mater Res A. 105:510–520. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Mohtashami Z, Singh MK, Salimiaghdam N,
Ozgul M and Kenney MC: MOTS-c, the most recent mitochondrial
derived peptide in human aging and age-related diseases. Int J Mol
Sci. 23:119912022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Jiang J, Chang X, Nie Y, Shen Y, Liang X,
Peng Y and Chang M: Peripheral administration of a cell-penetrating
MOTS-c analogue enhances memory and attenuates Aβ1-42-
or LPS-induced memory impairment through inhibiting
neuroinflammation. ACS Chem Neurosci. 12:1506–1518. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhai D, Ye Z, Jiang Y, Xu C, Ruan B, Yang
Y, Lei X, Xiang A, Lu H, Zhu Z, et al: MOTS-c peptide increases
survival and decreases bacterial load in mice infected with MRSA.
Mol Immunol. 92:151–160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Jiang J, Chang X, Nie Y, Xu L, Yang L,
Peng Y and Chang M: Orally administered MOTS-c analogue ameliorates
dextran sulfate sodium-induced colitis by inhibiting inflammation
and apoptosis. Eur J Pharmacol. 939:1754692023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xiao J, Zhang Q, Shan Y, Ye F, Zhang X,
Cheng J, Wang X, Zhao Y, Dan G, Chen M and Sai Y: The
mitochondrial-derived peptide (MOTS-c) interacted with Nrf2 to
defend the antioxidant system to protect dopaminergic neurons
against rotenone exposure. Mol Neurobiol. 60:5915–5930. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yang L, Li M, Liu Y, Bai Y, Yin T, Chen Y,
Jiang J and Liu S: MOTS-c is an effective target for treating
cancer-induced bone pain through the induction of AMPK-mediated
mitochondrial biogenesis. Acta Biochim Biophys Sin (Shanghai).
56:1323–1339. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Yin Y, Li Y, Ma B, Ren C, Zhao S and Li J,
Gong Y, Yang H and Li J: Mitochondrial-derived peptide MOTS-c
suppresses ovarian cancer progression by attenuating USP7-mediated
LARS1 deubiquitination. Adv Sci (Weinh). 11:e24056202024.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li Y, Li Z, Ren Y, Lei Y, Yang S, Shi Y,
Peng H, Yang W, Guo T, Yu Y and Xiong Y: Mitochondrial-derived
peptides in cardiovascular disease: Novel insights and therapeutic
opportunities. J Adv Res. 64:99–115. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Okada AK, Teranishi K, Lobo F, Isas JM,
Xiao J, Yen K, Cohen P and Langen R: The mitochondrial-derived
peptides, HumaninS14G and small humanin-like peptide 2, exhibit
chaperone-like activity. Sci Rep. 7:78022017. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Nashine S and Kenney MC: Effects of
mitochondrial-derived peptides (MDPs) on mitochondrial and cellular
health in AMD. Cells. 9:11022020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Shin JH, Kim HW, Rhyu IJ, Song KJ and Kee
SH: Axin expression reduces staurosporine-induced
mitochondria-mediated cell death in HeLa cells. Exp Cell Res.
318:2022–2033. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Emser SV, Schaschl H, Millesi E and
Steinborn R: Extension of mitogenome enrichment based on single
long-range PCR: mtDNAs and putative mitochondrial-derived peptides
of five rodent hibernators. Front Genet. 12:6858062021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Monteiro JP, Bennett M, Rodor J,
Caudrillier A, Ulitsky I and Baker AH: Endothelial function and
dysfunction in the cardiovascular system: The long non-coding road.
Cardiovasc Res. 115:1692–1704. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kinlay S, Libby P and Ganz P: Endothelial
function and coronary artery disease. Curr Opin Lipidol.
12:383–389. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Rhee M, Lee J, Lee EY, Yoon KH and Lee SH:
Lipid variability induces endothelial dysfunction by increasing
inflammation and oxidative stress. Endocrinol Metab (Seoul).
39:511–520. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Pober JS and Sessa WC: Evolving functions
of endothelial cells in inflammation. Nat Rev Immunol. 7:803–815.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lüscher TF and Barton M: Biology of the
endothelium. Clin Cardiol. 20 (11 Suppl 2):II-3-10. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Choi BJ, Prasad A, Gulati R, Best PJ,
Lennon RJ, Barsness GW, Lerman LO and Lerman A: Coronary
endothelial dysfunction in patients with early coronary artery
disease is associated with the increase in intravascular lipid core
plaque. Eur Heart J. 34:2047–2054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Libby P, Ridker PM and Maseri A:
Inflammation and atherosclerosis. Circulation. 105:1135–1143. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Choi BJ, Matsuo Y, Aoki T, Kwon TG, Prasad
A, Gulati R, Lennon RJ, Lerman LO and Lerman A: Coronary
endothelial dysfunction is associated with inflammation and vasa
vasorum proliferation in patients with early atherosclerosis.
Arterioscler Thromb Vasc Biol. 34:2473–2477. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kashiwagi M, Kitabata H, Ozaki Y, Imanishi
T and Akasaka T: Fatty streak assessed by optical coherence
tomography: Early atherosclerosis detection. Eur Heart J Cardiovasc
Imaging. 14:1092013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: A marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Madamanchi NR, Vendrov A and Runge MS:
Oxidative stress and vascular disease. Arterioscler Thromb Vasc
Biol. 25:29–38. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Galle J, Hansen-Hagge T, Wanner C and
Seibold S: Impact of oxidized low density lipoprotein on vascular
cells. Atherosclerosis. 185:219–226. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
van Dijk RA, Virmani R, von der Thusen JH,
Schaapherder AF and Lindeman JHN: The natural history of aortic
atherosclerosis: A systematic histopathological evaluation of the
peri-renal region. Atherosclerosis. 210:100–106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Widmer RJ, Flammer AJ, Herrmann J,
Rodriguez-Porcel M, Wan J, Cohen P, Lerman LO and Lerman A:
Circulating humanin levels are associated with preserved coronary
endothelial function. Am J Physiol Heart Circ Physiol.
304:H393–H397. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Coradduzza D, Cruciani S, Di Lorenzo B, De
Miglio MR, Zinellu A, Maioli M, Medici S, Erre GL and Carru C:
Plasma humanin and non-coding RNAs as biomarkers of endothelial
dysfunction in rheumatoid arthritis: A pilot study. Noncoding RNA.
11:52025.PubMed/NCBI
|
|
84
|
Balan AI, Halatiu VB and Scridon A:
Oxidative stress, inflammation, and mitochondrial dysfunction: A
link between obesity and atrial fibrillation. Antioxidants (Basel).
13:1172024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kusminski CM and Scherer PE: Mitochondrial
dysfunction in white adipose tissue. Trends Endocrinol Metab.
23:435–443. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Teodoro JS, Nunes S, Rolo AP, Reis F and
Palmeira CM: Therapeutic options targeting oxidative stress,
mitochondrial dysfunction and inflammation to hinder the
progression of vascular complications of diabetes. Front Physiol.
9:18572019. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Cai H and Harrison DG: Endothelial
dysfunction in cardiovascular diseases: The role of oxidant stress.
Circ Res. 87:840–844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Bachar AR, Scheffer L, Schroeder AS,
Nakamura HK, Cobb LJ, Oh YK, Lerman LO, Pagano RE, Cohen P and
Lerman A: Humanin is expressed in human vascular walls and has a
cytoprotective effect against oxidized LDL-induced oxidative
stress. Cardiovasc Res. 88:360–366. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hannun YA and Obeid LM: Principles of
bioactive lipid signalling: Lessons from sphingolipids. Nat Rev Mol
Cell Biol. 9:139–150. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cai H, Liu Y, Men H and Zheng Y:
Protective mechanism of humanin against oxidative stress in
aging-related cardiovascular diseases. Front Endocrinol (Lausanne).
12:6831512021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Chiba T, Yamada M, Hashimoto Y, Sato M,
Sasabe J, Kita Y, Terashita K, Aiso S, Nishimoto I and Matsuoka M:
Development of a femtomolar-acting humanin derivative named
colivelin by attaching activity-dependent neurotrophic factor to
its N terminus: Characterization of colivelin-mediated
neuroprotection against Alzheimer's disease-relevant insults in
vitro and in vivo. J Neurosci. 25:10252–10261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Urban C, Hayes HV, Piraino G, Wolfe V,
Lahni P, O'Connor M, Phares C and Zingarelli B: Colivelin, a
synthetic derivative of humanin, ameliorates endothelial injury and
glycocalyx shedding after sepsis in mice. Front Immunol.
13:9842982022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Kirkman DL, Robinson AT, Rossman MJ, Seals
DR and Edwards DG: Mitochondrial contributions to vascular
endothelial dysfunction, arterial stiffness, and cardiovascular
diseases. Am J Physiol Heart Circ Physiol. 320:H2080–H2100. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Thummasorn S, Shinlapawittayatorn K,
Chattipakorn SC and Chattipakorn N: High-dose humanin analogue
applied during ischemia exerts cardioprotection against
ischemia/reperfusion injury by reducing mitochondrial dysfunction.
Cardiovasc Ther. 35:2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Rentrop KP and Feit F: Reperfusion therapy
for acute myocardial infarction: Concepts and controversies from
inception to acceptance. Am Heart J. 170:971–980. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Dabravolski SA, Nikiforov NG, Starodubova
AV, Popkova TV and Orekhov AN: The role of mitochondria-derived
peptides in cardiovascular diseases and their potential as
therapeutic targets. Int J Mol Sci. 22:87702021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Thummasorn S, Apaijai N, Kerdphoo S,
Shinlapawittayatorn K, Chattipakorn SC and Chattipakorn N: Humanin
exerts cardioprotection against cardiac ischemia/reperfusion injury
through attenuation of mitochondrial dysfunction. Cardiovasc Ther.
34:404–414. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Arrigo M, Price S, Baran DA, Pöss J,
Aissaoui N, Bayes-Genis A, Bonello L, François B, Gayat E, Gilard
M, et al: Optimising clinical trials in acute myocardial infarction
complicated by cardiogenic shock: A statement from the 2020
critical care clinical trialists workshop. Lancet Respir Med.
9:1192–1202. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Muzumdar RH, Huffman DM, Calvert JW, Jha
S, Weinberg Y, Cui L, Nemkal A, Atzmon G, Klein L, Gundewar S, et
al: Acute humanin therapy attenuates myocardial ischemia and
reperfusion injury in mice. Arterioscler Thromb Vasc Biol.
30:1940–1948. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
McDermott-Roe C, Ye J, Ahmed R, Sun XM,
Serafin A, Ware J, Bottolo L, Muckett P, Cañas X, Zhang J, et al:
Endonuclease G is a novel determinant of cardiac hypertrophy and
mitochondrial function. Nature. 478:114–118. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Rizzi E, Guimaraes DA, Ceron CS, Prado CM,
Pinheiro LC, Martins-Oliveira A, Gerlach RF and Tanus-Santos JE:
β1-Adrenergic blockers exert antioxidant effects, reduce matrix
metalloproteinase activity, and improve renovascular
hypertension-induced cardiac hypertrophy. Free Radic Biol Med.
73:308–317. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Maillet M, van Berlo JH and Molkentin JD:
Molecular basis of physiological heart growth: Fundamental concepts
and new players. Nat Rev Mol Cell Biol. 14:38–48. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Lu J, McKinsey TA, Nicol RL and Olson EN:
Signal-dependent activation of the MEF2 transcription factor by
dissociation from histone deacetylases. Proc Natl Acad Sci USA.
97:4070–4075. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Dai DF, Johnson SC, Villarin JJ, Chin MT,
Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW II, Kang YJ, Prolla
TA, et al: Mitochondrial oxidative stress mediates angiotensin
II-induced cardiac hypertrophy and Galphaq overexpression-induced
heart failure. Circ Res. 108:837–846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Blasco N, Cámara Y, Núñez E, Beà A, Barés
G, Forné C, Ruíz-Meana M, Girón C, Barba I, García-Arumí E, et al:
Cardiomyocyte hypertrophy induced by Endonuclease G deficiency
requires reactive oxygen radicals accumulation and is inhibitable
by the micropeptide humanin. Redox Biol. 16:146–156. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Blasco N, Beà A, Barés G, Girón C,
Navaridas R, Irazoki A, López-Lluch G, Zorzano A, Dolcet X, Llovera
M and Sanchis D: Involvement of the mitochondrial nuclease EndoG in
the regulation of cell proliferation through the control of
reactive oxygen species. Redox Biol. 37:1017362020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Eghbali M, Blumenfeld OO, Seifter S,
Buttrick PM, Leinwand LA, Robinson TF, Zern MA and Giambrone MA:
Localization of types I, III and IV collagen mRNAs in rat heart
cells by in situ hybridization. J Mol Cell Cardiol. 21:103–113.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kong P, Christia P and Frangogiannis NG:
The pathogenesis of cardiac fibrosis. Cell Mol Life Sci.
71:549–574. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Edgley AJ, Krum H and Kelly DJ: Targeting
fibrosis for the treatment of heart failure: A role for
transforming growth factor-β. Cardiovasc Ther. 30:e30–e40. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Piera-Velazquez S, Li Z and Jimenez SA:
Role of endothelial-mesenchymal transition (EndoMT) in the
pathogenesis of fibrotic disorders. Am J Pathol. 179:1074–1080.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Zeisberg EM and Kalluri R: Origins of
cardiac fibroblasts. Circ Res. 107:1304–1312. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Travers JG, Kamal FA, Robbins J, Yutzey KE
and Blaxall BC: Cardiac fibrosis: The fibroblast awakens. Circ Res.
118:1021–1040. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liguori TTA, Liguori GR, Moreira LFP and
Harmsen MC: Fibroblast growth factor-2, but not the adipose
tissue-derived stromal cells secretome, inhibits TGF-β1-induced
differentiation of human cardiac fibroblasts into myofibroblasts.
Sci Rep. 8:166332018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Biernacka A and Frangogiannis NG: Aging
and cardiac fibrosis. Aging Dis. 2:158–173. 2011.PubMed/NCBI
|
|
115
|
Sangaralingham SJ, Wang BH, Huang L, Kumfu
S, Ichiki T, Krum H and Burnett JC Jr: Cardiorenal fibrosis and
dysfunction in aging: Imbalance in mediators and regulators of
collagen. Peptides. 76:108–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Biernacka A, Dobaczewski M and
Frangogiannis NG: TGF-β signaling in fibrosis. Growth Factors.
29:196–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Qin Q, Mehta H, Yen K, Navarrete G,
Brandhorst S, Wan J, Delrio S, Zhang X, Lerman LO, Cohen P and
Lerman A: Chronic treatment with the mitochondrial peptide humanin
prevents age-related myocardial fibrosis in mice. Am J Physiol
Heart Circ Physiol. 315:H1127–H1136. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Zavadzkas JA, Plyler RA, Bouges S, Koval
CN, Rivers WT, Beck CU, Chang EI, Stroud RE, Mukherjee R and
Spinale FG: Cardiac-restricted overexpression of extracellular
matrix metalloproteinase inducer causes myocardial remodeling and
dysfunction in aging mice. Am J Physiol Heart Circ Physiol.
295:H1394–H1402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Juhaszova M, Zorov DB, Yaniv Y, Nuss HB,
Wang S and Sollott SJ: Role of glycogen synthase kinase-3beta in
cardioprotection. Circ Res. 104:1240–1252. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hirotani S, Zhai P, Tomita H, Galeotti J,
Marquez JP, Gao S, Hong C, Yatani A, Avila J and Sadoshima J:
Inhibition of glycogen synthase kinase 3beta during heart failure
is protective. Circ Res. 101:1164–1174. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Ibanez B, James S, Agewall S, Antunes MJ,
Bucciarelli-Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA,
Halvorsen S, et al: 2017 ESC guidelines for the management of acute
myocardial infarction in patients presenting with ST-segment
elevation: The task force for the management of acute myocardial
infarction in patients presenting with ST-segment elevation of the
European society of cardiology (ESC). Eur Heart J. 39:119–177.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Choo EH, Kim PJ, Chang K, Ahn Y, Jeon DS,
Lee JM, Kim DB, Her SH, Park CS, Kim HY, et al: The impact of
no-reflow phenomena after primary percutaneous coronary
intervention: a time-dependent analysis of mortality. Coron Artery
Dis. 25:392–398. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Wong DT, Puri R, Richardson JD, Worthley
MI and Worthley SG: Myocardial ‘no-reflow’-diagnosis,
pathophysiology and treatment. Int J Cardiol. 167:1798–1806. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Çakmak T, Yaşar E, Çakmak E, Tekin S,
Karakuş Y, Türkoğlu C and Yüksel F: Evaluation of coronary flow
level with mots-C in patients with STEMI undergoing primary PCI.
Arq Bras Cardiol. 120:e202203582023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Marulanda J, Alqarni S and Murshed M:
Mechanisms of vascular calcification and associated diseases. Curr
Pharm Des. 20:5801–5810. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Zhang L, Yao J, Yao Y and Boström KI:
Contributions of the endothelium to vascular calcification. Front
Cell Dev Biol. 9:6208822021. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Liu W, Zhang Y, Yu CM, Ji QW, Cai M, Zhao
YX and Zhou YJ: Current understanding of coronary artery
calcification. J Geriatr Cardiol. 12:668–675. 2015.PubMed/NCBI
|
|
128
|
McCullough PA, Chinnaiyan KM, Agrawal V,
Danielewicz E and Abela GS: Amplification of atherosclerotic
calcification and Mönckeberg's sclerosis: A spectrum of the same
disease process. Adv Chronic Kidney Dis. 15:396–412. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Rasheed A and Cummins CL: Beyond the foam
cell: The role of LXRs in preventing atherogenesis. Int J Mol Sci.
19:23072018. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Andrews J, Psaltis PJ, Bartolo BAD,
Nicholls SJ and Puri R: Coronary arterial calcification: A review
of mechanisms, promoters and imaging. Trends Cardiovasc Med.
28:491–501. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhang X, Xiao J, Li R, Qin X, Wang F, Mao
Y, Liang W, Sheng X, Guo M, Song Y and Ji X: Metformin alleviates
vascular calcification induced by vitamin D3 plus nicotine in rats
via the AMPK pathway. Vascul Pharmacol. 81:83–90. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Xu M, Liu L, Song C, Chen W and Gui S:
Ghrelin improves vascular autophagy in rats with vascular
calcification. Life Sci. 179:23–29. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Li KX, Du Q, Wang HP and Sun HJ:
Death-associated protein kinase 3 deficiency alleviates vascular
calcification via AMPK-mediated inhibition of endoplasmic reticulum
stress. Eur J Pharmacol. 852:90–98. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wei M, Gan L, Liu Z, Liu L, Chang JR, Yin
DC, Cao HL, Su XL and Smith WW: Mitochondrial-derived peptide
MOTS-c attenuates vascular calcification and secondary myocardial
remodeling via adenosine monophosphate-activated protein kinase
signaling pathway. Cardiorenal Med. 10:42–50. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Honda J, Kimura T, Sakai S, Maruyama H,
Tajiri K, Murakoshi N, Homma S, Miyauchi T and Aonuma K: The
glucagon-like peptide-1 receptor agonist liraglutide improves
hypoxia-induced pulmonary hypertension in mice partly via
normalization of reduced ET(B) receptor expression. Physiol Res. 67
(Suppl 1):S175–S184. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Boccellino M, Di Domenico M, Donniacuo M,
Bitti G, Gritti G, Ambrosio P, Quagliuolo L and Rinaldi B:
AT1-receptor blockade: Protective effects of irbesartan in
cardiomyocytes under hypoxic stress. PLoS One. 13:e02022972018.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Qin Q, Delrio S, Wan J, Jay Widmer R,
Cohen P, Lerman LO and Lerman A: Downregulation of circulating
MOTS-c levels in patients with coronary endothelial dysfunction.
Int J Cardiol. 254:23–27. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Shen C, Wang J, Feng M, Peng J, Du X, Chu
H and Chen X: The mitochondrial-derived peptide MOTS-c attenuates
oxidative stress injury and the inflammatory response of H9c2 cells
through the Nrf2/ARE and NF-κB pathways. Cardiovasc Eng Technol.
13:651–661. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Li H, Ren K, Jiang T and Zhao GJ: MOTS-c
attenuates endothelial dysfunction via suppressing the MAPK/NF-κB
pathway. Int J Cardiol. 268:402018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Chen M, Fu H, Zhang J, Huang H and Zhong
P: CIRP downregulation renders cardiac cells prone to apoptosis in
heart failure. Biochem Biophys Res Commun. 517:545–550. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zhong P, Peng J, Hu Y, Zhang J and Shen C:
Mitochondrial derived peptide MOTS-c prevents the development of
heart failure under pressure overload conditions in mice. J Cell
Mol Med. 26:5369–5378. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Hage C, Wärdell E, Linde C, Donal E, Lam
CSP, Daubert C, Lund LH and Månsson-Broberg A: Circulating
neuregulin1-β in heart failure with preserved and reduced left
ventricular ejection fraction. ESC Heart Fail. 7:445–455. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Hedhli N, Huang Q, Kalinowski A, Palmeri
M, Hu X, Russell RR and Russell KS: Endothelium-derived neuregulin
protects the heart against ischemic injury. Circulation.
123:2254–2262. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Brero A, Ramella R, Fitou A, Dati C,
Alloatti G, Gallo MP and Levi R: Neuregulin-1beta1 rapidly
modulates nitric oxide synthesis and calcium handling in rat
cardiomyocytes. Cardiovasc Res. 88:443–452. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Li S, Wang M, Ma J, Pang X, Yuan J, Pan Y,
Fu Y and Laher I: MOTS-c and exercise restore cardiac function by
activating of NRG1-ErbB signaling in diabetic rats. Front
Endocrinol (Lausanne). 13:8120322022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Shankar-Hari M, Phillips GS, Levy ML,
Seymour CW, Liu VX, Deutschman CS, Angus DC, Rubenfeld GD and
Singer M; Sepsis Definitions Task Force, : Developing a new
definition and assessing new clinical criteria for septic shock:
For the third international consensus definitions for sepsis and
septic shock (sepsis-3). JAMA. 315:775–787. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Hollenberg SM and Singer M:
Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol.
18:424–434. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Shen Q, Yuan Y, Li Z, Ling Y, Wang J, Gao
M, Wang P, Li M, Lai L and Jin J: Berberine ameliorates septic
cardiomyopathy through protecting mitochondria and upregulating
Notch1 signaling in cardiomyocytes. Front Pharmacol.
15:15023542024. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Ehrman RR, Sullivan AN, Favot MJ, Sherwin
RL, Reynolds CA, Abidov A and Levy PD: Pathophysiology,
echocardiographic evaluation, biomarker findings, and prognostic
implications of septic cardiomyopathy: A review of the literature.
Crit Care. 22:1122018. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Ravikumar N, Sayed MA, Poonsuph CJ, Sehgal
R, Shirke MM and Harky A: Septic cardiomyopathy: From basics to
management choices. Curr Probl Cardiol. 46:1007672021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Carbone F, Liberale L, Preda A, Schindler
TH and Montecucco F: Septic cardiomyopathy: From pathophysiology to
the clinical setting. Cells. 11:28332022. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Khalid N, Patel PD, Alghareeb R, Hussain A
and Maheshwari MV: The effect of sepsis on myocardial function: A
review of pathophysiology, diagnostic criteria, and treatment.
Cureus. 14:e261782022.PubMed/NCBI
|
|
154
|
Liu YC, Yu MM, Shou ST and Chai YF:
Sepsis-induced cardiomyopathy: Mechanisms and treatments. Front
Immunol. 8:10212017. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wu J, Xiao D, Yu K, Shalamu K, He B and
Zhang M: The protective effect of the mitochondrial-derived peptide
MOTS-c on LPS-induced septic cardiomyopathy. Acta Biochim Biophys
Sin (Shanghai). 55:285–294. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Liu C, Shen YJ, Tu QB, Zhao YR, Guo H,
Wang J, Zhang L, Shi HW and Sun Y: Pedunculoside, a novel
triterpene saponin extracted from Ilex rotunda, ameliorates
high-fat diet induced hyperlipidemia in rats. Biomed Pharmacother.
101:608–616. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Su Z, Li K, Luo X, Zhu Y, Mai SY, Zhu Q,
Yang B, Zhou X and Tao H: Aromatic acids and leucine derivatives
produced from the deep-sea actinomycetes streptomyceschumphonensis
SCSIO15079 with antihyperlipidemic activities. Mar Drugs.
20:2592022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Bai X, Wang H, Li J, Xu J and Cai P:
Correlation analysis of the risk of ischemic stroke with related
risk factors in a health examination population. Pak J Med Sci.
40:2533–2537. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Huang L, Liu Z, Zhang H, Li D, Li Z, Huang
J, He J, Lu L, Wen H, Yuan H, et al: The association between serum
lipid profile levels and hypertension grades: A cross-sectional
study at a health examination center. High Blood Press Cardiovasc
Prev. 32:87–98. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Hannun YA and Obeid LM: Sphingolipids and
their metabolism in physiology and disease. Nat Rev Mol Cell Biol.
19:175–191. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Russo SB, Ross JS and Cowart LA:
Sphingolipids in obesity, type 2 diabetes, and metabolic disease.
Handb Exp Pharmacol. 373–401. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Zhang X, Zhang Y, Wang P, Zhang SY, Dong
Y, Zeng G, Yan Y, Sun L, Wu Q, Liu H, et al: Adipocyte
hypoxia-inducible factor 2α suppresses atherosclerosis by promoting
adipose ceramide catabolism. Cell Metab. 30:937–951.e5. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Mehta HH, Xiao J, Ramirez R, Miller B, Kim
SJ, Cohen P and Yen K: Metabolomic profile of diet-induced obesity
mice in response to humanin and small humanin-like peptide 2
treatment. Metabolomics. 15:882019. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Yen K, Miller B, Kumagai H, Silverstein A
and Cohen P: Mitochondrial-derived microproteins: From discovery to
function. Trends Genet. 41:132–145. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Merry TL, Chan A, Woodhead JST, Reynolds
JC, Kumagai H, Kim SJ and Lee C: Mitochondrial-derived peptides in
energy metabolism. Am J Physiol Endocrinol Metab. 319:E659–E666.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Kim SJ, Miller B, Kumagai H, Silverstein
AR, Flores M and Yen K: Mitochondrial-derived peptides in aging and
age-related diseases. Geroscience. 43:1113–1121. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Gao Y, Wei X, Wei P, Lu H, Zhong L, Tan J,
Liu H and Liu Z: MOTS-c functionally prevents metabolic disorders.
Metabolites. 13:1252023. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Verma S, Goand UK, Husain A, Katekar RA,
Garg R and Gayen JR: Challenges of peptide and protein drug
delivery by oral route: Current strategies to improve the
bioavailability. Drug Dev Res. 82:927–944. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Mitragotri S, Burke PA and Langer R:
Overcoming the challenges in administering biopharmaceuticals:
Formulation and delivery strategies. Nat Rev Drug Discov.
13:655–672. 2014. View Article : Google Scholar : PubMed/NCBI
|