Introduction
Messenger RNA (mRNA) is a key component in the
regulation of gene expression. Before mRNA is formed, pre-messenger
RNA (pre-mRNA) is directly transcribed from DNA, but does not have
the ability to synthesize proteins. The introns in the pre-mRNA
need to be removed for it to become functional, a process carried
out by the spliceosome (1). The
spliceosome, a multiprotein complex of five small nuclear
ribonucleoproteins (snRNPs), recognizes regulatory sequences near
intron-exon junctions. By removing introns and splicing exons, it
ensures the formation of functional, mature mRNA (2,3).
U5-snRNP is one of the core small nuclear ribonucleoproteins of the
spliceosome. After U5 recruits U4/U6, they assemble to form the
U4/U6-U5 tri-snRNP, which constitutes the precatalytic spliceosome
complex (4). The complexity of
regulatory elements that control proper splicing makes this process
not only crucial to maintain tissue homeostasis, but also highly
susceptible to genetic and somatic mutations associated with
diseases (5).
Elongation factor Tu GTP binding domain containing 2
(EFTUD2) is an essential protein of U5-snRNP, which is involved in
binding GTP and maintaining the normal function of the spliceosome
(6). EFTUD2, also known as Snu114,
has primary functions in the growth and development of the
organism. The EFTUD2 gene is distributed in almost all types of
human cells and its mutations can cause biological dysfunctions in
various systems. Thus, the present study reviewed current research
on EFTUD2 mutations and their clinical implications, focusing
particularly on developmental abnormalities, innate immune
responses and cancer progression, aiming to offer new insights into
the diagnosis and treatment of EFTUD2-related diseases.
Expression and structure of EFTUD2
The EFTUD2 gene, located on human chromosome
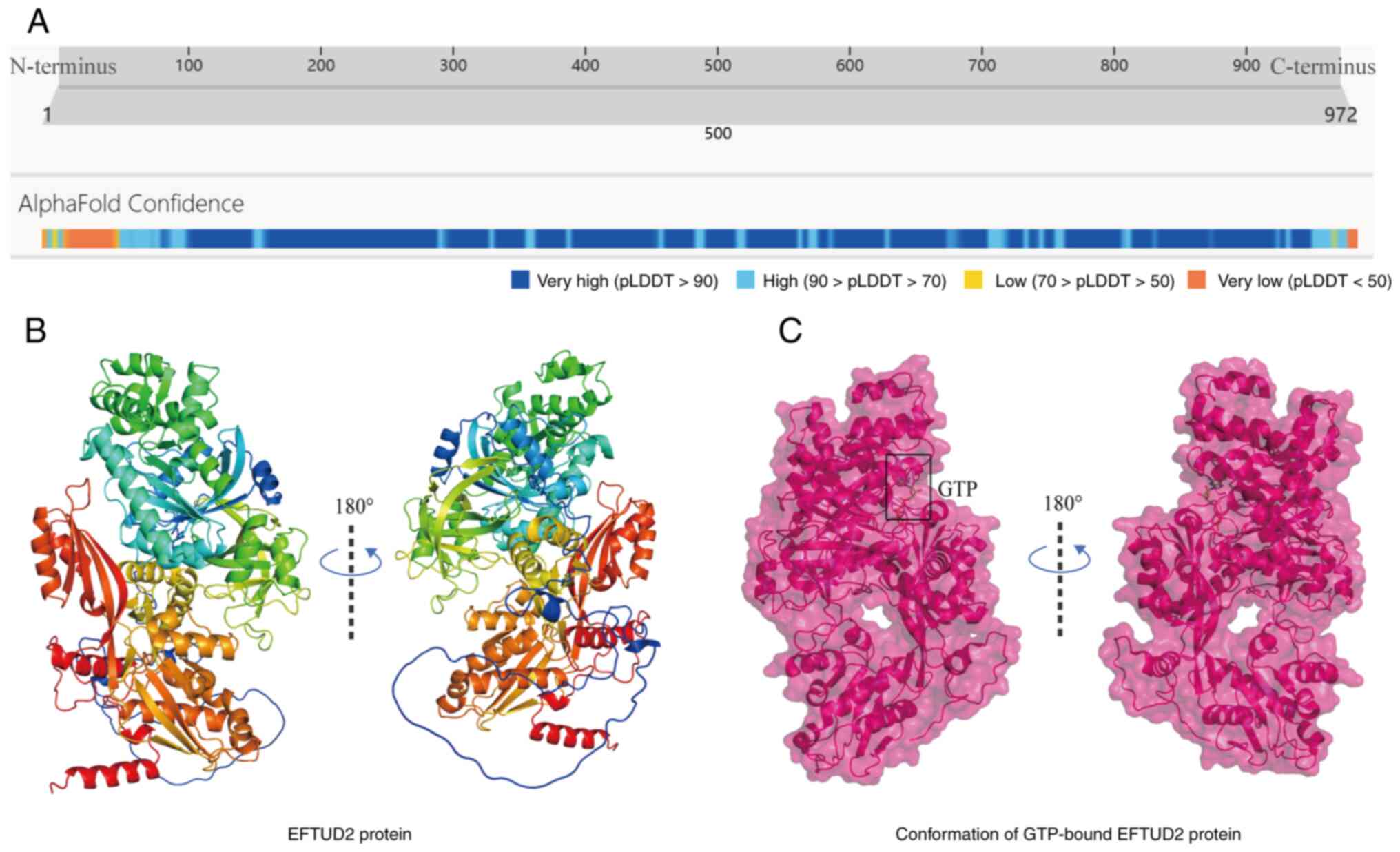
17q21.31, encodes a ubiquitously expressed 972-amino acid protein
with a molecular weight of 109.436 kDa (Fig. 1A) (7). EFTUD2 is also known as MFDGA,
U5-116KD and Snu114. The post-translational modifications of EFTUD2
include ubiquitination at Lys352, Lys405, Lys409, Lys581 and
Lys790; one glycosylation site according to GlyGen; and one
O-linked glycan modification site (8). Mature EFTUD2 contains six functional
domains: Elongation Factor G C/N-terminus, Domain III, Domain IV,
Elongation Factor Tu Domain 2 and Elongation Factor Tu GTP-binding
domain. The monomeric structure of EFTUD2 is shown in Fig. 1B. EFTUD2 shows the highest
expression levels in human testis and appendix tissues. Regarding
subcellular localization, EFTUD2 is predominantly found in the
nucleus, with additional expression observed in the cytoplasm and
mitochondria (9). EFTUD2 is also a
highly conserved spliceosomal GTPase and an essential component of
the spliceosomal complex in cells (6). Plaschka et al (10) discovered through cryo-electron
microscopy that EFTUD2 binds to GTP but does not appear to
hydrolyze GTP to facilitate conformational changes in the
spliceosome (Fig. 1C). Therefore,
EFTUD2 is more likely to act as a component of a platform that
supports precursor mRNA splicing.
Physiological functions of EFTUD2
Role of EFTUD2 in splicing
The spliceosome is a large ribonucleoprotein (RNP)
complex composed of five snRNPs (U1, U2, U4, U5 and U6) and
numerous protein factors, which is assembled de novo on each
intron (11). Pre-mRNA introns
have minimal conserved structural information. The spliceosome
recognizes key sequences, such as the 5′ splice site and 3′ splice
site and removes introns through splicing to form mature mRNA
(3). Specifically, U1 and U2 first
bind to the splice sites of the intron (12) (Fig. 2A
and C), followed by the recruitment of the U4/U6/U5 tri-snRNP,
thereby assembling the precatalytic spliceosome complex (4) (Fig. 2B
and H).
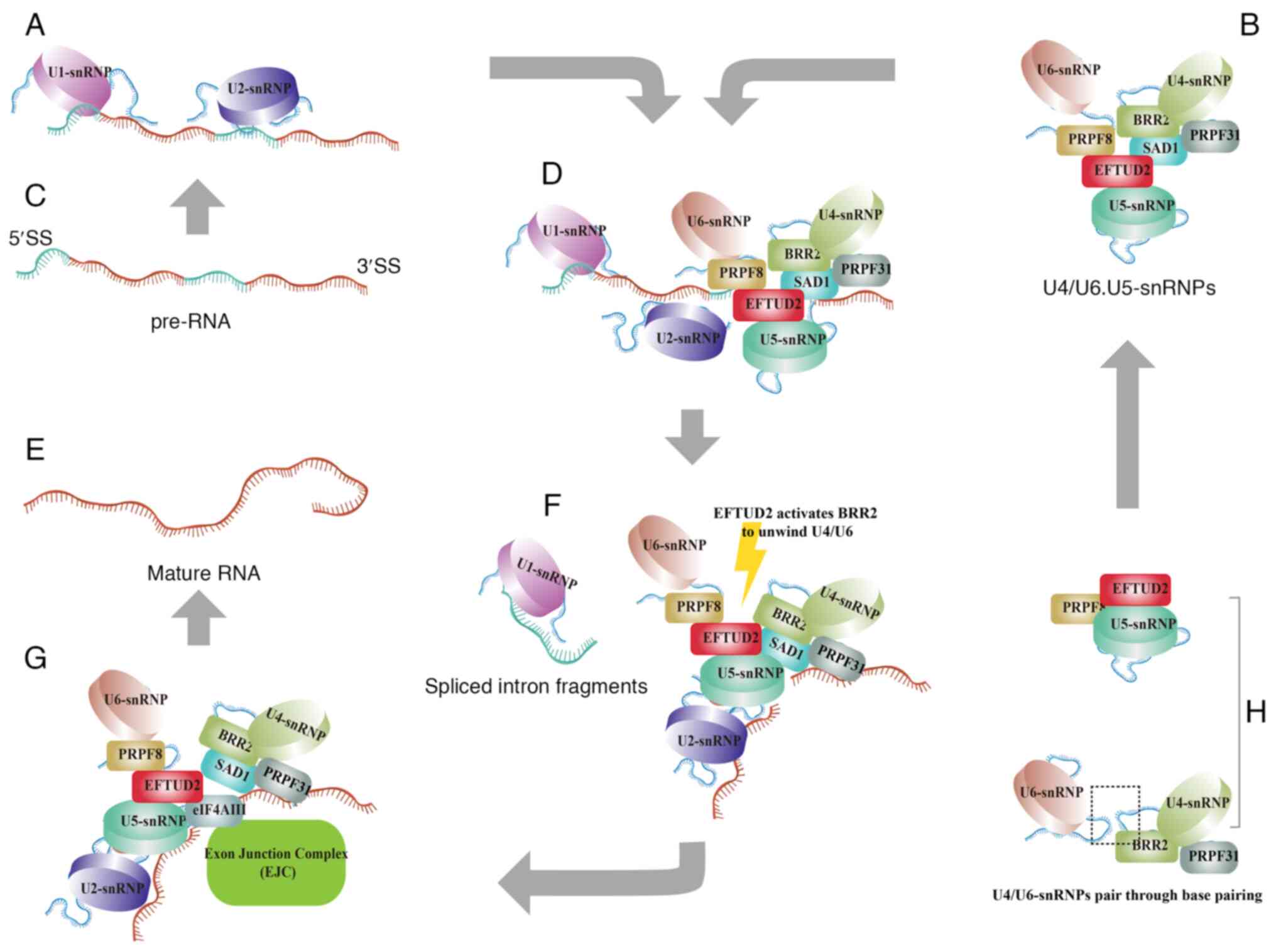 | Figure 2.EFTUD2 regulates the removal of
introns and the junction of exons. (A) After the production of
pre-mRNA, U1 and U2 snRNPs recognize and bind to the 5′ and 3′ ends
of the exons, respectively. (B) U4 and U6 snRNPs form a complex
through base pairing and then the U5 snRNP, which contains EFTUD2,
associates with the U4/U6 complex to form the U4/U6.U5 tri-snRNP
complex. (C) Precursor RNA. (D) The U4/U6.U5 tri-snRNP complex
subsequently binds to the pre-mRNA already interacted with U1 and
U2 snRNPs, initiating the activation of the intron splicing
process. (E) Mature RNA. (F) Activation of the intron splicing
process. (G) During exon splicing, EFTUD2 interacts with the EJC
complex and catalyzes the joining of exons. (H) U4/U6 snRNPs and U5
snRNP. The figure was created using Adobe Illustrator (version,
28.3; Adobe Systems, Inc.). EFTUD2, elongation factor Tu GTP
binding domain containing 2; snRNP, small nuclear
ribonucleoprotein; EJC, exon junction complex; BRR2, small nuclear
ribonucleoprotein U200. |
EFTUD2 is located in the central region of the
tri-snRNP complex, where it interacts with the N-terminal domain of
Pre-mRNA Processing Factor 8 (13). EFTUD2 also plays a central role by
interacting with Sad1 and UNC84 domain containing 1, which binds to
small nuclear ribonucleoprotein U200 (BRR2) and U4/U6-PRPF31. This
interaction helps stabilize the association between U5 and U4/U6
snRNPs, ensuring proper spliceosome assembly (14) (Fig.
2D). Then, EFTUD2 regulates the unwinding of U4/U6 by
controlling BRR2′s helicase activity, which promotes the
spliceosome's transition to an active state (15,16)
(Fig. 2F).
The exon junction complex (EJC), which contains
eukaryotic translation initiation factor 4A3 (eIF4AIII), is a group
of proteins involved in the splicing process, specifically at the
exon-exon junctions (17). The
RecA1 domain of eIF4AIII directly interacts with EFTUD2, while the
EJC recognizes the upstream 5′-exon sequence and binds to EFTUD2
(18) (Fig. 2G). This indicates that EFTUD2 not
only serves as a central component of the spliceosome complex, but
also actively participates in binding the exon-exon junction
(Fig. 2E). Overall, EFTUD2
orchestrates the events required for the correct removal of introns
and junctions of exons, thereby influencing gene expression
regulation at the post-transcriptional level.
Role of EFTUD2 in embryonic
development
Park et al (19) found that EFTUD2 is maternally
expressed and remains constant throughout development in
Xenopus embryos. EFTUD2 is enriched in the anterior neural
plate and neural crest formation regions during the neurula stage.
While at tailbud stage 29/30, EFTUD2 transcripts are most
abundant in the pharyngeal arches and head. Following EFTUD2
knockdown, the expression of key neural crest development markers
SRY-box transcription factor 9 and SRY-Box transcription factor 2
is reduced in Xenopus embryos (19). Thus, the decreased expression of
EFTUD2 inhibits the neural crest development of embryos.
A study reported that, compared with heterozygous
mutants, EFTUD2 homozygous mutant embryos exhibit an almost
complete absence of the midbrain in neural crest cell mutants by
embryonic day 11.5 (20). As
embryonic development progresses, neural crest cell-specific EFTUD2
homozygous mutant embryos exhibit severe cranial malformations. By
the mid to late embryonic stages, most of these embryos did not
survive and the surviving ones displayed exencephaly. Additionally,
neural crest cell-specific EFTUD2 homozygous mutant embryos show
abnormal trigeminal ganglion formation (20).
However, EFTUD2 heterozygous mutant embryos exhibit
developmental delay before organogenesis, but recover by birth
(21). Notably, EFTUD2 homozygous
mutant embryos are unable to survive post-implantation, a result
consistent with the previously findings by Beauchamp et al
(20,21). Further research revealed that the
reduction in EFTUD2 levels led to selective splicing inhibition of
double min 2 protein in embryos, resulting in the accumulation of
nuclear P53 and increased expression of P53 target genes. Enhanced
P53 activity causes abnormal midbrain morphology (20). Whether EFTUD2 affects embryonic
development through the regulation of the P53 pathway or other
mechanisms remains to be further investigated.
Role of the EFTUD2 in the innate
immune response
The innate immune response is the body's first line
of defense, preventing infections and targeting invading pathogens
(22). Multiple studies have shown
that EFTUD2 plays a role in regulating innate immunity (23–25)
(Fig. 3).
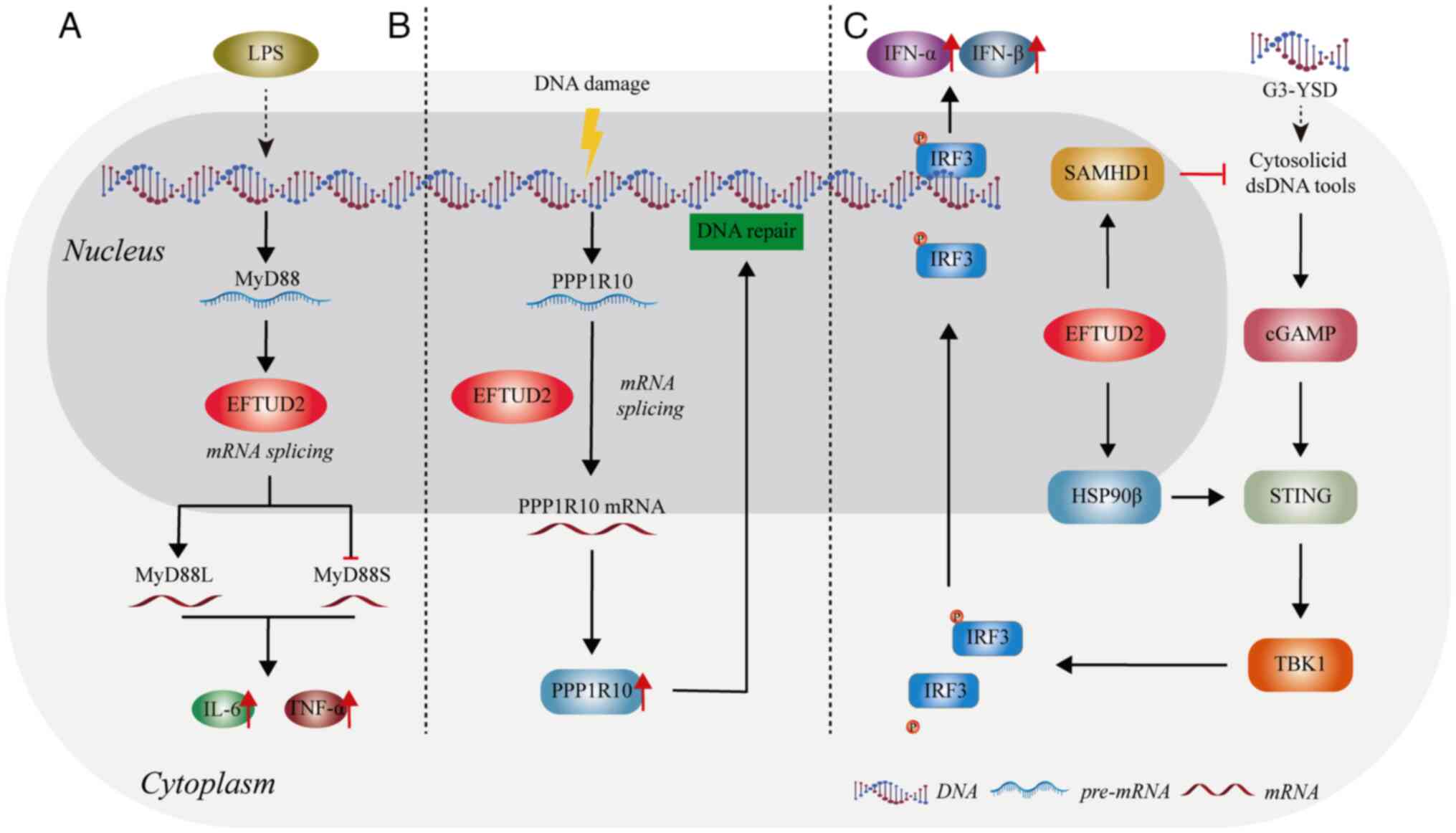 | Figure 3.The role of EFTUD2 in the innate
immune response. (A) EFTUD2 modulates the activation of the innate
immune response by regulating the proportions of MyD88 splice
variants. (B) EFTUD2 promotes DNA damage repair by regulating the
mRNA splicing of PPP1R10, thereby inhibiting inflammatory
damage. (C) EFTUD2 is involved in activating the cGAS-STING
pathway. The figure was created using Adobe Illustrator (version,
28.3; Adobe Systems, Inc.). EFTUD2, elongation factor Tu GTP
binding domain containing 2; MyD88, myeloid differentiation primary
response 88; LPS, lipopolysaccharide; IFN, interferon; IRF,
interferon regulatory factor; SAMHD1, sterile alpha motif and
histidine-aspartate domain-containing protein 1; cGAS, cyclic
GMP-AMP synthase; STING, stimulator of interferon response cGAMP
interactor; TBK1, tank binding kinase 1. |
Enhancement of immune effects on
macrophages
Macrophages are important players in innate
immunity, recognizing and effectively responding to invading
pathogens, thereby providing an early defense against external
attacks (26). De Arras et
al found that after lipopolysaccharide (LPS) stimulation,
EFTUD2 regulates macrophage activation by splicing myeloid
differentiation primary response gene 88 (MyD88) pre-mRNA into two
forms: MyD88L and MyD88S (23) (Fig.
3A). MyD88 is an adaptor protein that functions downstream of
most Toll-like receptors (TLRs) (27). The full-length MyD88L encodes a
critical signaling adaptor protein in multiple TLR response
pathways. By contrast, the shorter spliced form, MyD88S,
which lacks one exon, encodes an in-frame protein that acts as a
negative regulator of TLR signaling, preventing downstream signal
activation (28). When EFTUD2 is
inhibited, a marked increase in the inhibitory spliced form
MyD88S is observed, along with a concomitant reduction in
the production of cytokines interleukin 6 (IL-6) and tumor necrosis
factor α (TNFα) (23). Therefore,
EFTUD2 can promote the production of IL-6 and TNFα by macrophages
by reducing the proportion of MyD88S mRNA (Fig. 3A).
White et al (24) found that EFTUD2 boosts the
production of protein phosphatase 1 regulatory subunit 10 (PPP1R10)
in human macrophages by altering its mRNA splicing. The increase in
PPP1R10 protein levels of allows the repair of DNA damage in human
macrophages, potentially preventing immune damage triggered by
cytoplasmic DNA sensors (24)
(Fig. 3B). Current research shows
that EFTUD2 primarily regulates immune factors through its splicing
function. However, the exact mechanisms remain unclear and requires
further investigation.
Regulation of the cyclic GMP-AMP
synthase (cGAS)-stimulator of interferon response cGAMP interactor
(STING) pathway
The cyclic cGAS-STING pathway is one of the crucial
innate immunity pathways. As a highly conserved innate immune
signaling mechanism in mammals, activation of the cGAS-STING
pathway is characterized by complex transcriptomic changes
(29). Sun et al (25) reveal that EFTUD2 is predicted to be
highly relevant to the cytosolic DNA sensing pathway and shows a
high expression correlation with cGAS and STING. Upon
overexpression of EFTUD2, the number of induced and
repressed genes following cGAS-STING activation markedly increases
and decreases, respectively. This indicates that EFTUD2 plays a
regulatory role in the transcriptomic changes mediated by
cGAS-STING pathway activation.
Further research found that overexpression of
EFTUD2 led to the upregulation of heat shock protein 90 β
(HSP90β) and sterile alpha motif and histidine-aspartate
domain-containing protein 1 (SAMHD1) (25). SAMHD1 is involved in regulating the
availability of intracellular nucleic acids and participates in the
formation of cGAMP (30). The
chaperone protein HSP90β, as a novel STING-interacting protein,
modulates STING to promote the activation of tank binding kinase 1
(TBK1) through the aforementioned pathway to phosphorylate
interferon regulatory factor 3 (IRF3), facilitating the release of
interferon (IFN)α/β (31)
(Fig. 3C). Overall, EFTUD2 might
control the cGAS-STING pathway to regulate innate immunity.
EFTUD2 mutations
Clinical studies have extensively reported
EFTUD2 mutations and their manifestations in humans.
According to previous studies, EFTUD2 mutations primarily
cause developmental defects (32–35)
(Table I). The process from
EFTUD2 genomic mutations to abnormal protein expression is
illustrated in Fig. 4. The main
types of EFTUD2 mutations include frameshift mutations,
missense mutations, splice site mutations and nonsense mutations.
The clinical symptoms associated with different mutations are shown
in Table II.
 | Table I.EFTUD2 mutation sites and mutation
types. |
Table I.
EFTUD2 mutation sites and mutation
types.
| First author/s,
year | EFTUD2 mutation
site | Mutation type | Developmental
defects in the patient | (Refs.) |
|---|
| Sarkar et
al, 2015 | c.933dupC
(p.S312fs) | Frameshift | MFDM (Guion-Almeida
type) | (32) |
| Smigiel et
al, 2015 | c.1435dup
(p.Thr479AsnfsX2) | Frameshift | MFDM (Guion-Almeida
type) | (33) |
| Matsuo et
al, 2017 | c.2698_2701del | Frameshift | MFDM (Guion-Almeida
type) | (34) |
| Narumi-Kishimoto
et al, 2020 | c.2624dupT
(p.Ile875fs) | Frameshift | MFDM (Guion-Almeida
type) | (35) |
| McDermott et
al, 2017 | c.944delG
(p.Ser315fs) | Frameshift | TAPVD, MFDM | (36) |
| Wang et al,
2021 | c.2314del
(p.Gln772ArgfsTer21) | Frameshift | EA/TEF | (37) |
| Khattar and Suhrie,
2023 | c.969del | Frameshift | MFDM, EA/TEF | (38) |
| Smigiel et
al, 2015 | c.1859A>T
(p.Lys620Met) | Missense | MFDM (Guion-Almeida
type) | (33) |
| Lines et al,
2012 | c.784C>T
(p.Arg262Trp) | Missense | MFDM (Guion-Almeida
type) | (40) |
| Bukowska-Olech
et al, 2020 | c.491A>G
(p.Asp164Gly) | Missense | FD | (39) |
| Bukowska-Olech
et al, 2020 | c.779T>A
(p.Ile260Asn) | Missense | FD | (39) |
| Lacour et
al, 2019 | c.2333C>A
(p.Pro778His) | Missense | MFDM | (41) |
| Luquetti et
al, 2013 | c.2637G>A
(p.Glu794Lys) | Missense | MFDM (de
novo variant) | (42) |
| Luquetti et
al, 2013 | c.1458C>G
(p.Gln401Glu) | Missense | MFDM (de
novo variant) | (42) |
| Lacour et
al, 2019 | c.2466+1G>A
(IVS24+1G>A) | Splice Site | MFDM | (41) |
| Luquetti et
al, 2013 | c.504-2G>T | Splice Site | MFDM | (42) |
| Voigt et al,
2013 | c.994+1G>C | Splice Site |
Oculo-Auriculo-Vertebral Spectrum
(OAVS) | (44) |
| Voigt et al,
2013 | c.2562-1G>C | Splice Site | MFDM, seizures | (44) |
| Khattar and Suhrie,
2023 | c.1058+1G>A | Splice Site | EA/TEF | (38) |
| Kim et al,
2020 | c.271+1G>A | Splice Site | MFDM | (43) |
| Voigt et al,
2013 | c.351-1G>A
(p.Asp117Glufs*8) | Splice Site | Oto-facial
syndrome | (44) |
| Voigt et al,
2013 | c.594T>G
(p.Tyr198*) | Nonsense | MFDM, intellectual
disability | (44) |
| Rengasamy
Venugopalan et al, 2017 | c.259C>T
(p.Gln87*) | Nonsense | MFDM (de
novo variant) | (45) |
| Sarkar et
al, 2015 | c.1732C>T
(p.R578X) | Nonsense | MFDM | (32) |
| Yang et al,
2022 | c.1012G>T
(p.E338*) | Nonsense | MFDM, gonadal
mosaicism | (46) |
| Table II.Clinical symptoms of patients with
EFTUD2 mutations. |
Table II.
Clinical symptoms of patients with
EFTUD2 mutations.
| First author/s,
year | EFTUD2 mutation
site | Clinical symptoms
of patients | (Refs.) |
|---|
| Sarkar et
al, 2015 | c.933dupC
(p.S312fs) | Malar hypoplasia,
mandibular hypoplasia, microcephaly, abnormal external ears,
hearing loss, developmental delay | (32) |
| Smigiel et
al, 2015 | c.1435dup
(p.Thr479AsnfsX2) | Cleft palate,
microcephaly, facial asymmetry, palpebral fissures downslanting,
absent/sparse lateral lower eyelashes, hyperplastic supraorbital
ridges, malar hypoplasia, broad base of nose, microtia, low set
ears, preauricular tags, dysplastic ears, micrognathia, proximally
placed thumbs, brachydactyly, esophageal atresia and
tracheoesophageal fistula, hearing loss, feeding problems,
gastrostomy, tracheostomy, psychomotor delay, speech delay, somatic
delay and microsomia | (33) |
| Matsuo et
al, 2017 | c.2698_2701del | Microcephaly, malar
hypoplasia, mandibular hypoplasia, deafness, epilepsy and
developmental delay | (34) |
| Narumi-Kishimoto
et al, 2020 | c.2624dupT
(p.Ile875fs) | Micrognathia, malar
hypoplasia, microcephaly, abnormality of the pinna, ossicular
abnormalities, hearing impairment, cleft palate, seizures,
esophageal atresia, scoliosis/kyphosis, growth failure,
intellectual disability and choanal atresia | (35) |
| McDermott et
al, 2017 | c.944delG
(p.Ser315fs) | Total anomalous
pulmonary venous drainage, tracheoesophageal fistula, facial palsy
or asymmetry and esophageal Atresia | (36) |
| Wang et al,
2021 | c.2314del
(p.Gln772ArgfsTer21) | EA/TEF, atrial
septal defect, bilateral clubfoot, hydrocele and renal
pyelectasis | (37) |
| Khattar and Suhrie,
2023 | c.969del | EA/TEF,
micrognathia, microcephaly, accessory ear tags, mitral valve
stenosis, cleft palate, left ear microtia, right preauricular tag,
exotropia, amblyopia, astigmatism and mild left kidney
pelviectasis | (38) |
| Smigiel et
al, 2015 | c.1859A>T
(p.Lys620Met) | Microcephaly,
facial asymmetry, palpebral fissures downslanting, lacrimal duct
anomalies, hypertelorism, malar hypoplasia, broad base of nose,
preauricular tags, dysplastic ears, micrognathia, proximally placed
thumbs, camptodactyly, choanal atresia, hearing loss, psychomotor
delay, speech delay, somatic delay and microsomia | (33) |
| Lines et al,
2012 | c.784C>T
(p.Arg262Trp) | Hyperplastic
supraorbital ridges, hypertelorism, malar hypoplasia, broad base of
nose, low set ears, dysplastic ears, micrognathia, proximally
placed thumbs, choanal atresia, hearing loss, ophthalmology
problems, astigmatism, myopia, ptosis, strabismus feeding problems,
gastrostomy, tracheostomy, psychomotor delay, speech delay | (40) |
| Bukowska-Olech
et al, 2020 | c.491A>G
(p.Asp164Gly) | Trigonocephaly,
upturned nose and preaxial polydactyly | (39) |
| Bukowska-Olech
et al, 2020 | c.779T>A
(p.Ile260Asn) | Intellectual
impairment, delayed psychomotor development, delayed speech
development, epilepsy, microcephaly, trigonocephaly, midface
hypoplasia, malar hypoplasia, micrognathia, buccal tags,
preauricular tag, preauricular pit, low-set ears, dysplastic ears,
conductive hearing loss, upslanting palpebral fissures,
downslanting palpebral fissures, short nose and atrial septal
defect | (39) |
| Lacour et
al, 2019 | c.2333C>A
(p.Pro778His) | Hemifacial
microsomia, cleft lip and palate, mild microcephaly, dysplastic
ears and hearing loss | (41) |
| Luquetti et
al, 2013 | c.2637G>A
(p.Glu794Lys) | Facial asymmetry,
choanal atresia, epibulbar dermoid, cleft of left zygomatic arch,
bilateral microtia, preauricular skin tags, small external auditory
canal, hearing loss, incompletely formed lateral semicircular canal
and dilated vestibule, mandibular hypoplasia, malar hypoplasia,
micrognathia, cleft palate, thumb abnormalities, developmental
delay | (42) |
| Luquetti et
al, 2013 | c.1458C>G
(p.Gln401Glu) | Microcephaly,
facial asymmetry, choanal atresia, cleft of zygomatic arch,
bilateral microtia, preauricular skin tags, atretic external
auditory canal, hearing loss, dysplastic ossicles, mandibular
hypoplasia, malar hypoplasia, micrognathia, developmental delay,
seizures, malformed ossicles, mandibular asymmetry, thumb
abnormalities, cervical spine abnormalities, developmental delay,
seizures | (42) |
| Lacour et
al, 2019 | c.2466+1G>A
(IVS24+1G>A) | Left hemifacial
microsomia, left ear canal atresia with third-degree microtia,
presence of a lobule in anomalous position, the upper half of the
partial pinna located posteriorly, metopic craniosynostosis with
trigonocephaly, VSD, PFO and mild diffuse atrophy of the brain | (41) |
| Luquetti et
al, 2013 | c.504-2G>T | Microcephaly,
facial asymmetry, choanal atresia, cleft of zygomatic arch,
bilateral microtia, preauricular skin tags, atretic external
auditory canal, hearing loss, dysplastic ossicles, mandibular
hypoplasia, malar hypoplasia, micrognathia, developmental delay,
seizures | (42) |
| Voigt et al,
2013 | c.994+1G>C | Polyhydramnios,
facial asymmetry, upslanting palpebral fissures, microtia/with
squared earlobe, a-/hypoplasia of external ear canal, hearing loss,
cleft palate, reduced mouth opening, micrognathia, malformations
tracheostomy, esophageal atresia, CHD, scoliosis left of zygomatic
bone, clinodactyly V | (44) |
| Voigt et al,
2013 | c.2562-1G>C | Epilepsy,
hyperplastic supraorbital ridges, Frontal bossing, Microtia/with
squared earlobe, preauricular tag, preauricular pit, a-/hypoplasia
of external ear canal, hearing loss, nasal speech, reduced mouth
opening, micrognathia | (44) |
| Khattar and Suhrie,
2023 | c.1058+1G>A | EA/TEF,
microcephaly, micrognathia, hyperopia and astigmatism,
microtia | (38) |
| Kim et al,
2020 | c.271+1G>A | Abnormal
echogenicity in the pulmonary artery area, tricuspid valve
insufficiency, | (43) |
| Voigt et al,
2013 | c.351-1G>A
(p.Asp117Glufs*8) | Polyhydramnios,
facial asymmetry, upslanting palpebral fissures, microtia/with
squared earlobe, A-/hypoplasia of external ear canal, hearing loss,
cleft of zygomatic bone, choanal atresia, small middle ear
cavity | (44) |
| Voigt et al,
2013 | c.594T>G
(p.Tyr198*) | Polyhydramnios,
upslanting palpebral fissures, microtia/with squared earlobe,
A-/hypoplasia of external ear canal, cleft palate, micrognathia,
esophageal atresia, inner/middle ear malformations | (44) |
| Rengasamy
Venugopalan et al, 2017 | c.259C>T
(p.Gln87*) | Gross facial
asymmetry, micrognathia, airway obstruction, choanal atresia, left
ear microtia, bilateral absence of ear canals and conductive
hearing loss, speech articulation problems and microcephaly | (45) |
| Sarkar et
al, 2015 | c.1732C>T
(p.R578X) | Malar hypoplasia,
mandibular hypoplasia, microcephaly, abnormal external ears,
hearing loss, developmental delay, auditory canal defects, inner
ear abnormalities | (32) |
| Yang et al,
2022 | c.1012G>T
(p.E338*) | Recurrent pregnancy
loss | (46) |
Frameshift mutations
Frameshift mutations are caused by insertions or
deletions. These mutations disrupt the reading frame of the gene.
As a result, they frequently produce premature stop codons, leading
to the creation of truncated, non-functional proteins. Sarkar et
al (32) identified the
c.933dupC (p.S312fs) mutation in patients with developmental
defects, this mutation is categorized as an insertion/frameshift
mutation. It occurs because of the duplication of a nucleotide at
position 933, resulting in a shift in the reading frame. Smigiel
et al (33) also identified
the c.1435dup (p.Thr479AsnfsX2) mutation in patients with
developmental defects. Similarly, the deletion mutation
c.2698_2701del, found in a patient with ventriculomegaly, leads to
a frameshift and premature stop codon (34). Another mutation, c.2624dupT
(p.Ile875fs), was linked to Guion-Almeida type mandibulofacial
dysostosis with microcephaly (MFDM), resulting in a truncated
EFTUD2 protein (35).
Additionally, a heterozygous c.944delG (p.Ser315fs) frameshift
mutation was reported in a male infant with total anomalous
pulmonary venous drainage (TAPVD) and his mother (36). A novel frameshift mutation,
c.2314del (p.Gln772ArgfsTer21), was also detected in a patient with
esophageal atresia/tracheoesophageal fistula (EA/TEF) (37). Khattar and Suhrie (38) detected EFTUD2 mutations in
two patients with EA/TEF, respectively NM_004247.3: c.969del and
NM_001258353: c.969del, which are the same mutation occurring at
the same position in different EFTUD2 transcripts. The
occurrence of similar mutations in EFTUD2 across different
diseases suggests that frameshift mutations in EFTUD2 might
have a widespread impact on the pathogenesis of these conditions.
Further research into the molecular mechanisms is needed to
determine whether there are underlying connections.
Missense mutations
Missense mutations cause a single nucleotide change,
replacing one amino acid with another, with varying effects on
protein function. Bukowska-Olech et al (39) reported two missense mutations in
patients with facial dysostoses: c.491A>G (p.Asp164Gly) and
c.779T>A (p.Ile260Asn). These two mutations are both classified
as missense mutations, leading to the substitution of aspartic acid
(Asp) with glycine (Gly) and isoleucine (Ile) with asparagine
(Asn), respectively, further resulting in the structural or
functional alteration of EFTUD2. In patients with Guion-Almeida
type MFDM, the missense mutations c.1859A>T (p.Lys620Met)
(33) and c.784C>T
(p.Arg262Trp) (40) were
identified, which altered the protein structure. Additionally,
Lacour et al (41)
discovered a missense mutation, c.2333C>A (p.Pro778His), in exon
23 of EFTUD2 in a patient with MFDM. Luquetti et al
(42) also identified two de
novo variants associated with MFDM: c.2637G>A (p.Glu794Lys)
and c.1458C>G (p.Gln401Glu). From the aforementioned, it is
evident that missense mutations in EFTUD2 typically lead to
abnormal development of the human mandible. Whether this mutation
will cause other defects remains to be further investigated.
Splice site mutations
Splice site mutations occur at exon-intron
boundaries, disrupting normal RNA splicing and potentially leading
to exon skipping or intron retention. In a child with MFDM, Kim
et al (43) identified a
novel splice donor site variant, c.271+1G>A in EFTUD2.
Minigene assays demonstrated that this variant led to the erroneous
integration of a 118 bp fragment from Intervening Sequence 3 (IVS3)
of EFTUD2 in the c.271+1G>A variant clones. This
integration produced a truncated EFTUD2 protein, reducing its
length by 11.7%. Lacour et al (41) identified a novel de novo
splice site mutation, c.2466+1G>A (IVS24+1G>A), at the splice
donor site of EFTUD2 intron 24. Luquetti et al
(42) found the EFTUD2
splice site mutation c.504-2G>T at the acceptor site near exon
504, while Voigt et al (44) reported a c.994+1G>C mutation in
patients with oculo-auriculo-vertebral spectrum (OAVS). In a
patient with generalized seizures, the c.2562-1G>C mutation was
identified at the splice acceptor site. Similarly, a splice site
mutation in intron 4 of EFTUD2, c.351-1G>A
(p.Asp117Glufs*8), was detected in a patient with Oto-facial
syndrome (44). Another splice
site mutation, c.1058+1G>A, was detected in a patient with
EA/TEF (38). Splice site
mutations in EFTUD2 are associated with facial developmental
defects such as MFDM, OAVS and Oto-facial syndrome. Compared with
missense mutations, splice site mutations of EFTUD2 are more
likely to result in a broader range of craniofacial developmental
abnormalities.
Nonsense mutations
Nonsense mutations introduce premature stop codons,
truncating the protein and typically resulting in non-functional
proteins. The c.259C>T (p.Gln87*) mutation of EFTUD2 was
detected in a patient with mandibulofacial dysostosis (MFD)
(45), while another patient with
MFDM carried the c.1732C>T (p.R578X) mutation (32), both of which caused premature
termination of EFTUD2 protein synthesis. Additionally, Voigt et
al (44) identified a novel
heterozygous mutation, c.594T>G (p.Tyr198*), in a patient with
Nager syndrome. A de novo nonsense mutation, c.1012G>T
(p.E338*), in exon 12 of EFTUD2 was discovered through
whole-genome sequencing in a couple with recurrent miscarriages.
This mutation produces a truncated EFTUD2 protein, missing 634
amino acids. Zebrafish models confirmed that this mutation causes
EFTUD2 loss of function, affecting hindbrain development and heart
formation (46). Notably, besides
causing MFDM, nonsense mutations in EFTUD2 can also lead to
miscarriages. Further research is needed to determine whether these
miscarriages are related to fetal defects caused by EFTUD2
mutations.
In summary, the mechanisms of EFTUD2
mutations have deepened our understanding of the gene; however, to
intervene in EFTUD2 mutations or compensate for the
subsequent outcomes caused by EFTUD2 mutations represents a
significant research challenge for the future.
Role of EFTUD2 in tumors
EFTUD2, as an important component of the
spliceosome, is involved in pro-mRNA pruning and splicing, thus its
altered expression inevitably plays a crucial role in tumorigenesis
and development. Studies found that the expression level of EFTUD2
was markedly increased in tumor tissues (47,48).
An elevated level of EFTUD2 is also associated markedly with the
prognosis of a variety of tumors and has the potential to be used
as a tumor independent prognostic biomarker (Table III).
| Table III.Research progress for EFTUD2 in
tumors. |
Table III.
Research progress for EFTUD2 in
tumors.
| First author/s,
year | Tumor type | EFTUD2
expression | Mechanisms | Effects | Function | (Refs.) |
|---|
| Tu et al,
2020; Lv et al, 2022; Zhou, et al, 2022; Zhou et
al, 2021; Chi et al, 2020 | Hepatocellular
carcinoma | UP | Regulation of
methylation; reduction of YTHDF3 ubiquitination; promotion of the
STAT3 pathway | Enhanced lactate
production; Promotion of cell cycle progression; Inhibition of
apoptosis; Promotion of EMT; Immune infiltration; Reduced
prognosis | Biomarker | (47–49,51,97) |
| Beyer et al,
2023 | Endometrial
Cancer | UP | - | Reduced
prognosis | Biomarker | (64) |
| Sato et al,
2015 | Breast Cancer | UP | Binding with
SNW1 | Inhibition of
apoptosis | Biomarker | (61) |
| Chen et al,
2021; Zhang et al, 2023 | Bladder Cancer | UP | - | Reduced
prognosis | Diagnostic
Biomarker | (63,98) |
| Lv et al,
2019 | Colorectal
Cancer | UP | Activation of TLR4
signaling and NF-κB; TLR4/MD-2/MyD88 pathway | Reduced prognosis;
The occurrence of colitis-associated cancer | Biomarker | (52) |
Hepatocellular carcinoma (HCC)
EFTUD2 is closely associated with the progression of
HCC. Studies have reported that EFTUD2 is upregulated in HCC
tissues and HCC patients with high levels of EFTUD2 have shorter
overall and recurrence-free survival (47,48).
Knockdown of EFTUD2 markedly inhibits HCC cell viability and
cell cycle progression, promotes apoptosis and suppresses
metastasis. When the expression level of EFTUD2 is reduced, it
arrests the cell cycle of liver cancer cells and hinders the
transition from the G1 to S phase (47,48).
Zhou et al (49) discovered
that EFTUD2 binds to YTH domain family protein 3 (YTHDF3), thereby
inhibiting YTHDF3′s ubiquitination. This inhibition leads to an
increase in YTHDF3 levels. In turn, the elevated YTHDF3 can
suppress the degradation of phosphofructokinase (PFKL) mRNA via m6A
modification, thus maintaining PFKL mRNA levels. Through
this mechanism, EFTUD2 enhances tumor glycolysis in HCC, which
promotes the proliferation, migration and invasion of HCC cells
(49) (Fig. 5A). Further research showed that
EFTUD2 enhances the expression of signal transducer and activator
of transcription 3 (STAT3) and cytokine IL-6. IL-6 participates in
STAT3 protein phosphorylation, thereby promoting the expression of
Myeloid cell leukemia 1 (MCL-1) and vimentin, ultimately leading to
EMT and the inhibition of apoptosis (47,50)
(Fig. 5A). Zhou et al
(51) found that EFTUD2 correlates
positively with levels of B cells, CD4+ T cells, CD8+ T cells,
neutrophils, macrophages and dendritic cells in the tumor
microenvironment. This suggests that EFTUD2 is involved in
regulating immune infiltration in HCC by promoting the formation of
a tumor-favorable immune microenvironment. Thus, EFTUD2 is
considered to be a potential therapeutic target for liver cancer
(48,51).
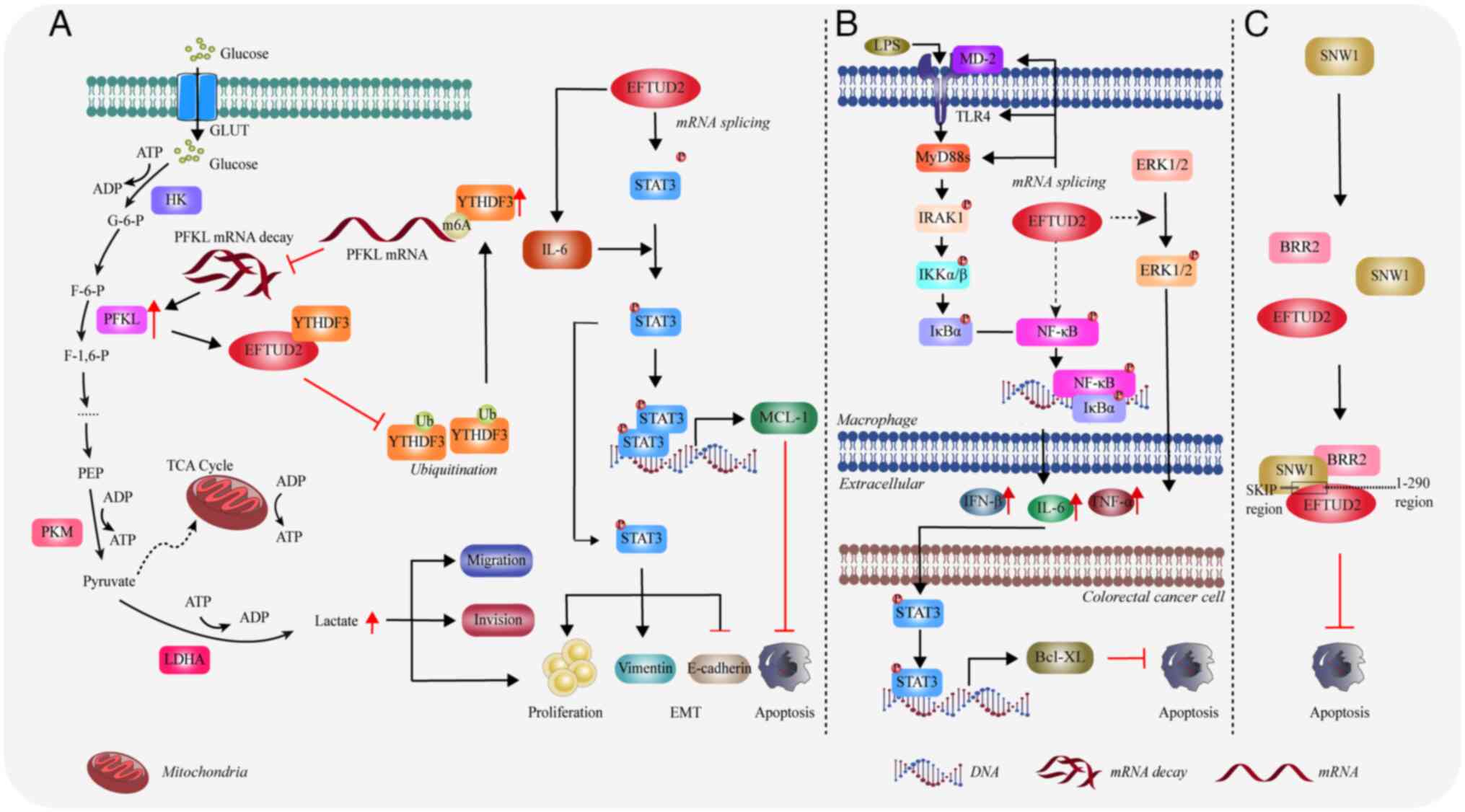 | Figure 5.EFTUD2 promotes the development and
progression of various tumors. (A) The mechanism by which EFTUD2
promotes the development of hepatocellular carcinoma. (B) The
mechanism by which EFTUD2 promotes the development of colorectal
cancer through the regulation of macrophage inflammatory response.
(C) The role of EFTUD2 in the progression of breast cancer. The
figure was created using Adobe Illustrator (version, 28.3; Adobe
Systems, Inc.). EFTUD2, elongation factor Tu GTP binding domain
containing 2; MyD88, myeloid differentiation primary response 88;
GLUT, glucose transporter; ADP, adenosine diphosphate; HK,
Hexokinase; PFKL, phosphofructokinase L; YTHDF3, YTH domain family
protein 3; LDHA, lactate dehydrogenase A; IL, interleukin; STAT3,
signal transducer and activator of transcription 3; IRAK1,
interleukin 1 receptor associated kinase 1; MCL-1, myeloid cell
leukemia-1; ERK, extracellular signal-regulated kinase; TLR,
Toll-like receptor; SNW1, SNW domain containing 1; BRR2, small
nuclear ribonucleoprotein U200; NF-κB, nuclear factor kappa B; TNF,
tumor necrosis factor. |
Colorectal cancer
Lv et al (52) discovered that EFTUD2 protein levels
are abnormally increased in colorectal cancer and reduced EFTUD2
levels leads to a marked decrease in both the number and size of
colorectal tumors, but an increase in low-grade dysplasia. The
binding of TLR ligands, such as LPS produced by gut microbiota,
activates the Toll-like receptor (TLR4)/nuclear factor κB (NF-κB)
inflammatory signaling pathways, which are critical factors in the
development of colorectal cancer (52,53).
EFTUD2 can enhance this pathway by regulating the splicing of mRNAs
for components and kinases, including membrane-bound TLR4,
full-length myeloid differentiation protein-2 (MD-2) and MyD88L,
thereby activating macrophages (52). These activated macrophages then
produce and release pro-tumor cytokines such as IL-6, IFN-β and
TNF-α (52,54–56)
(Fig. 5B). Additionally, EFTUD2
also promotes the activation of the mitogen activated protein
kinase (MAPK)/extracellular regulated kinase pathway in macrophages
and mouse colon tissues, leading to an increased release of factors
such as IL-6 and IFN-β (52)
(Fig. 5B). These cytokines promote
the proliferation of intestinal epithelial cells by activating
STAT3 and its downstream target gene Bcl-XL (52) (Fig.
5B). Bcl-2-like protein 1 3 (Bcl-XL) is an anti-apoptotic
protein that plays a critical role in mediating the survival of
colorectal epithelial cells (57).
Therefore, EFTUD2 deficiency can induce apoptosis in colon cancer
cells by inhibiting the secretion of cytokines through the
suppression of the TLR/NF-κB signaling pathway and by suppressing
the expression of BCL-XL.
Several studies have explored tumor-related
inflammatory marker characteristics, including those of colorectal
cancer, through blood leukocyte levels (58–60).
Whether EFTUD2 contributes to colorectal cancer progression by
enhancing this inflammatory response remains to be investigated.
Additionally, as aforementioned in sections ‘Enhancement of immune
effects on macrophages’ and ‘Regulation of the cGAS-STING pathway’,
studies have reported that EFTUD2 is involved in regulating immune
responses, such as those related to macrophages (23) and the cGAS-STING pathway (25). While macrophages, as immune cells,
have been highlighted in the development of colorectal cancer, the
role of other immune cells remains unclear. Therefore, further
investigation into the inflammatory characteristics of EFTUD2 in
colorectal cancer progression is crucial for improving treatment
and prognosis.
Breast cancer
A study reported that EFTUD2 expression is markedly
elevated in breast cancer cells (61). Through immunoprecipitation
analysis, researchers found that the 1–260 region of EFTUD2
interacts with SNW domain containing 1 (SNW1) 174–335 region (SKIP
domain). SNW1 is a highly conserved protein that functions as a
splicing factor in RNA transcription and splicing and its
deficiency can lead to splicing defects (62).
Additionally, EFTUD2 connects the C-terminus of BRR2
to the SKIP domain of SNW1 through its N-terminus, thereby forming
a complex protein structure (Fig.
5C). Researchers expressed EFTUD2 and SNW1
deletion mutants in breast cancer cells to disrupt the
EFTUD2-SNW1-BRR2 complex. They found that >50% of cells with
these mutants experienced apoptosis (61) (Fig.
5C). Thus, the interaction between EFTUD2 and SNW1 is crucial
for the survival of breast cancer cells, because its disruption
will lead to increased apoptosis.
Other types of cancers
Investigators found that EFTUD2 had the highest risk
score in the development of a prognostic risk score model for
bladder cancer, indicating its significant predictive value for
patient outcomes (63). In
addition, a clinical study has shown that high expression of EFTUD2
in endometrial cancer is predictive of poor prognosis (64). In multivariate Cox regression
analysis, EFTUD2 was identified as an independent marker for
progression-free survival in endometrial cancer and could serve as
a negative prognostic indicator for patients (64). Research on EFTUD2 in bladder and
endometrial cancer is currently limited to data analysis. Further
cellular and in vivo experiments are required to explore the
mechanisms by which EFTUD2 influences the development and
progression of this tumor.
In summary, the evidence linking EFTUD2 to poor
prognosis in cancers suggests that it could serve as a valuable
biomarker to predict patient outcomes. Furthermore, the role of
EFTUD2 in shaping the tumor microenvironment through immune
regulation presents potential therapeutic avenues for targeting
EFTUD2 in cancer treatment. Nonetheless, more research is needed to
fully understand its molecular mechanisms across different cancers
and to explore its potential as a therapeutic target.
Role of EFTUD2 in non-neoplastic
diseases
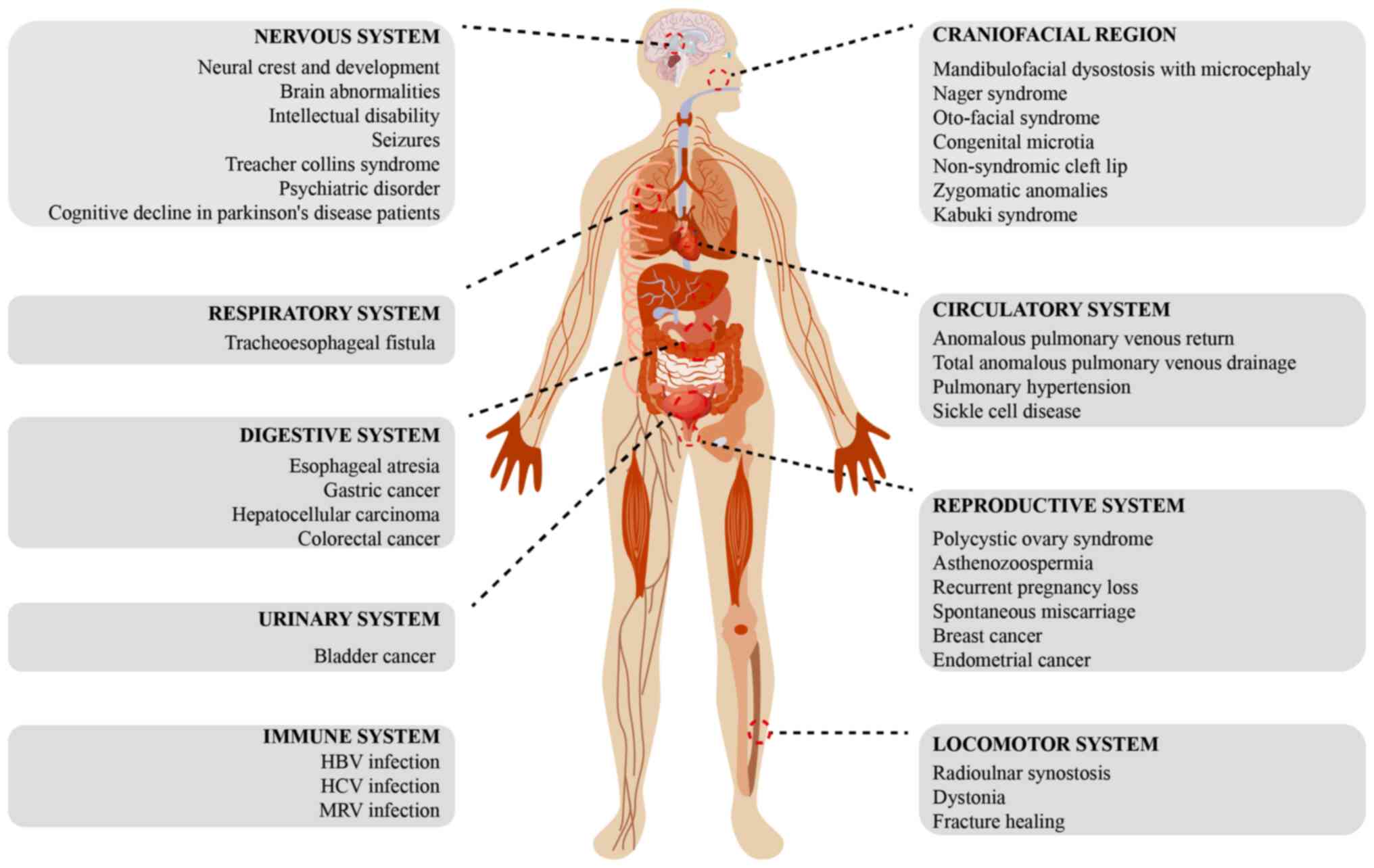
Mutations in EFTUD2 can disrupt the normal function
of the spliceosome, leading to RNA splicing errors, which manifest
as various systemic defects and other associated diseases in the
body (Fig. 6).
MFD
MFD, commonly known as Treacher Collins syndrome, is
a rare congenital disorder characterized by underdeveloped
craniofacial bones (19,65). The etiology of MFD is
multifactorial and recent studies emphasize that mutations in genes
encoding major spliceosome core components are associated with
various forms of this disease (66–68).
MFD encompasses different subtypes, including MFDM and the MFDM
Guion-Almeida type. The first description of MFDM emerged from a
cohort of four unrelated Brazilian patients (69), marking an important step in
recognizing the genetic foundation of this disorder. Since then,
the identification of EFTUD2 mutations through clinical
genomic sequencing has become increasingly prevalent, underscoring
the critical role of the gene in MFD pathology. Patients with
EFTUD2 mutations consistently exhibit significant mandibular
hypoplasia characteristics (Table
II) (35,40,70–73).
Vincent et al (74)
conducted genetic analysis of EFTUD2 in 11 suspected cases
of MFDM or MFD Guion-Almeida type. Among the cohort, four patients
exhibited molecular abnormalities in EFTUD2, including
missense mutations, nonsense mutations, duplications and deletions.
All patients carrying EFTUD2 mutations presented with
microcephaly, hypoplasia of the zygomatic and mandibular bones,
hearing loss, downslanting palpebral fissures and microtia
(Table II). Bukowska-Olech et
al (39) reported two female
patients carrying novel heterozygous variants in EFTUD2,
both presenting with mandibulofacial dysostosis of the
Guion-Almeida type (Table II).
Similarly, Luquetti et al (42) confirmed the presence of three novel
variants in EFTUD2 through Sanger sequencing, all of which
were de novo variants. Two patients had missense mutations
and one patient had a splice site mutation. All three patients
exhibited significant mandibular hypoplasia (Table II). This pattern of symptoms
reinforces the fact that EFTUD2 mutations play a critical
role in craniofacial development. EFTUD2 is increasingly
becoming a key gene that should be checked for mutations in the
diagnosis or research on MFDM. Increasing types of EFTUD2
mutations are being discovered, highlighting its significant role
in craniofacial development and other developmental defect
diseases. Especially in fetal screening, it provides a valuable
reference to detect developmental defects.
Nervous system
Intellectual disability and epilepsy
A case of a patient with mild intellectual
disability was reported, in which the individual was identified to
have a nonsense mutation in EFTUD2 (44). In a study involving 12 patients
with congenital anomalies and/or intellectual disabilities and
their trios, researchers identified two EFTUD2 mutations
through whole-exome sequencing (75). Matsuo et al (34), reported a case of a patient with
epilepsy with early closure of the anterior fontanelle and patent
ductus arteriosus at birth (Table
II). The patient was found to have an EFTUD2 mutation.
An individual carrying an EFTUD2 mutation was reported to
suffer an initial seizure at 8 months old. Furthermore, an EEG
indicated the presence of occasional spikes in the right frontal
region (35). Another patient with
generalized seizures and moderate intellectual disability was also
found to have an EFTUD2 splice site mutation (Table II) (44). Various signs indicate that
EFTUD2 mutations are probably an important factor in brain
abnormalities, such as intellectual disabilities or epilepsy caused
by developmental defects. However, no experimental studies have
been reported so far and the specific mechanisms remain to be
further explored.
Psychiatric disorders
Park et al (19) constructed a quantitative model
based on whole-genome sequencing information, which identified
EFTUD2 as an RNA binding protein (RBP) involved in diseases
such as psychiatric disorders through RNA-RBP interaction
profiling. Dysregulation of EFTUD2 plays a critical role in
psychiatric disorders, such as attention deficit hyperactivity
disorder and schizophrenia, by affecting target gene expression
(76). The report indicated that
variations in EFTUD2 and its downstream targets are associated with
neurological diseases. However, the specific targets and mechanisms
through which EFTUD2 is involved in intellectual disabilities have
not been clarified and thus require further investigation.
Cognitive impairment in patients with
Parkinson's disease
Santiago and Potashkin (77) investigated 10 RNA biomarkers,
including EFTUD2 and compared their expression levels
between patients with Parkinson's disease (PD) and healthy
controls. The level of EFTUD2 in cognitively normal PD
patients (PD-CN) was markedly higher than in cognitively impaired
PD patients (PD-MCI). The researchers performed a receiver
operating characteristic (ROC) curve analysis on EFTUD2,
which yielded an area under the curve (AUC) value of 0.64,
suggesting that EFTUD2 has some predictive value for PD-MCI.
Current research is limited to clinical data analysis and whether
EFTUD2 has a definitive impact on the progression of this disease
requires further in-depth studies.
Circulatory system
Anomalous pulmonary venous return
A male infant with TAPVD was reported to have an
EFTUD2 mutation (Table II)
(36). Notably, the researchers
observed that the infant's mother had mild facial asymmetry.
Genomic sequencing revealed a heterozygous frameshift mutation in
EFTUD2 in both the mother and two of her infants (36). That study was the first to report a
case of EFTUD2 haploinsufficiency presenting with TAPVD. The
affected infants and their mother did not exhibit the classic
phenotypic features of MFDM and the diagnosis was made solely
through exome sequencing.
Pulmonary hypertension
Wang et al (78) extracted data from 58 healthy
controls and 135 patients with pulmonary hypertension from the Gene
Expression Omnibus datasets, identifying EFTUD2 as a
differentially expressed hub gene. The authors used a hypoxic
pulmonary hypertension rat model to confirm the significant
upregulation of EFTUD2 in the pulmonary arteries of hypoxic
pulmonary hypertension rats. The specific mechanisms of EFTUD2 in
pulmonary arterial hypertension remain to be further
elucidated.
Sickle cell disease
In a bioinformatic analysis of Sickle Cell Disease,
Liu et al (79) conducted a
genome-wide association study (GWAS) on individuals with high and
low hemoglobin (Hb) F levels. The GWAS data revealed single
nucleotide polymorphisms (SNPs) in EFTUD2 associated with
high Hb F (79), hinting that
EFTUD2 is a potential new candidate locus. However, larger
sample studies are required to confirm the role of EFTUD2 in
γ-globin regulation.
Digestive system
Researchers reported that esophageal atresia in
patients was caused either by deletions in EFTUD2 or novel
heterozygous loss-of-function mutations in EFTUD2. These
patients presented with severe micrognathia, upper airway
obstruction, esophageal atresia, tracheoesophageal fistula and
choanal atresia (Table II)
(33,35,44,80).
Wang et al (37) also
reported a novel de novo frameshift deletion in the
EFTUD2 gene in a patient with EA/TEF. The patient's
phenotype included EA/TEF, bilateral talipes equinovarus,
hydrocele, atrial septal defect and renal pelvis dilatation
(Table II). Khattar and
Suhrie (38) also performed
exome sequencing analysis on nine patients with EA/TEF and detected
EFTUD2 mutations in three patients. These studies indicate
that mutations in EFTUD2 can lead to severe gastrointestinal
defects.
Reproductive system
Polycystic ovary syndrome
In a screening study for characteristic genes of
polycystic ovary syndrome (PCOS), Heidarzadehpilehrood et al
(81) identified EFTUD2 as
a hub gene associated with PCOS. In addition, Hou et al
(82) identified EFTUD2 as
a hub gene in the protein-protein interaction network of
differential genes in PCOS. However, the specific function and
mechanism of EFTUD2 in PCOS require further investigation.
Recurrent pregnancy loss and
spontaneous miscarriage
Yang et al (46) reported a study involving a
non-consanguineous couple with a history of four consecutive
clinical miscarriages at 10 weeks of gestation. A novel de
novo nonsense mutation was identified in exon 12 of
EFTUD2 (Table II). The
same mutation was found in 13.5% of sperm cells from the male
partner, suggesting gonadal mosaicism. The researchers further
confirmed the loss of EFTUD2 gene function using a zebrafish
model. Embryos carrying the EFTUD2 mutation exhibited a
significant reduction in the hindbrain neuronal marker paired box
2, as well as defects in the heart marker myosin light chain 7.
Additionally, these embryos showed the common small head phenotype
associated with EFTUD2 mutations (46). It is noteworthy that the couple
successfully conceived by selecting and implanting embryos without
EFTUD2 mutations and the pregnancy was confirmed through
human chorionic gonadotropin testing and ultrasound (46).
Asthenozoospermia
Li and Chen (83)
identified EFTUD2 as one of the genes with the highest
connectivity degree in the differential gene network related to
asthenozoospermia. They found that EFTUD2 expression was
downregulated in patients with asthenozoospermia. Further in
vivo and in vitro studies are needed to confirm the
potential mechanisms underlying these findings.
Locomotor system
Limb deformities
In a sequencing study of 12 patients with Nager
syndrome with limb anomalies, three were found to have
EFTUD2 mutations (84).
Zarate et al (85)
identified a heterozygous loss-of-function mutation in
EFTUD2 located on 17q21.31 in a patient with a rare syndrome
characterized as the Guion-Almeida type. Compared with the
previously mentioned three patients, this patient had a complete
deletion of EFTUD2 and exhibited a more pronounced phenotype
of proximal radioulnar synostosis. This indicated the crucial role
of EFTUD2 in skeletal development. However, the precise mechanisms
of EFTUD2′s effect on radial bone development are not well
understood and additional research is required to clarify EFTUD2′s
role in limb anomalies.
Dystonia
Zech et al (86) identified 14 pathogenic or
potentially pathogenic CNVs, with EFTUD2 being one of the
most clinically relevant genes. The patients exhibited phenotypic
features such as segmental dystonia with juvenile onset, facial
dysmorphism, hearing impairment and autism spectrum disorder. A
heterozygous single exon deletion mutation in EFTUD2 (exon
11, 124bp) was also discovered, leading to MFDM and
neurodevelopmental disorders with phenotypic variability.
Infectious diseases
Anti-Hepatitis B virus (HBV) infection
A study of 379 chronic HBV-infected individuals
identified a SNP in EFTUD2, EFTUD2-rs3809756, which is
associated with increased susceptibility to HBV infection (87). Individuals with the rs3809756-CC
genotype have a higher risk of HBV infection compared with those
with the rs3809756-AA genotype. Located in the promoter region of
EFTUD2, the rs3809756 A>C polymorphism might reduce
promoter activity (87). Following
IFN-α treatment, EFTUD2 knockout HepG2 cells showed a
5.43-fold increase in HBV DNA, a 2.80-fold increase in Hepatitis B
surface antigen (HBsAg) and a 3.29-fold increase in Hepatitis B
e-Antigen (HBeAg). The percentage of HBcAg-positive cells in
HepG2.2.15 cells increased by 15% (88). Upon viral entry, HBV DNA is
recognized through the combined action of retinoic acid-inducible
gene I (RIG-I) and TLR3 (89,90).
This pathogen recognition triggers downstream signaling pathways,
leading to the expression of interferon-stimulated Genes (ISGs)
(91,92). Research shows that EFTUD2 promotes
the expression of key ISGs, such as MX dynamin-like GTPase 1 (MX1),
2′-5′-oligoadenylate synthetase and protein kinase R, by regulating
mRNA splicing (88,93) to impede HBV infection (Fig. 7A). These proteins enhance the
effects of type I IFNs and play a critical role in the host's
innate immune defense. EFTUD2 might become a key target to
exploring the therapeutic potential against HBV infection in future
research.
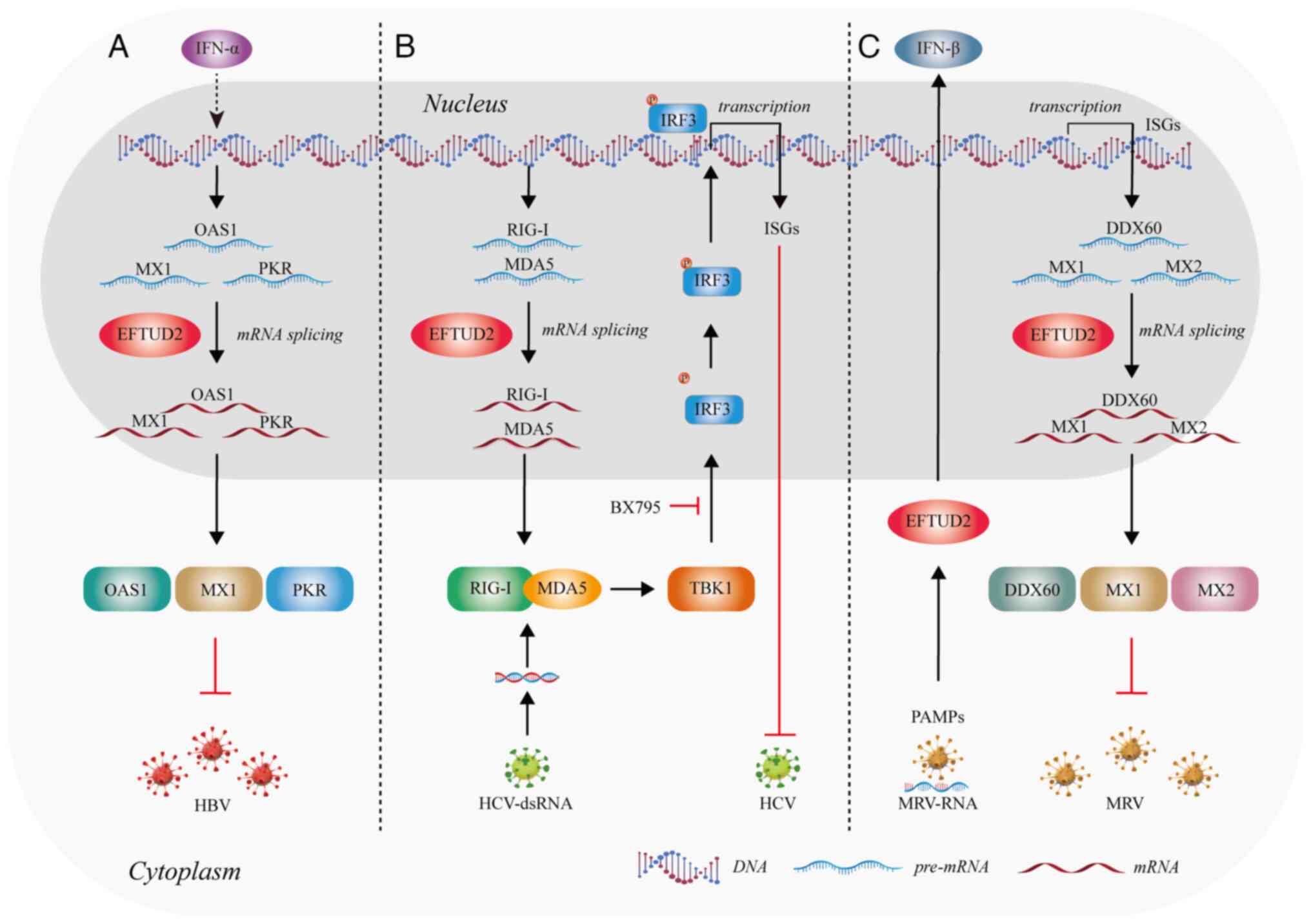 | Figure 7.EFTUD2 is involved in antiviral
processes by regulating splicing. (A) EFTUD2 participates in the
anti-HBV response by controlling molecular splicing. (B) EFTUD2
regulates the splicing of antiviral proteins to inhibit HCV
infection. (C) EFTUD2 participates in multiple antiviral mechanisms
in response to MRV infection. The figure was created using Adobe
Illustrator (version, 28.3; Adobe Systems, Inc.). EFTUD2,
elongation factor Tu GTP binding domain containing 2; HBV,
Hepatitis B virus; HCV, Hepatitis C virus; MRV, mammalian reovirus;
IFN, interferon; OAS1, 2′-5′ oligoadenylate synthetase 1; MX1, MX
dynamin-like GTPase 1; PKR, protein kinase R; IRF3, interferon
regulatory factor 3; RIG-1, retinoic acid-inducible gene I; MDA5,
melanoma differentiation-associated protein 5; TBK1, tank binding
kinase 1; DDX60, DEAD-box helicase 60; PAMPs, pathogen-associated
molecular patterns. |
Anti-Hepatitis C Virus (HCV) and
anti-Mammalian Reovirus (MRV) infection
Zhu et al (91) found that EFTUD2 inhibits HCV
infection at 12 h post-infection and reaches a plateau at 24 h,
indicating that EFTUD2 restricts HCV infection during the later
stages of viral entry. EFTUD2 can also regulate the expression of
RIG-I-like receptors RIG-I and melanoma differentiation-associated
protein 5 (MDA5) through mRNA splicing. RIG-I and MDA5 can detect
and recognize HCV RNA (94). This
leads to the binding of RIG-I and MDA5 and activates the kinase
TBK1, which then phosphorylates IRF3. Once phosphorylated, IRF3
forms dimers that translocate into the nucleus, subsequently
inducing the expression of IFN genes and ISGs (95,96)
(Fig. 7B). Therefore, EFTUD2 is
essential for the activation of IRF3 and the expression of ISGs in
TBK1-mediated anti-HCV responses downstream of RIG-I/MDA5
effectors.
EFTUD2 also restricted MRV replication in both
single and multiple replication cycles (93), by regulating the recognition of
viral pathogen-associated molecular patterns and the subsequent
production of IFNs (93) (Fig. 7C). In addition, EFTUD2 elevates the
basal mRNA levels of three ISGs:, MX1 and MX dynamin-like GTPase 2
to inhibit MRV infection (93)
(Fig. 7C).
Overall, EFTUD2 exerts antiviral immune activity by
regulating the expression of ISGs and cytokines through mRNA
splicing. Exploring the innate immune mechanisms of EFTUD2 holds
great potential for developing antiviral therapies.
In conclusion, EFTUD2 plays a crucial role in
various non-neoplastic diseases, affecting craniofacial
development, the nervous system, circulatory function, digestion,
reproduction, the musculoskeletal system and immune responses to
infections. Mutations in EFTUD2 are associated with a range of
disorders, including craniofacial deformities, neurological
impairments, circulatory dysfunction, reproductive issues and
immune response regulation. Given its critical role in multiple
pathological processes, EFTUD2 is a key gene for future clinical
research and mechanistic exploration. Further studies on the
molecular mechanisms through which EFTUD2 mutations lead to disease
will help uncover potential therapeutic strategies and assess the
feasibility of EFTUD2 as a treatment target.
Conclusion and perspectives
Collectively, EFTUD2 plays a vital role in fetal
development and is frequently implicated in gene mutations linked
to developmental defects. It is involved in the development and
maintenance of nearly all bodily systems, including the
craniofacial, nervous and respiratory systems. Thus, EFTUD2 shows
potential as a target for gene therapy in the early treatment of
fetal developmental defects.
In addition to its role in development, EFTUD2 is
highly expressed in several cancers, including hepatocellular
carcinoma, colorectal cancer, breast cancer and bladder cancer, in
which it promotes tumor progression. However, research on EFTUD2 in
cancer is still in its early stages. As a core component of the
spliceosome, EFTUD2 holds significant potential for future
research. Uncovering its molecular functions could provide new
insights into the prevention and treatment of diseases such as
cancer and developmental defects, particularly from the perspective
of RNA splicing.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82004007) and the Zhejiang
Provincial Traditional Chinese Medicine Science and Technology
Program (grant no. 2025ZL234).
Availability of data and materials
Not applicable.
Authors' contributions
AY was responsible for literature collection and
screening, table creation, reviewing the text, data visualization
and writing the original draft. QZ was responsible for literature
collection, literature review and writing the original draft. YC
was responsible for literature data organization and table
creation, figure creation and writing the first draft. JW was
responsible for writing guidance, literature resource acquisition,
funding support, manuscript review and editing. Data authentication
is not applicable. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
EFTUD2
|
elongation factor Tu GTP binding
domain containing 2
|
|
GTP
|
guanosine-5′-triphosphate
|
|
snRNP
|
small nuclear ribonucleoproteins
|
|
MFD
|
mandibulofacial dysostosis
|
|
MFDM
|
mandibulofacial dysostosis with
microcephaly
|
|
EJC
|
exon junction complex
|
|
MX
|
myxovirus resistance
|
|
MyD88
|
myeloid differentiation primary
response 88
|
|
HSP90
|
heat shock protein 90β
|
|
SAMHD1
|
sterile alpha motif and
histidine-aspartate domain-containing protein 1
|
|
OAVS
|
oculo-auriculo-vertebral spectrum
|
|
PFKL
|
phosphofructokinase L
|
|
YTHDF3
|
YTH domain family protein 3
|
|
BRR2
|
small nuclear ribonucleoprotein
U200
|
References
|
1
|
Gilbert W: Why genes in pieces? Nature.
271:5011978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jurica MS and Moore MJ: Pre-mRNA splicing:
Awash in a sea of proteins. Mol Cell. 12:5–14. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Will CL and Luhrmann R: Spliceosome
structure and function. Cold Spring Harb Perspect Biol.
3:a0037072011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boesler C, Rigo N, Anokhina MM, Tauchert
MJ, Agafonov DE, Kastner B, Urlaub H, Ficner R, Will CL and
Lührmann R: A spliceosome intermediate with loosely associated
tri-snRNP accumulates in the absence of Prp28 ATPase activity. Nat
Commun. 7:119972016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saez B, Walter MJ and Graubert TA:
Splicing factor gene mutations in hematologic malignancies. Blood.
129:1260–1269. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Guo R, Zheng L, Park JW, Lv R, Chen H,
Jiao F, Xu W, Mu S, Wen H, Qiu J, et al: BS69/ZMYND11 reads and
connects histone H3.3 lysine 36 trimethylation-decorated chromatin
to regulated pre-mRNA processing. Mol Cell. 56:298–310. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zody MC, Garber M, Adams DJ, Sharpe T,
Harrow J, Lupski JR, Nicholson C, Searle SM, Wilming L, Young SK,
et al: DNA sequence of human chromosome 17 and analysis of
rearrangement in the human lineage. Nature. 440:1045–1049. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zahn-Zabal M, Michel PA, Gateau A, Nikitin
F, Schaeffer M, Audot E, Gaudet P, Duek PD, Teixeira D, Rech de
Laval V, et al: The neXtProt knowledgebase in 2020: Data, tools and
usability improvements. Nucleic Acids Res. 48(D1): D328–D334.
2020.PubMed/NCBI
|
|
9
|
Fagerberg L, Hallstrom BM, Oksvold P,
Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S,
Danielsson A, Edlund K, et al: Analysis of the human
tissue-specific expression by genome-wide integration of
transcriptomics and antibody-based proteomics. Mol Cell Proteomics.
13:397–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Plaschka C, Newman AJ and Nagai K:
Structural basis of nuclear pre-mRNA splicing: Lessons from yeast.
Cold Spring Harb Perspect Biol. 11:a0323912019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Papasaikas P and Valcarcel J: The
spliceosome: The ultimate RNA chaperone and sculptor. Trends
Biochem Sci. 41:33–45. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gozani O, Feld R and Reed R: Evidence that
sequence-independent binding of highly conserved U2 snRNP proteins
upstream of the branch site is required for assembly of
spliceosomal complex A. Genes Dev. 10:233–243. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Misra B, Wagner R and Boneval H: Injuries
of hepatic veins and retrohepatic vena cava. Am Surg. 49:55–60.
1983.PubMed/NCBI
|
|
14
|
Agafonov DE, Kastner B, Dybkov O, Hofele
RV, Liu WT, Urlaub H, Lührmann R and Stark H: Molecular
architecture of the human U4/U6.U5 tri-snRNP. Science.
351:1416–1420. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Laggerbauer B, Achsel T and Luhrmann R:
The human U5-200kD DEXH-box protein unwinds U4/U6 RNA duplices in
vitro. Proc Natl Acad Sci USA. 95:4188–4192. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maeder C, Kutach AK and Guthrie C:
ATP-dependent unwinding of U4/U6 snRNAs by the Brr2 helicase
requires the C terminus of Prp8. Nat Struct Mol Biol. 16:42–48.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Boehm V and Gehring NH: Exon junction
complexes: Supervising the gene expression assembly line. Trends
Genet. 32:724–735. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang X, Yan C, Hang J, Finci LI, Lei J
and Shi Y: An atomic structure of the human spliceosome. Cell.
169:918–929. e142017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park BY, Tachi-Duprat M, Ihewulezi C,
Devotta A and Saint-Jeannet JP: The Core splicing factors EFTUD2,
SNRPB and TXNL4A are essential for neural crest and craniofacial
development. J Dev Biol. 10:292022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Beauchamp MC, Djedid A, Bareke E, Merkuri
F, Aber R, Tam AS, Lines MA, Boycott KM, Stirling PC, Fish JL, et
al: Mutation in Eftud2 causes craniofacial defects in mice via
mis-splicing of Mdm2 and increased P53. Hum Mol Genet. 30:739–757.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Beauchamp MC, Djedid A, Daupin K, Clokie
K, Kumar S, Majewski J and Jerome-Majewska LA: Loss of function
mutation of Eftud2, the gene responsible for mandibulofacial
dysostosis with microcephaly (MFDM), leads to pre-implantation
arrest in mouse. PLoS One. 14:e02192802019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Janeway CA Jr and Medzhitov R: Innate
immune recognition. Annu Rev Immunol. 20:197–216. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
De Arras L, Laws R, Leach SM, Pontis K,
Freedman JH, Schwartz DA and Alper S: Comparative genomics RNAi
screen identifies Eftud2 as a novel regulator of innate immunity.
Genetics. 197:485–496. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
White CR, Dungan M and Carrithers MD:
Activation of human macrophage sodium channels regulates RNA
processing to increase expression of the DNA repair protein
PPP1R10. Immunobiology. 224:80–93. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun J, Li L, Hu J, Gao Y, Song J, Zhang X
and Hu H: Time-course RNA-Seq profiling reveals isoform-level gene
expression dynamics of the cGAS-STING pathway. Comput Struct
Biotechnol J. 20:6490–6500. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang S, Zhao M and Jia S: Macrophage: Key
player in the pathogenesis of autoimmune diseases. Front Immunol.
14:10803102023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: Update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mendoza-Barbera E, Corral-Rodriguez MA,
Soares-Schanoski A, Velarde M, Macieira S, Messerschmidt A,
López-Collazo E and Fuentes-Prior P: Contribution of globular death
domains and unstructured linkers to MyD88.IRAK-4 heterodimer
formation: An explanation for the antagonistic activity of MyD88s.
Biochem Biophys Res Commun. 380:183–187. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu MM and Shu HB: Innate immune response
to cytoplasmic DNA: Mechanisms and diseases. Annu Rev Immunol.
38:79–98. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maelfait J, Bridgeman A, Benlahrech A,
Cursi C and Rehwinkel J: Restriction by SAMHD1 Limits
cGAS/STING-dependent innate and adaptive immune responses to HIV-1.
Cell Rep. 16:1492–1501. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sato S, Li K, Sakurai N, Hashizume M,
Baidya S, Nonaka H, Noguchi K, Ishikawa K, Obuse C and Takaoka A:
Regulation of an adaptor protein STING by Hsp90β to enhance innate
immune responses against microbial infections. Cell Immunol.
356:1041882020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sarkar A, Emrick LT, Smith EM, Austin EG,
Yang Y, Hunter JV, Scaglia F and Lalani SR: Novel de novo mutations
in EFTUD2 detected by exome sequencing in mandibulofacial
dysostosis with Microcephaly syndrome. Am J Med Genet A.
167A:914–918. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smigiel R, Bezniakow N, Jakubiak A, Błoch
M, Patkowski D, Obersztyn E and Sasiadek MM: Phenotype analysis of
Polish patients with mandibulofacial dysostosis type Guion-Almeida
associated with esophageal atresia and choanal atresia caused by
EFTUD2 gene mutations. J Appl Genet. 56:199–204. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Matsuo M, Yamauchi A, Ito Y, Sakauchi M,
Yamamoto T, Okamoto N, Tsurusaki Y, Miyake N, Matsumoto N and Saito
K: Mandibulofacial dysostosis with microcephaly: A case presenting
with seizures. Brain Dev. 39:177–181. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Narumi-Kishimoto Y, Ozawa H, Yanagi K,
Kawai T, Okamura K, Hata K, Kaname T and Matsubara Y: A novel
EFTUD2 mutation identified an adult male with mandibulofacial
dysostosis Guion-Almeida type. Clin Dysmorphol. 29:186–188. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McDermott JH, Study DD and Clayton-Smith
J: Sibling recurrence of total anomalous pulmonary venous drainage.
Eur J Med Genet. 60:265–267. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang J, Ahimaz PR, Hashemifar S, Khlevner
J, Picoraro JA, Middlesworth W, Elfiky MM, Que J, Shen Y and Chung
WK: Novel candidate genes in esophageal atresia/tracheoesophageal
fistula identified by exome sequencing. Eur J Hum Genet.
29:122–130. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Khattar D and Suhrie KR: Esophageal
atresia with or without tracheoesophageal fistula: Comorbidities,
genetic evaluations and neonatal outcomes. Cureus.
15:e347792023.PubMed/NCBI
|
|
39
|
Bukowska-Olech E, Materna-Kiryluk A,
Walczak-Sztulpa J, Popiel D, Badura-Stronka M, Koczyk G, Dawidziuk
A and Jamsheer A: Targeted Next-generation sequencing in the
diagnosis of facial dysostoses. Front Genet. 11:5804772020.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lines MA, Huang L, Schwartzentruber J,
Douglas SL, Lynch DC, Beaulieu C, Guion-Almeida ML, Zechi-Ceide RM,
Gener B, Gillessen-Kaesbach G, et al: Haploinsufficiency of a
spliceosomal GTPase encoded by EFTUD2 causes mandibulofacial
dysostosis with microcephaly. Am J Hum Genet. 90:369–377. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lacour JC, McBride L, St Hilaire H,
Mundinger GS, Moses M, Koon J, Torres JI and Lacassie Y: Novel de
novo EFTUD2 Mutations in 2 Cases With MFDM, initially suspected to
have alternative craniofacial diagnoses. Cleft Palate Craniofac J.
56:674–678. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Luquetti DV, Hing AV, Rieder MJ, Nickerson
DA, Turner EH, Smith J, Park S and Cunningham ML: ‘Mandibulofacial
dysostosis with microcephaly’ caused by EFTUD2 mutations: Expanding
the phenotype. Am J Med Genet A. 161A:108–113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kim SY, Lee DH, Han JH and Choi BY: Novel
splice site pathogenic variant of EFTUD2 is associated with
mandibulofacial dysostosis with microcephaly and extracranial
symptoms in Korea. Diagnostics (Basel). 10:2962020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Voigt C, Megarbane A, Neveling K, Czeschik
JC, Albrecht B, Callewaert B, von Deimling F, Hehr A, Falkenberg
Smeland M, König R, et al: Oto-facial syndrome and esophageal
atresia, intellectual disability and zygomatic anomalies-expanding
the phenotypes associated with EFTUD2 mutations. Orphanet J Rare
Dis. 8:1102013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rengasamy Venugopalan S, Farrow EG and
Lypka M: Whole-exome sequencing identified a variant in EFTUD2 gene
in establishing a genetic diagnosis. Orthod Craniofac Res. 20
(Suppl 1):S50–S56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang M, Sun H, Liu Y and Hu T: Whole exome
sequencing revealed a heterozygous elongation factor Tu GTP-binding
domain containing 2 (EFTUD2) mutation in a couple experiencing
recurrent pregnancy loss. Chin Med J (Engl). 135:1108–1110. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tu M, He L, You Y, Li J, Yao N, Qu C,
Huang W, Xu L, Luo R and Hong J: EFTUD2 maintains the survival of
tumor cells and promotes hepatocellular carcinoma progression via
the activation of STAT3. Cell Death Dis. 11:8302020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lv C, Li XJ, Hao LX, Zhang S, Song Z, Ji
XD and Gong B: Over-activation of EFTUD2 correlates with tumor
propagation and poor survival outcomes in hepatocellular carcinoma.
Clin Transl Oncol. 24:93–103. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zhou R, Ni W, Qin C, Zhou Y, Li Y, Huo J,
Bian L, Zhou A and Li J: A functional loop between YTH domain
family protein YTHDF3 mediated m(6)A modification and
phosphofructokinase PFKL in glycolysis of hepatocellular carcinoma.
J Exp Clin Cancer Res. 41:3342022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Johnson DE, O'Keefe RA and Grandis JR:
Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev
Clin Oncol. 15:234–248. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou W, Chen Y, Luo R, Li Z, Jiang G and
Ou X: Identification of biomarkers related to immune cell
infiltration in hepatocellular carcinoma using gene co-expression
network. Pathol Oncol Res. 27:6016932021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lv Z, Wang Z, Luo L, Chen Y, Han G, Wang
R, Xiao H, Li X, Hou C, Feng J, et al: Spliceosome protein Eftud2
promotes colitis-associated tumorigenesis by modulating
inflammatory response of macrophage. Mucosal Immunol. 12:1164–1173.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fukata M, Chen A, Vamadevan AS, Cohen J,
Breglio K, Krishnareddy S, Hsu D, Xu R, Harpaz N, Dannenberg AJ, et
al: Toll-like receptor-4 promotes the development of
colitis-associated colorectal tumors. Gastroenterology.
133:1869–1881. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Grivennikov S, Karin E, Terzic J, Mucida
D, Yu GY, Vallabhapurapu S, Scheller J, Rose-John S, Cheroutre H,
Eckmann L and Karin M: IL-6 and Stat3 are required for survival of
intestinal epithelial cells and development of colitis-associated
cancer. Cancer Cell. 15:103–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Popivanova BK, Kitamura K, Wu Y, Kondo T,
Kagaya T, Kaneko S, Oshima M, Fujii C and Mukaida N: Blocking
TNF-alpha in mice reduces colorectal carcinogenesis associated with
chronic colitis. J Clin Invest. 118:560–570. 2008.PubMed/NCBI
|
|
56
|
Matsumoto S, Hara T, Mitsuyama K, Yamamoto
M, Tsuruta O, Sata M, Scheller J, Rose-John S, Kado S and Takada T:
Essential roles of IL-6 trans-signaling in colonic epithelial
cells, induced by the IL-6/soluble-IL-6 receptor derived from
lamina propria macrophages, on the development of
colitis-associated premalignant cancer in a murine model. J
Immunol. 184:1543–1551. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ramesh P, Lannagan TRM, Jackstadt R,
Atencia Taboada L, Lansu N, Wirapati P, van Hooff SR, Dekker D,
Pritchard J, Kirov AB, et al: BCL-XL is crucial for progression
through the adenoma-to-carcinoma sequence of colorectal cancer.
Cell Death Differ. 28:3282–3296. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hashimoto K, Nishimura S, Shinyashiki Y,
Ito T and Akagi M: Characterizing inflammatory markers in highly
aggressive soft tissue sarcomas. Medicine (Baltimore).
101:e306882022. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo G, Wang Y, Zhou Y, Quan Q, Zhang Y,
Wang H, Zhang B and Xia L: Immune cell concentrations among the
primary tumor microenvironment in colorectal cancer patients
predicted by clinicopathologic characteristics and blood indexes. J
Immunother Cancer. 7:1792019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yu L, Jiang R, Chen W, Liu Y, Wang G, Gong
X and Wang Y: Novel prognostic indicator combining inflammatory
indicators and tumor markers for gastric cancer. World J Surg
Oncol. 21:502023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sato N, Maeda M, Sugiyama M, Ito S, Hyodo
T, Masuda A, Tsunoda N, Kokuryo T, Hamaguchi M, Nagino M and Senga
T: Inhibition of SNW1 association with spliceosomal proteins
promotes apoptosis in breast cancer cells. Cancer Med. 4:268–277.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Albers M, Diment A, Muraru M, Russell CS
and Beggs JD: Identification and characterization of Prp45p and
Prp46p, essential pre-mRNA splicing factors. RNA. 9:138–150. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Chen F, Wang Q and Zhou Y: The
construction and validation of an RNA binding protein-related
prognostic model for bladder cancer. BMC Cancer. 21:2442021.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Beyer S, Muller L, Mitter S, Keilmann L,
Meister S, Buschmann C, Kraus F, Topalov NE, Czogalla B, Trillsch
F, et al: High RIG-I and EFTUD2 expression predicts poor survival
in endometrial cancer. J Cancer Res Clin Oncol. 149:4293–4303.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Wieczorek D: Human facial dysostoses. Clin
Genet. 83:499–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wood KA, Eadsforth MA, Newman WG and
O'Keefe RT: The Role of the U5 snRNP in genetic disorders and
cancer. Front Genet. 12:6366202021. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Griffin C and Saint-Jeannet JP:
Spliceosomopathies: Diseases and mechanisms. Dev Dyn.
249:1038–1046. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lehalle D, Wieczorek D, Zechi-Ceide RM,
Passos-Bueno MR, Lyonnet S, Amiel J and Gordon CT: A review of
craniofacial disorders caused by spliceosomal defects. Clin Genet.
88:405–415. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Guion-Almeida ML, Zechi-Ceide RM,
Vendramini S and Ju Nior AT: A new syndrome with growth and mental
retardation, mandibulofacial dysostosis, microcephaly and cleft
palate. Clin Dysmorphol. 15:171–174. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Abell K, Hopkin RJ, Bender PL, Jackson F,
Smallwood K, Sullivan B, Stottmann RW, Saal HM and Weaver KN:
Mandibulofacial dysostosis with microcephaly: An expansion of the
phenotype via parental survey. Am J Med Genet A. 185:413–423. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Silva JB, Soares D, Leao M and Santos H:
Mandibulofacial dysostosis with microcephaly: A syndrome to
remember. BMJ Case Rep. 12:e2298312019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yu KPT, Luk HM, Gordon CT, Fung G, Oufadem
M, Garcia-Barcelo MM, Amiel J, Chung BHY, Lo IFM and Tiong YT:
Mandibulofacial dysostosis Guion-Almeida type caused by novel
EFTUD2 splice site variants in two Asian children. Clin Dysmorphol.
27:31–35. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Huang L, Vanstone MR, Hartley T, Osmond M,
Barrowman N, Allanson J, Baker L, Dabir TA, Dipple KM, Dobyns WB,
et al: Mandibulofacial dysostosis with microcephaly: Mutation and
database update. Hum Mutat. 37:148–154. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Vincent M, Genevieve D, Ostertag A, Marlin
S, Lacombe D, Martin-Coignard D, Coubes C, David A, Lyonnet S,
Vilain C, et al: Treacher collins syndrome: A clinical and
molecular study based on a large series of patients. Genet Med.
18:49–56. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Need AC, Shashi V, Hitomi Y, Schoch K,
Shianna KV, McDonald MT, Meisler MH and Goldstein DB: Clinical
application of exome sequencing in undiagnosed genetic conditions.
J Med Genet. 49:353–361. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Park CY, Zhou J, Wong AK, Chen KM,
Theesfeld CL, Darnell RB and Troyanskaya OG: Genome-wide landscape
of RNA-binding protein target site dysregulation reveals a major
impact on psychiatric disorder risk. Nat Genet. 53:166–173. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Santiago JA and Potashkin JA: Blood
biomarkers associated with cognitive decline in early stage and
drug-naive Parkinson's disease patients. PLoS One. 10:e01425822015.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang S, Sun D, Liu C, Guo Y, Ma J, Ge RL
and Cui S: Weighted gene co-expression network analysis reveals the
hub genes associated with pulmonary hypertension. Exp Biol Med
(Maywood). 248:217–231. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu L, Pertsemlidis A, Ding LH, Story MD,
Steinberg MH, Sebastiani P, Hoppe C, Ballas SK and Pace BS:
Original research: A case-control genome-wide association study
identifies genetic modifiers of fetal hemoglobin in sickle cell
disease. Exp Biol Med (Maywood). 241:706–718. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Gordon CT, Petit F, Oufadem M,
Decaestecker C, Jourdain AS, Andrieux J, Malan V, Alessandri JL,
Baujat G, Baumann C, et al: EFTUD2 haploinsufficiency leads to
syndromic oesophageal atresia. J Med Genet. 49:737–746. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Heidarzadehpilehrood R, Pirhoushiaran M,
Binti Osman M, Abdul Hamid H and Ling KH: Weighted gene
co-expression network analysis (WGCNA) Discovered novel long
non-coding RNAs for polycystic ovary syndrome. Biomedicines.
11:5182023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hou Y, Wang Y, Xu S, Qi G and Wu X:
Bioinformatics identification of microRNAs involved in polycystic
ovary syndrome based on microarray data. Mol Med Rep. 20:281–291.
2019.PubMed/NCBI
|
|
83
|
Li L and Chen S: Screening, identification
and interaction analysis of key MicroRNAs and genes in
Asthenozoospermia. Int J Med Sci. 18:1670–1679. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Czeschik JC, Voigt C, Alanay Y, Albrecht
B, Avci S, Fitzpatrick D, Goudie DR, Hehr U, Hoogeboom AJ,
Kayserili H, et al: Clinical and mutation data in 12 patients with
the clinical diagnosis of Nager syndrome. Hum Genet. 132:885–898.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zarate YA, Bell C and Schaefer GB:
Radioulnar synostosis and brain abnormalities in a patient with
17q21.31 microdeletion involving EFTUD2. Cleft Palate Craniofac J.
52:237–239. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zech M, Boesch S, Skorvanek M, Necpál J,
Švantnerová J, Wagner M, Dincer Y, Sadr-Nabavi A, Serranová T,
Rektorová I, et al: Clinically relevant copy-number variants in
exome sequencing data of patients with dystonia. Parkinsonism Relat
Disord. 84:129–134. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Tian A, Li Y, Fan H, Hu P, Xu R, Yuan H,
Cai J, Zhang W, Yue M, Li J, et al: Association of elongation
factor Tu GTP-binding Domain-containing 2 Gene (EFTUD2)
polymorphism with the risk of hepatitis B virus infection. Immunol
Invest. 51:1485–1497. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hu P, Li Y, Zhang W, Liu R, Peng L, Xu R,
Cai J, Yuan H, Feng T, Tian A, et al: The spliceosome factor EFTUD2
promotes IFN Anti-HBV effect through mRNA splicing. Mediators
Inflamm. 2023:25462782023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Sumpter R Jr, Loo YM, Foy E, Li K,
Yoneyama M, Fujita T, Lemon SM and Gale M Jr: Regulating
intracellular antiviral defense and permissiveness to hepatitis C
virus RNA replication through a cellular RNA helicase, RIG-I. J
Virol. 79:2689–2699. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wang N, Liang Y, Devaraj S, Wang J, Lemon
SM and Li K: Toll-like receptor 3 mediates establishment of an
antiviral state against hepatitis C virus in hepatoma cells. J
Virol. 83:9824–9834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhu C, Xiao F, Hong J, Wang K, Liu X, Cai
D, Fusco DN, Zhao L, Jeong SW, Brisac C, et al: EFTUD2 is a novel
innate immune regulator restricting hepatitis C virus infection
through the RIG-I/MDA5 pathway. J Virol. 89:6608–6618. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Metz P, Reuter A, Bender S and
Bartenschlager R: Interferon-stimulated genes and their role in
controlling hepatitis C virus. J Hepatol. 59:1331–1341. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Boudreault S, Lemay G and Bisaillon M: U5
snRNP core proteins are key components of the defense response
against viral infection through their roles in programmed cell
death and interferon induction. Viruses. 14:27102022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Kawai T, Takahashi K, Sato S, Coban C,
Kumar H, Kato H, Ishii KJ, Takeuchi O and Akira S: IPS-1, an
adaptor triggering RIG-I- and Mda5-mediated type I interferon
induction. Nat Immunol. 6:981–988. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Lau DT, Fish PM, Sinha M, Owen DM, Lemon
SM and Gale M Jr: Interferon regulatory factor-3 activation,
hepatic interferon-stimulated gene expression and immune cell
infiltration in hepatitis C virus patients. Hepatology. 47:799–809.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Schoggins JW, Wilson SJ, Panis M, Murphy
MY, Jones CT, Bieniasz P and Rice CM: A diverse range of gene
products are effectors of the type I interferon antiviral response.
Nature. 472:481–485. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chi Q, Geng X, Xu K, Wang C and Zhao H:
Potential targets and molecular mechanism of miR-331-3p in
hepatocellular carcinoma identified by weighted gene coexpression
network analysis. Biosci Rep. 40:BSR202001242020. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhang ZG, Shi ZD, Dong JJ, Chen YA, Cao
MY, Li YT, Ma WM, Hao L, Pang K, Zhou JH, et al: Novel potential
urinary biomarkers for effective diagnosis and prognostic
evaluation of high-grade bladder cancer. Transl Cancer Res.
12:1992–2007. 2023. View Article : Google Scholar : PubMed/NCBI
|