Introduction
Systemic Epstein-Barr virus (EBV)-positive T-cell
lymphoproliferative disease (LPD) of childhood is an extremely rare
and distinct clinicopathological entity, which has recently been
categorized into the spectrum of EBV-related T-cell
lymphoproliferative disorders of childhood according to the World
Health Organization (WHO) classification (1). The majority of these cases usually
occur with an apparent primary EBV infection, but may occasionally
be associated with severe chronic active EBV infection in children
or young adults, with an aggressive clinical course (1–3). In
this study, we describe a case of systemic EBV-positive T-cell LPD
in a 23-year-old female with primary EBV infection. We identify the
cytological features of the peripheral blood and review the
clinicopathological features of this disease.
Case report
A previously healthy 23-year-old female without an
immunocompromised status presented with an acute onset of high
fever at an outpatient clinic. Laboratory examinations revealed a
number of atypical lymphocytes within the peripheral blood,
elevated liver and renal function and disseminated intravascular
coagulation syndrome. The patient was referred to the Department of
Hematology at Shiga University of Medical Science, Shiga, Japan.
This study was approved by the Ethics Committee at Shiga University
of Medical Science. Informed consent was obtained from the patient
prior to the study.
A physical examination revealed hepatosplenomegaly,
hemorrhage in the bilateral conjunctiva, mild lymphadenopathy of
the cervical lymph nodes, and pleural effusion and ascites.
Laboratory examinations demonstrated markedly elevated white blood
cells (61,800 cells/μl; range, 3,000–8,000), activated
partial thromboplastin time (91.9 sec; range, 25–40), D-dimer (68.8
μg/ml; <1.0), aspartate aminotransferase (11,376 U/l;
range, 13–33), alanine aminotransferase (2,254 U/l; range, 6–27),
lactate dehydrogenase (19,179 U/l; range, 119–229), blood urea
nitrogen (65.2 mg/dl; range, 8–22), creatinine (3.37 mg/dl; range,
0.4–0.7) and soluble IL-2 receptor levels (51,500 U/ml; range,
124–466). The patient’s red blood cell count (4.39×104
cells/μl; range, 3.8–4.8) was within the normal range, while
the platelet concentration was decreased (64×103
platelets/μl; range, 150–400). Additionally, the patient’s
EBV-anti-VCA-IgG antibody titer was increased 160-fold, and
anti-VCA-IgM, anti-VCA-IgA, anti-EA-DR-IgG, anti-EA-DR-IgA and
anti-EBNA antibody titers were less than 10-fold. EBV-DNA was
detected at 2.8×106 copies/106 cells in the
patient’s serum.
Peripheral blood smears stained with Giemsa were
taken. These demonstrated a number of atypical lymphocytes, which
had large irregular-shaped nuclei with lobated and moderately
coarse chromatin and inconspicuous nucleoli. They also revealed an
abundant radial or peripheral basophilic cytoplasm with small
azurophilic granules (Fig. 1). Flow
cytometry of the peripheral blood demonstrated a marked increase
(96%) of HLA-DR+ CD8+ T lymphocytes.
Formalin-fixed, paraffin-embedded tissue blocks of
the bone marrow and liver were cut into 3-μm sections,
deparaffinized and rehydrated. Each section was stained with
hematoxylin and eosin (H&E) and then used for immunostaining
and in situ hybridization. Immunostaining and in situ
hybridization were conducted using an autostainer (BenchMark XT
system; Ventana Medical System Inc., Tucson, AZ, USA) according to
the manufacturer’s instructions. The primary antibodies used were:
rabbit monoclonal antibody against CD4 (SP35; Ventana Medical
Systems Inc.) and mouse monoclonal antibodies against β F1 (8A3;
Thermo Scientific, Waltham, MA, USA), CD3 (PS1; Novocastra
Laboratories, Ltd., Newcastle upon Tyne, UK), CD8 (1A5; Novocastra
Laboratories), CD20 (L26; Novocastra Laboratories), CD56 (CD564;
Novocastra Laboratories), granzyme B (11F; Novocastra Laboratories)
and TIA-1 (TIA-1; GeneTex Inc., Irvine, CA, USA). For in
situ hybridization, an INFORM EBV-encoded small RNA (EBER) 1
probe (Ventana Medical Systems Inc.) was used.
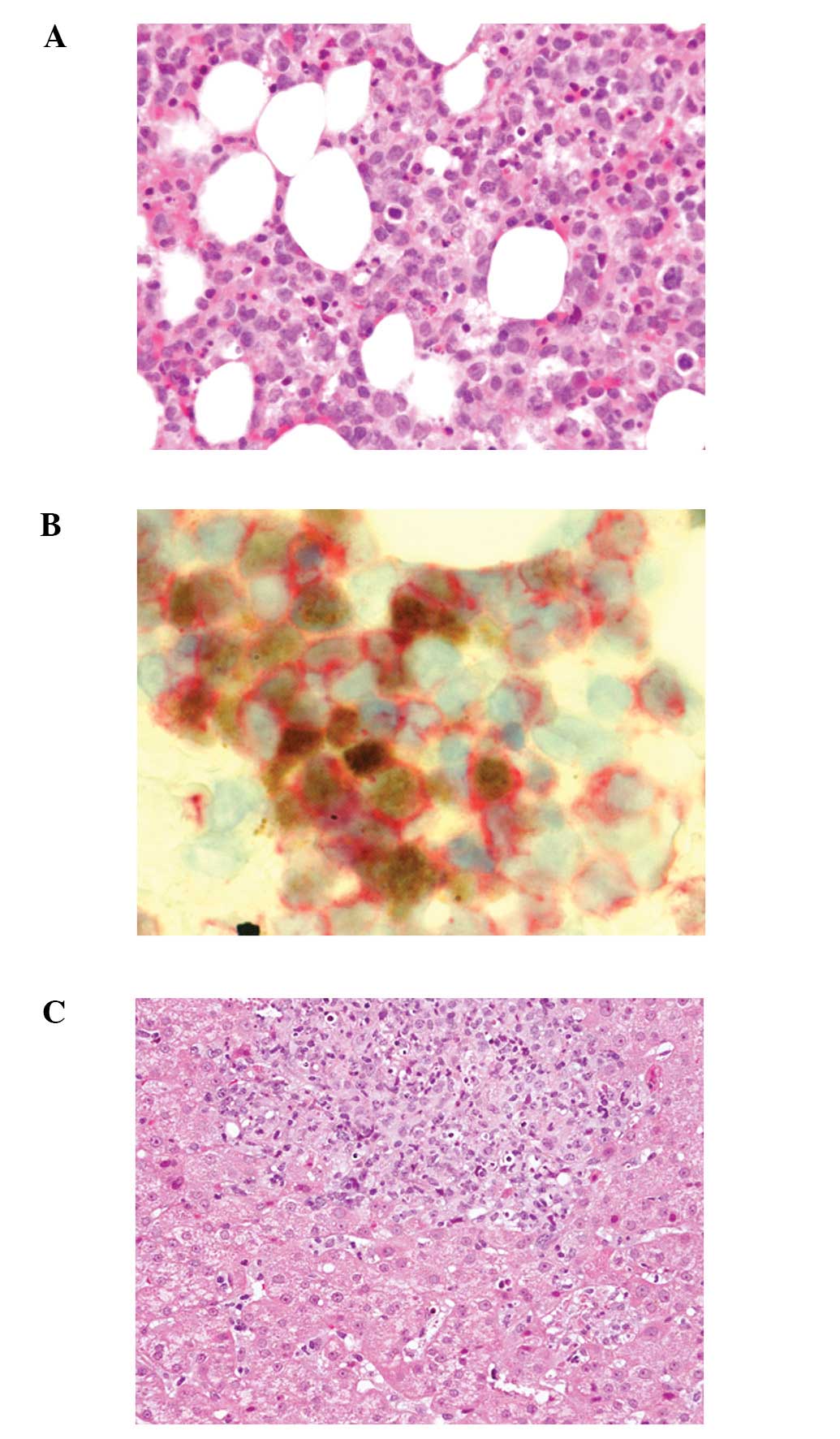
Bone marrow aspiration revealed marked proliferation
of small-sized lymphocytes with slightly enlarged and convoluted
nuclei, inconspicuous nucleoli (Fig.
2A) and hemophagocytosis. In situ hybridization for EBV
using the EBER1 probe demonstrated marked positivity in the
majority of small-sized lymphocytes. Double staining for EBER1 and
CD3 or CD8 revealed that these small lymphocytes expressed both
EBER1 and CD3 or CD8 (Fig. 2B).
Furthermore, these lymphocytes also expressed β F1, granzyme B and
TIA-1, but were negative for CD4 and CD56.
A liver biopsy demonstrated proliferation of
small-sized lymphocytes in the portal area with sinusoidal
infiltration (Fig. 2C); these
lymphocytes were positive for CD3, CD8, β F1, granzyme B, TIA-1 and
EBER1.
An analysis of T-cell receptor (TCR) gene
rearrangement was conducted using southern blot analysis. The
results demonstrated monoclonal rearrangements of the TCR β chain
Cβ and γ chain Jγ regions. EBV DNA was also detected in the
atypical lymphocytes of the peripheral blood. Subsequently, an
ultimate diagnosis of systemic EBV-positive T-cell LPD of childhood
was confirmed.
The patient underwent continuous hemodiafiltration,
and was initially administered etoposide, cyclosporine and
prednisolone, followed by cyclophosphamide, hydroxydaunorubicin,
oncovin and prednisolone (CHOP) therapy. The patient succumbed to
multiorgan failure 20 weeks after chemotherapy.
Discussion
More than 90% of EBV-associated LPD is of B-cell
origin and occurs in the setting of immunosuppression or following
an organ transplant. However, EBV may also infect T lymphocytes.
Two major types of EBV-positive T-cell LPDs of childhood have been
defined by WHO classification. The first is systemic EBV-positive
T-cell LPD of childhood, and the second is hydroa vacciniforme-like
lymphoma demonstrating an indolent clinical course (1,2).
Systemic EBV-positive T-cell LPD of childhood following acute EBV
infection is extremely rare. The majority of previous studies have
reported single cases; however, the study by Quintanilla-Martinez
et al described the clinicopathological features of 5 cases
of this disease (2). In this study,
we reviewed the clinicopathological features of 17 cases with
systemic EBV-positive T-cell LPD of childhood following acute EBV
infection, including the present case (Table I) (2,4–12). We
revealed that clonal systemic proliferation of EBV-infected T-cells
that appear morphologically innocuous with an activated cytotoxic
phenotype expressing CD8 and/or CD4 is central to the disease. We
also identified that there is a high prevalence in the Asian
population (11/15 cases), as well as in children and young adults
(median age, 12.7 years; range, 0–45 years); with the exception of
a recently reported case of a 45-year-old patient who complained of
chronic diarrhea (4). There also
appears to be a predilection for males (male to female ratio,11:6).
Our group also revealed that the most commonly involved sites are
the liver, spleen, lymph node and bone marrow, and the main
clinical presentations are hepatosplenomegaly, fever and
pancytopenia. Finally, we identified that almost all cases have an
aggressive clinical course, which results in mortality (2,4–12).
 | Table IClinicopathological features of
systemic EBV-positive T-cell LPD following primary EBV
infection. |
Table I
Clinicopathological features of
systemic EBV-positive T-cell LPD following primary EBV
infection.
| Case No. | Age (years) | Gender | Ethnic origin | Clinical
presentation | Phenotype | TCR status | Outcome | Refs. |
|---|
| 1 | 13 | M | Asian | Fever,
hepatosplenomegaly, jaundice, lymphadenopathy | CD8+ | βR | DOD | 4 |
| 2 | 7 | M | Asian | Fever,
hepatosplenomegaly, lymphadenopathy | CD8+ | βR | DOD | 4 |
| 3 | 2 | M | Asian | IM followed by
hepatic failure, pancytopenia, sepsis | CD8+ | NA | DOD | 5 |
| 4 | 16 | F | NA | IM followed by fever,
lymphadenopathy, VAHS | CD8+ | β and γR | DOD | 6 |
| 5 | 3 | F | Asian | Fever,
hepatosplenomegaly, lymphadenopathy | CD8+ | βR | DOD | 7 |
| 6 | 20 months | F | NA | Fever,
hepatosplenomegaly, skin rash | NA | βR | DOD | 8 |
| 7 | 1 | M | Asian | Fever,
hepatosplenomegaly, lymphadenopathy | CD8+ | NA | DOD | 9 |
| 8 | 1 | F | Asian | Fever,
hepatosplenomegaly, pancytopenia | CD4+ and
CD8+ | β and γR | Relapse 15
months | 10 |
| 9 | 5 | M | Asian | Fever,
hepatosplenomegaly, pancytopenia, VAHS | NA | β and γG | DOD | 11 |
| 10 | 1 | M | Asian | Fever,
hepatosplenomegaly, pancytopenia, VAHS | NA | β and γG | DOD | 11 |
| 11 | 37 | M | Caucasian | Fever,
hepatosplenomegaly, jaundice, pancytopenia | CD4+ | γR | DOD | 2 |
| 12 | 17 | M | Mexican | Hepatosplenomegaly,
symptoms of upper respiratory infection, jaundice,
pancytopenia | CD8+ | γR | DOD | 2 |
| 13 | 23 | M | Asian | Fever,
hepatosplenomegaly, jaundice, pancytopenia, lymphadenopathy | CD4+ and
CD8+ | γ R | DOD | 2 |
| 14 | 22 | F | Mexican | Fever,
hepatosplenomegaly, jaundice | CD4+ | γ G | NA | 2 |
| 15 | 27 months | M | Mexican | Fever,
hepatosplenomegaly, skin rash, pancytopenia | CD8+ | γ R | DOD | 2 |
| 16 | 45 | M | Asian | Diarrhea, weight
loss | NA | γ G | DOD | 12 |
| Present case | 23 | F | Asian | Fever,
hepatosplenomegaly, DIC, liver and renal failure | CD8+ | β and γ R | DOD | |
Ohshima et al categorized EBV-associated
T/NK-cell LPD in children and young adults into the following
groups; A1 (polymorphic and polyclonal), A2 (polymorphic and
generally monoclonal), A3 (monomorphic and monoclonal proliferation
of T or NK-cell origin) and B (monomorphic and monoclonal T-cell
LPD with an aggressive clinical course) (13). Group B is equivalent to systemic
EBV-positive T-cell LPD of childhood according to the WHO
classification, and corresponds to the case presented in this study
(13).
Cytological atypia of this disease is minimal
(1,2,12) and
has been demonstrated to delay diagnosis (12). In the present case, cytological
atypia of the proliferative lymphocytes was minimal; however,
double staining for EBER1 and CD3 or CD8 was useful for diagnosing
systemic EBV-positive T-cell LPD of childhood. The cytological
features of the atypical lymphocytes in the peripheral blood smear
were also analyzed in this case report, and it was identified that
the features observed in the present case were indistinguishable
from those of infectious mononucleosis (14).
In conclusion, we reviewed the clinicopathological
features of systemic EBV-positive T-cell LPD of childhood following
acute EBV infection in order to broaden our knowledge of this
disease and facilitate diagnosis. Cytological atypia of neoplastic
cells in this disease is minimal; however, double staining for
EBER1 and CD3 or CD8 is useful for diagnosis. Furthermore, the
cytomorphological features of atypical lymphocytes in the
peripheral blood are indistinguishable from those of infectous
mononucleosis. Since the clinical course of systemic EBV-positive
T-cell LPD of childhood is aggressive, further studies are required
to clarify early and accurate diagnostic and therapeutic strategies
for this disease.
References
|
1
|
Quintanilla-Martinez L, Kimura H and Jaffe
ES: EBV-positive T-cell lymphoproliferative disorders of childhood.
WHO Classification of Tumours of Haematopoietic and Lymphoid
Tissues. Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA,
Stein H, Thiele J and Vardiman JW: 4th edition. IARC Press; Lyon:
pp. 278–280. 2008
|
|
2
|
Quintanilla-Martinez L, Kumar S, Fend F,
et al: Fulminant EBV(+) T-cell lymphoproliferative disorder
following acute/chronic EBV infection: a distinct clinicopathologic
syndrome. Blood. 96:443–451. 2000.
|
|
3
|
Cohen JI, Kimura H, Nakamura S, Ko YH and
Jaffe ES: Epstein-Barr virus-associated lymphoproliferative disease
in non-immunocompromised hosts: a status report and summary of an
international meeting, 8–9 September 2008. Ann Oncol. 20:1472–1482.
2009.PubMed/NCBI
|
|
4
|
Su IJ, Lin KH, Chen CJ, et al:
Epstein-Barr virus-associated peripheral T-cell lymphoma of
activated CD8 phenotype. Cancer. 66:2557–2562. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mori M, Kurozumi H, Akagi K, Tanaka Y,
Imai S and Osato T: Monoclonal proliferation of T cells containing
Epstein-Barr virus in fatal mononucleosis. N Eng J Med. 327:581992.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gaillard F, Mechinaud-Lacroix F, Papin S,
et al: Primary Epstein-Barr virus infection with clonal T-cell
lymphoproliferation. Am J Clin Pathol. 98:324–333. 1992.PubMed/NCBI
|
|
7
|
Chan LC, Srivastava G, Pittaluga S, Kwong
YL, Liu HW and Yuen HL: Detection of clonal Epstein-Barr virus in
malignant proliferation of peripheral blood CD3+
CD8+ T cells. Leukemia. 6:952–956. 1992.PubMed/NCBI
|
|
8
|
Craig FE, Clare CN, Sklar JL and Banks PM:
T-cell lymphoma and the virus-associated hemophagocytic syndrome.
Am J Clin Pathol. 97:189–194. 1992.PubMed/NCBI
|
|
9
|
Tazawa Y, Nishinomiya F, Noguchi H, et al:
A case of fatal infectious mononucleosis presenting with fulminant
hepatic failure associated with an extensive CD8-positive
lymphocyte infiltration in the liver. Hum Pathol. 24:1135–1139.
1993. View Article : Google Scholar
|
|
10
|
Noma T, Kou K, Yoshizawa I, et al:
Monoclonal proliferation of Epstein-Barr virus-infected T-cells in
a patient with virus-associated haemophagocytic syndrome. Eur J
Pediatr. 153:734–738. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kawaguchi H, Miyashita T, Herbst H, et al:
Epstein-Barr virus-infected T lymphocytes in Epstein-Barr
virus-associated hemophagocytic syndrome. J Clin Invest.
92:1444–1450. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abdul-Ghafar J, Kim JW, Park KH and Cho
MY: Fulminant Epstein-Barr virus-associated T-cell
lymphoproliferative disorder in an immunocompetent middle-aged man
presenting with chronic diarrhea and gastrointestinal bleeding. J
Korean Med Sci. 26:1103–1107. 2011. View Article : Google Scholar
|
|
13
|
Ohshima K, Kimura H, Yoshino T, et al:
Proposed categorization of pathological states of EBV-associated
T/natural killer-cell lymphoproliferative disorder (LPD) in
children and young adults: overlap with chronic active EBV
infection and infantile fulminant EBV T-LPD. Pathol Int.
58:209–217. 2008. View Article : Google Scholar
|
|
14
|
Foucar K, Viswanatha DS and Wilson CA:
Infectious mononucleosis (Epstein-Barr virus). Non-Neoplastic
Disorders of Bone Marrow. Foucar K, Viswanatha DS and Wilson CA:
ARP Press; Washington, DC: pp. 338–341. 2008
|