Introduction
The identification of cancer-associated genes is
crucial for understanding the molecular mechanisms that are
involved in tumorigenesis and tumor development. These genes may
contribute to or drive cancer development by their involvement in
the functions or pathways that allow cancer cells to evade growth
control, metastasis or invasion (1).
In the past decade, a number of cancer-associated
genes have been identified using rapidly evolving biotechnologies,
particularly high-throughput techniques, including gene expression
microarrays and proteomics (2). For
example, known and novel breast cancer genes have been identified
using microarray-based comparative genomic hybridization (3). Colorectal cancer-associated genes have
also been identified using combined computational methods (4) and deep transcriptome sequencing
(5). Certain genes are well-known
biomarkers in particular cancers, including BRCA1 and BRCA2 in
breast cancer (6), PSA in prostate
cancer (7) and WFDC2 in ovarian
cancer (8). The associated
biological functions of these genes provide clues for the research
into cancer biology. However, the few cancer biomarkers that have
identified do not act as individual units to perform biological
functions. Through the development of sequencing techniques,
integrated omics profiles provide the opportunity to identify
additional cancer genes. Therefore, it is necessary to understand
the mechanisms underlying tumorigenesis at an integrative
level.
Public resources, including the public (GenBank,
UniGene, Swiss_Prot) and ontological [Gene Ontology,(GO) consortium
and Kyoto Encyclopedia of Genes and Genomes, (KEGG)] databases,
currently offer complementary information for cancer gene
identification (9). Furthermore,
the increasing omics studies of human cancer have produced useful
datasets, resulting in the establishment of cancer-associated
databases. The Cancer Genome Anatomy Project (CGAP) aims to
determine the gene expression profiles of normal, precancerous and
cancerous cells (http://cgap.nci.nih.gov). The Cancer Genome Project
uses human genome sequencing in order to identify the genes that
are involved in the development of human cancer (http://www.sanger.ac.uk/genetics/CGP).
The Cancer Genome Atlas (TCGA) accelerates our understanding of the
molecular basis of cancer (http://cancergenome.nih.gov). Other derived cancer
databases are also publicly available.
Among these online resources, the UniGene database
(NCBI GeneBank; http://www.ncbi.nlm.nih.gov/unigene) allows the use of
Digital Differential Display (DDD, http://www.ncbi.nlm.nih.gov/UniGene/ddd.cgi) for the
rapid identification of genes whose expressions are altered between
tissue types. The UniGene database includes expressed sequences
from diverse species-, organ- and disease-derived cDNA libraries.
The availability of various human cancer libraries in the UniGene
database provides a platform for the rapid identification of
selectively-expressed cancer genes.
The present study compares the expressed sequence
tags (ESTs) that were identified in 15 types of cancer using the
DDD tool. The differentially-expressed genes were bioinformatically
evaluated for potential biological functions and pathways, and
their expressions were evaluated in the human epididymis. These
genes may be involved in regulating the initiation, development and
progression of cancer, and may provide potential targets or markers
for cancer prognosis, diagnosis, prevention and treatment.
Materials and methods
DDD analysis
The DDD tool was used to screen the
selectively-expressed genes in various cancer types using the
UniGene database to compare the number of times that sequences from
the libraries were assigned to a particular UniGene cluster. The
selected 15 types of human cancer libraries were for adrenal,
bladder, breast, cervical, colorectal, esophageal, germ cell,
glioma, head and neck, kidney, liver, lung, ovarian, pancreatic
tumor and prostate cancer. The statistically significant
differences in the EST counts between the cancer groups were
determined using the Fisher’s exact test. P<0.05 was considered
to indicate a statistically significant difference.
Gene ontological analysis
All selectively-expressed cancer genes were broadly
classified into several catalogs according to the GO annotation
(www.geneontology.com) and the functions reported
in the literature.
Enrichment bioinformatics analysis
The protein identifiers (IDs) were uploaded to the
Database for Annotation, Visualization and Integrated Discovery
(DAVID; http://david.abcc.ncifcrf.gov) and
the enrichment analyses of the GO terms, including the biological
process, molecular function and cellular component, were performed
using the functional clustering annotation tools. The default
options with a high classification stringency were used. Finally,
the cluster names were extracted from the most biologically
relevant GO term that was assigned to that cluster.
Pathway analysis
Ingenuity pathway analysis v9.0 (IPA; www.ingenuity.com; Ingenuity Systems, Redwood City,
CA, USA) was used to analyze the pathways and networks that the
cancer-associated proteins were involved in. The following settings
were used: Reference set, ingenuity knowledge base (genes only);
network analysis, direct and indirect relationships; 35 molecules
per network; and 25 networks used per analysis. For all species,
tissues and cell lines were used for the analysis. The IPA used the
Fisher’s exact test to determine the significant pathways linked to
the input protein set compared with the whole ingenuity knowledge
base.
Further characteristics of
cancer-associated genes compared with the published literature
Highly-expressed human testicular and epididymal
genes from a previous study (10–12)
were compared with the present data. The overlapping epididymal
genes were further characterized using microarray annotation
(http://humanet.scbit.org/index.jsp).
Secretory proteins and cell surface proteins are considered to be
promising biomarkers. All cancer-associated proteins were compared
with the serum/plasma proteome (13) in order to select the secretory
proteins, and were also compared with the cell surfaceome (14) to select the cell surface proteins. A
GO annotation was used to further filter the results.
Results
Summary of selectively-expressed genes in
human cancers
The statistical comparison of the ESTs among the
various human cancer libraries was performed using the DDD tool. A
total of 15 human cancer types and 208 libraries(1,196,901 ESTs)
were selected (Table I). The genes
that were significantly expressed in the various cancer types were
termed the selectively-expressed cancer genes, and those that were
uniquely expressed in one given cancer type were termed the
uniquely-expressed cancer genes. A total of 1,904
selectively-expressed human cancer genes were identified in the 15
cancer types, of which 274 were uniquely-expressed cancer genes
(Fig. 1).
 | Table ISummary of libraries used in the
present study. |
Table I
Summary of libraries used in the
present study.
| Cancer type | Libraries | ESTs |
|---|
| Adrenal tumor | 5 | 12384 |
| Bladder
carcinoma | 2 | 8752 |
| Breast tumor | 16 | 88917 |
| Cervical tumor | 5 | 38138 |
| Colorectal tumor | 24 | 87951 |
| Esophageal tumor | 4 | 8672 |
| Germ cell tumor | 26 | 259108 |
| Glioma | 15 | 99668 |
| Head and neck
tumor | 24 | 81627 |
| Kidney tumor | 13 | 68473 |
| Liver tumor | 24 | 102783 |
| Lung tumor | 15 | 139981 |
| Ovarian tumor | 15 | 61470 |
| Pancreatic tumor | 9 | 90266 |
| Prostate cancer | 11 | 48711 |
Bioinformatics analysis
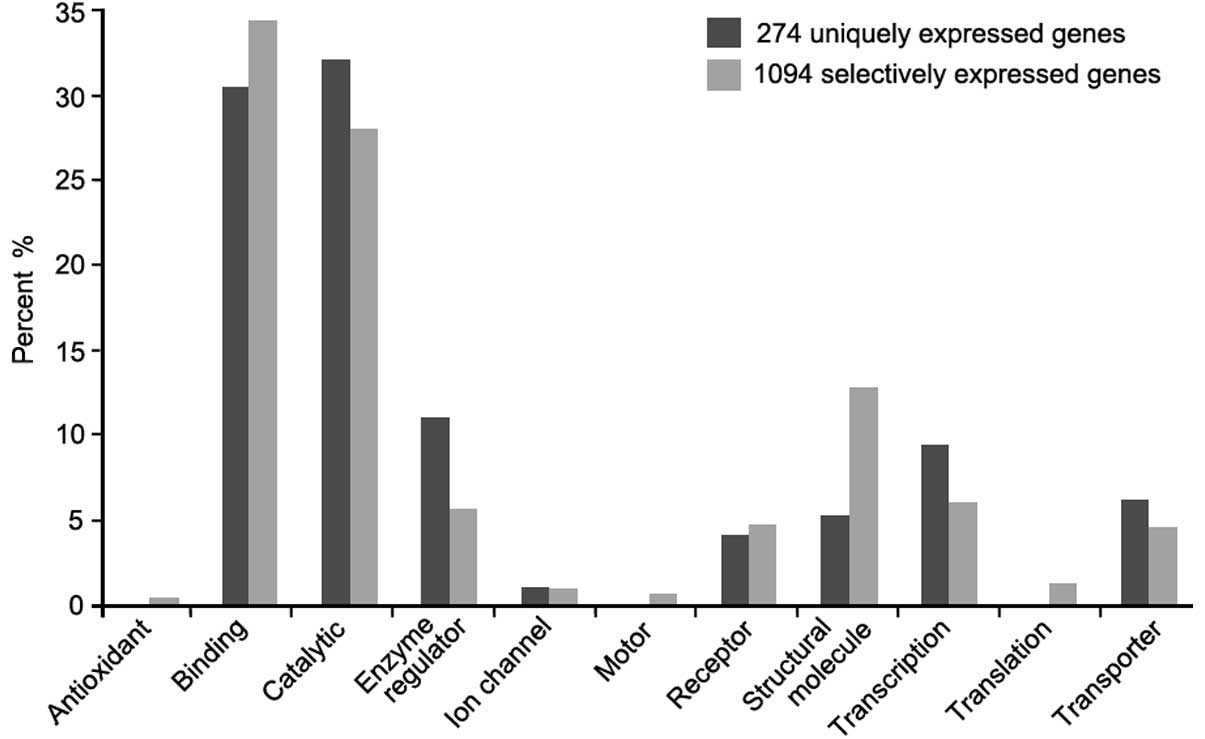
The functional classification analysis showed that
the majority of the selectively-expressed cancer genes were
associated with binding (34%) and catalytic (27%) activity. Within
the 274 uniquely-expressed cancer genes, non-related proteins were
associated with antioxidant, motor or translation regulator
activity functions (Fig. 2).
The domain analysis using the DAVID functional
annotation tool from the PIR-superfamily classification system
identified 13 and nine statistically significant domains in the
selectively- and uniquely-expressed cancer genes, respectively. The
common domains among the genes were cytochrome P450 CYP2D6
(PIRSF000045), serpin (PIRSF001630) and apolipoprotein A-I
(PIRSF002367).
To map the major functional categories, all
selectively-expressed cancer genes were grouped into several
functional clusters using the functional annotation clustering tool
(DAVID). The analysis revealed seven major functional clusters,
including i) ribosome, ii) extracellular region, iii) cell
adhesion, iv) acute-phase response, v) peptidase inhibitor
activity, vi) regulation of cell death and vii) vasculature
development.
An analysis of the pathway and the network was
performed using IPA. A total of 21 statistically significant
pathways were present in the selectively-expressed cancer genes
(Table II) and 25 networks were
generated. Certain functions were linked to more than three of the
25 networks and mainly included cancer development, cell death, the
cell cycle, genetic disorder formation and cellular assembly and
organization. The cancer category consists of numerous
subcategories of which tumorigenesis is the largest.
| Table IIEnriched pathways associated with
selectively-expressed cancer genes. |
Table II
Enriched pathways associated with
selectively-expressed cancer genes.
| Pathwaysa | EST count | Percentage | P-value |
|---|
| hsa03010:
Ribosome | 76 | 4.33 |
1.50×10−53 |
| hsa04610: Complement
and coagulation cascades | 33 | 1.88 |
3.60×10−11 |
| hsa04512:
ECM-receptor interaction | 28 | 1.59 |
9.53×10−06 |
| hsa00010:
Glycolysis/Gluconeogenesis | 22 | 1.25 |
2.21×10−05 |
| hsa00360:
Phenylalanine metabolism | 12 | 0.68 |
4.83×10−05 |
| hsa04612: Antigen
processing and presentation | 23 | 1.31 |
1.31×10−03 |
| hsa04510: Focal
adhesion | 43 | 2.45 |
2.60×10−03 |
| hsa00982: Drug
metabolism | 18 | 1.02 |
3.06×10−03 |
| hsa04115: p53
signaling pathway | 19 | 1.08 |
3.55×10−03 |
| hsa03320: PPAR
signaling pathway | 19 | 1.08 |
4.21×10−03 |
| hsa05010: Alzheimer’s
disease | 34 | 1.94 |
1.16×10−02 |
| hsa00350: Tyrosine
metabolism | 13 | 0.74 |
1.25×10−02 |
| hsa00480:
Glutathione metabolism | 14 | 0.80 |
1.44×10−02 |
| hsa00030: Pentose
phosphate pathway | 9 | 0.51 |
1.49×10−02 |
| hsa00190: Oxidative
phosphorylation | 28 | 1.59 |
1.54×10−02 |
| hsa05130:
Pathogenic Escherichia coli infection | 15 | 0.85 |
1.87×10−02 |
| hsa03050:
Proteasome | 13 | 0.74 |
2.11×10−02 |
| hsa04530: Tight
junction | 28 | 1.59 |
2.24×10−02 |
| hsa05012:
Parkinson’s disease | 27 | 1.54 |
2.25×10−02 |
| hsa05416: Viral
myocarditis | 17 | 0.97 |
2.74×10−02 |
| hsa05215: Prostate
cancer | 20 | 1.14 |
2.94×10−02 |
Comparison analysis
Certain genes that are highly-expressed in the
testes or epididymis are promising markers for cancer (15,16).
Of the 1,904 selectively-expressed genes, 52 were highly-expressed
in the human testes and 30 in the epididymis. Of the 274
uniquely-expressed genes, 15 were highly-expressed in the human
testis and 22 in the epididymis. A total of 360 secretory and 272
surface proteins were identified as potential biomarkers that
corresponded with the selectively-expressed cancer genes.
Characteristics of highly-expressed
cancer genes in the human epididymis
Of the 274 highly-expressed genes in the human
epdidymis, 22 exhibited distinct spatial and temporal expression
patterns (Table III). According
to the microarray data analysis (http://humanet.scbit.org/index.jsp), three genes were
highly expressed in the caput epididiymis, seven in the corpus
epididymis and three in the cauda epididymis. Seven genes showed
higher expression levels in the aged males.
| Table IIISpatial and temporal expression of 22
genes that were highly-expressed in the human epididymis. |
Table III
Spatial and temporal expression of 22
genes that were highly-expressed in the human epididymis.
| Gene symbol | Accession no. | Spatial
expression | Temporal
expression | Broad
functions |
|---|
|
|
|---|
| Caput | Corpus | Cauda | Newborn | Young | Aged |
|---|
| ALDH3B2 | NM_001031615 | ++ | +++ | ++ | + | ++ | +++ | Aldehyde
dehydrogenase family |
| ANO9 | NM_001012302 | + | +++ | + | + | +++ | ++ | Ion transport |
| C1QL1 | NM_006688 | +++ | ++ | + | + | ++ | + | Locomotory
behavior |
| CYB561 | NM_182580 | ++ | +++ | +++ | + | +++ | ++ | Electron
transport |
| ECM1 | NM_004425 | + | +++ | ++ | + | ++ | +++ | Signal
transduction |
| EPN3 | NM_017957 | ++ | ++ | + | + | ++ | +++ | lipid binding |
| GABRP | NM_014211 | ++ | ++ | + | +++ | + | ++ | Ion transport |
| GNAS | NM_000516 | +++ | +++ | +++ | ++ | +++ | ++ | Signal
transduction |
| GP1BB | NM_000407 | ++ | + | + | + | ++ | + | Cell adhesion |
| GSN | NM_001127665 | + | ++ | +++ | + | +++ | ++ | Calcium ion
binding |
| NME2 | NM_001018137 | +++ | ++ | +++ | ++ | ++ | ++ | Transcription
regulation |
| NPC2 | NM_006432 | +++ | +++ | +++ | ++ | +++ | +++ | Lipid
metabolism |
| NPFF | NM_003717 | ++ | +++ | + | + | ++ | +++ | Receptor
binding |
| PKM2 | NM_002654 | +++ | +++ | ++ | ++ | +++ | +++ | Glycolysis |
| PKP3 | NM_007183 | ++ | +++ | + | + | ++ | ++ | Cell adhesion |
| RPL41 | NM_021104 | +++ | +++ | +++ | +++ | +++ | +++ |
Ribonucleoprotein |
| RPS24 | NM_001026 | +++ | ++ | ++ | ++ | +++ | ++ |
Ribonucleoprotein |
| SEMA4A | NM_022367 | ++ | +++ | + | + | ++ | +++ | Receptor
activity |
| TG | NM_003235 | ++ | ++ | + | + | ++ | +++ | Signal
transduction |
| USH1G | NM_173477 | ++ | + | +++ | ++ | ++ | ++ | Actin
cytoskeleton |
| WFDC2 | NM_006103 | + | +++ | ++ | + | ++ | +++ | Protease
inhibitor |
| ZNF750 | NM_024702 | +++ | ++ | +++ | + | +++ | ++ | Transcription
regulation |
Discussion
The identification of genomic markers that are
associated with the progression of cancer is a key target in the
field of cancer research. Genes that are selectively-expressed in
cancer cells are promising targets for the development of new
diagnostic and therapeutic markers (17). An ideal way to identify and
characterize cancer-associated genes and their biological functions
is to consolidate data from multiple comparable studies in order to
perform an integrative analysis.
In the present study, cDNA libraries from 15 human
cancer types were integrated for a statistical comparison. Through
the utilization of the DDD tool, which screens statistically
differentially-expressed genes between different libraries, a total
of 1,904 selectively-expressed cancer genes were obtained,
including 274 uniquely-expressed cancer genes within various cancer
types. Certain genes are well-known to be specifically expressed in
certain cancers, including WFDC2, which is uniquely-expressed in
ovarian tumors and may be a promising marker in the diagnosis of
ovarian carcinoma (18). A
systematic bioinformatics analysis revealed significant biological
functions that were enriched among these genes. These enriched
functions or pathways will provide clues for further studies in
cancer biology and may lead to the development of new diagnostic
and therapeutic markers.
The functional characteristic analysis showed that
the selectively-expressed genes mainly performed catalytic and
binding activities. These genes shared common functional domains.
Cytochrome P450 is a key enzyme domain in cancer formation and
treatment (19), and the serpin
domain contains cancer-related functions that are involved in
tumorigenesis by regulating differentiation or inhibiting
proteinases (20). Apolipoprotein
A-I plays a significant role in the progression of ovarian cancer
(21) and cholangiocarcinoma
(22). These proteins are
associated with the processes involved in cancer development.
The functional clustering analysis identified the
main cellular components and biological process clusters that were
enriched in the cancer genes. The ribosomal cluster, which is
essential for protein synthesis, was the first to be identified in
the analysis. It has also been reported that the ribosomal proteins
contain certain extraribosomal functions, including those of
apoptosis, DNA repair and RNA splicing and modification (23). Alterations in ribosome biogenesis is
a cause of neoplastic transformation (24). Ribosomal protein expression has been
shown to be differentially regulated in human colorectum carcinoma,
in which ribosomal protein L7 has a neuroendocrine function
(25). Notably, the
selectively-expressed genes that corresponded to the ribosomal
proteins were not specifically expressed in certain cancer
types.
Cell adhesion molecules are known to play key roles
in cancer progression through alterations of cell-cell and
cell-extracellular matrix (ECM) adhesions, resulting in cancer cell
migration, invasion and proliferation. In the present study, 24
cadherins, including 20 protocadherins and five integrins, were
selectively expressed in the cancer cells. E-cadherin is expressed
in normal epithelial tissues, but its suboptimal expression has
been suggested to be associated with cancer invasion. E-cadherin
immunohistochemistry is useful in diagnosing breast cancer
(26). However, P-cadherin is
frequently highly expressed in high-grade breast tumors (27). Further studies of these cell
adhesion molecules may provide supporting information for the
mechanisms of cancer cell interactions and metastatic
activities.
The enriched functions of the cancer-associated
genes are involved in various stages of cancer development and
progression by participating in numerous cellular pathways or
networks. Through IPA analysis, 21 significant pathways were
identified, which corresponded to the enriched functional
clusters.
Notably, 37 of the 274 uniquely-expressed genes were
highly expressed in the human testes and epididymis. The
cancer/testis genes that displayed a restricted expression in the
testis and certain cancers are promising biomarkers. In total,
>200 cancer/testis genes have been identified in the
Cancer-Testis (CT) Antigens database (http://www.cta.lncc.br). The epididymis is subjected
to rare tumors through several potential mechanisms. Certain
selectively-expressed genes in the epididymis have spatial and
temporal expression patterns for sperm maturation. The alternative
expression levels of these genes in certain organs may act as an
indicator of cancer development, including WFDC2 in ovarian cancer
(28), ADAM29 and ADAM7 in melanoma
(29) and Eppin in prostate cancer
(30). However, to the best of our
knowledge, no studies have reported the association between the
highly-expressed epididymal genes and cancer. In the present study,
it was hypothesized that certain genes that were strictly expressed
in the epididymis may play specific roles in cancer biology.
In conclusion, the present study identified and
characterized human cancer-associated genes and their biological
functions. The genes that were differentially expressed in various
cancer types mainly functioned as ribosomal proteins, enzymes,
receptors, secretory proteins and cell adhesion molecules. The
results provide a new insight into cancer biology and a new
perspective into the involvement of highly-expressed epididymal
genes in cancer biomarkers.
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gray JW and Collins C: Genome changes and
gene expression in human solid tumors. Carcinogenesis. 21:443–452.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Klijn C, Holstege H, de Ridder J, Liu X,
Reinders M, Jonkers J and Wessels L: Identification of cancer genes
using a statistical framework for multiexperiment analysis of
nondiscretized array CGH data. Nucleic Acids Res. 36:e132008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li BQ, Huang T, Liu L, Cai YD and Chou KC:
Identification of colorectal cancer related genes with mRMR and
shortest path in protein-protein interaction network. PLoS One.
7:e333932012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu Y, Wang X, Wu F, Huang R, Xue F, Liang
G, Tao M, Cai P and Huang Y: Transcriptome profiling of the cancer,
adjacent non-tumor and distant normal tissues from a colorectal
cancer patient by deep sequencing. PLoS One. 7:e410012012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diamond JR, Borges VF, Eckhardt SG and
Jimeno A: BRCA in breast cancer: from risk assessment to
therapeutic prediction. Drug News Perspect. 22:603–608. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Balk SP, Ko YJ and Bubley GJ: Biology of
prostate-specific antigen. J Clin Oncol. 21:383–391. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hellström I, Raycraft J, Hayden-Ledbetter
M, Ledbetter JA, Schummer M, McIntosh M, Drescher C, Urban N and
Hellström KE: The HE4 (WFDC2) protein is a biomarker for ovarian
carcinoma. Cancer Res. 63:3695–3700. 2003.PubMed/NCBI
|
|
9
|
Fu-Jun L, Hai-Yan W and Jian-Yuan L: A new
analysis of testicular proteins through integrative bioinformatics.
Mol Biol Rep. 39:3965–3970. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu F, Jin S, Li N, Liu X, Wang H and Li
J: Comparative and functional analysis of testis-specific genes.
Biol Pharm Bull. 34:28–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fu-Jun L and Xiao-Fang S: Comparative
analysis of human reproductive proteomes identifies candidate
proteins of sperm maturation. Mol Biol Rep. 39:10257–10263. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li JY, Wang HY, Liu J, Liu Q, Zhang JS,
Wan FC, Liu FJ, Jin SH and Zhang YL: Transcriptome analysis of a
cDNA library from adult human epididymis. DNA Res. 15:115–122.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li SJ, Peng M, Li H, Liu BS, Wang C, Wu
JR, Li YX and Zeng R: Sys-BodyFluid: a systematical database for
human body fluid proteome research. Nucleic Acids Res. 37(database
issue): D907–D912. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
da Cunha JP, Galante PA, de Souza JE, de
Souza RF, Carvalho PM, Ohara DT, Moura RP, Oba-Shinja SM, Marie SK,
Silva WA Jr, et al: Bioinformatics construction of the human cell
surfaceome. Proc Natl Acad Sci USA. 106:16752–16757.
2009.PubMed/NCBI
|
|
15
|
Liu FJ, Hua XF and Wang WJ: A new
bioinformatics insight into human cancer-associated proteins. Oncol
Rep. 27:1932–1936. 2012.PubMed/NCBI
|
|
16
|
Fu-Jun L, Shao-Hua J and Xiao-Fang S:
Differential proteomic analysis of pathway biomarkers in human
breast cancer by integrated bioinformatics. Oncol Lett.
4:1097–1103. 2012.PubMed/NCBI
|
|
17
|
Lo HW, Day CP and Hung MC: Cancer-specific
gene therapy. Adv Genet. 54:235–255. 2005.
|
|
18
|
Langmár Z, Németh M, Vleskó G, Király M,
Hornyák L and Bösze P: HE4 - a novel promising serum marker in the
diagnosis of ovarian carcinoma. Eur J Gynaecol Oncol. 32:605–610.
2011.PubMed/NCBI
|
|
19
|
Rodriguez-Antona C and Ingelman-Sundberg
M: Cytochrome P450 pharmacogenetics and cancer. Oncogene.
25:1679–1691. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chang WS, Chang NT, Lin SC, Wu CW and Wu
FY: Tissue-specific cancer-related serpin gene cluster at human
chromosome band 3q26. Genes Chromosomes Cancer. 29:240–255. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su F, Kozak KR, Imaizumi S, Gao F, Amneus
MW, Grijalva V, Ng C, Wagner A, Hough G, Farias-Eisner G,
Anantharamaiah GM, Van Lenten BJ, Navab M, Fogelman AM, Reddy ST
and Farias-Eisner R: Apolipoprotein A-I (apoA-I) and apoA-I mimetic
peptides inhibit tumor development in a mouse model of ovarian
cancer. Proc Natl Acad Sci USA. 107:19997–20002. 2010. View Article : Google Scholar
|
|
22
|
Wang X, Dai S, Zhang Z, Liu L, Wang J,
Xiao X, He D and Liu B: Characterization of apolipoprotein A-I as a
potential biomarker for cholangiocarcinoma. Eur J Cancer Care
(Engl). 18:625–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lai MD and Xu J: Ribosomal proteins and
colorectal cancer. Curr Genomics. 8:43–49. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ruggero D and Pandolfi PP: Does the
ribosome translate cancer? Nat Rev Cancer. 3:179–192. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kasai H, Nadano D, Hidaka E, Higuchi K,
Kawakubo M, Sato TA and Nakayama J: Differential expression of
ribosomal proteins in human normal and neoplastic colorectum. J
Histochem Cytochem. 51:567–574. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Singhai R, Patil VW, Jaiswal SR, Patil SD,
Tayade MB and Patil AV: E-Cadherin as a diagnostic biomarker in
breast cancer. N Am J Med Sci. 3:227–233. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Albergaria A, Ribeiro AS, Vieira AF, Sousa
B, Nobre AR, Seruca R, Schmitt F and Paredes J: P-cadherin role in
normal breast development and cancer. Int J Dev Biol. 55:811–822.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Montagnana M, Danese E, Giudici S, Franchi
M, Guidi GC, Plebani M and Lippi G: HE4 in ovarian cancer: from
discovery to clinical application. Adv Clin Chem. 55:1–20. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei X, Moncada-Pazos A, Cal S,
Soria-Valles C, Gartner J, Rudloff U, Lin JC, Rosenberg SA,
López-Otín C and Samuels Y; NISC Comparative Sequencing Program.
Analysis of the disintegrin-metalloproteinases family reveals
ADAM29 and ADAM7 are often mutated in melanoma. Hum Mutat.
32:E2148–E2175. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Izumi K, Zheng Y and Miyamoto H: Eppin
expression in prostate cancer. Eur Urol. 59:1071–1072. 2011.
View Article : Google Scholar : PubMed/NCBI
|