Introduction
Thyroid-like follicular carcinoma of the kidney
(TLFCK) is microscopically similar to thyroid follicular carcinoma
(1). TLFCK is a rare pathological
type of renal tumor, with potential origins in the kidney cells.
This emerging entity has not been included in the World Health
Organization Classification of Tumors: Pathology and Genetics of
Tumors of the Urinary System and Male Genital Organs, and its
biological behavior has not yet been determined (2). TLFCK was documneted by Jung et
al for the first time in 2006 (1). To date there have only been 13
complete case reports and clinical physician’s understanding of its
clinical features and pathological characteristics remains
inadequate. The existing cases of TLFCK normally occur in young and
middle-aged females (eight of 13 cases). The majority of the
patients are without obvious clinical symptoms, certain patients
present with hematuresis or waist pain. All patients were treated
with surgery. Radical nephrectomy is capable of achieving
successful patient outcomes. In the present study, two patients
with TLFCK are reported who were treated at The First Affiliated
Hospital of Fujian Medical University (Fushou, Fujian, China)
between 2011 and 2013. The clinical manifestations, diagnosis,
pathology and treatment of these patients and other patients
described in the literature are discussed. Written informed consent
was obtained from the patients.
Case report
Patient 1
A 65-year-old male was admitted for repeated
hematuria during urination for four years and right back pain for
seven days. The patient had no tumor family history. The physical
examination upon admission showed normal vital signs, and normal
findings of the heart, lungs, liver and spleen. Percussion
tenderness over the right kidney region was noticed. Plain magnetic
resonance imaging revealed a mass in the right kidney, which was
possibly a renal carcinoma with involvement of the renal fascia.
Color Doppler ultrasonography confirmed the presence of a solid
mass with hypoechogenicity in the right renal hilum. Enhanced
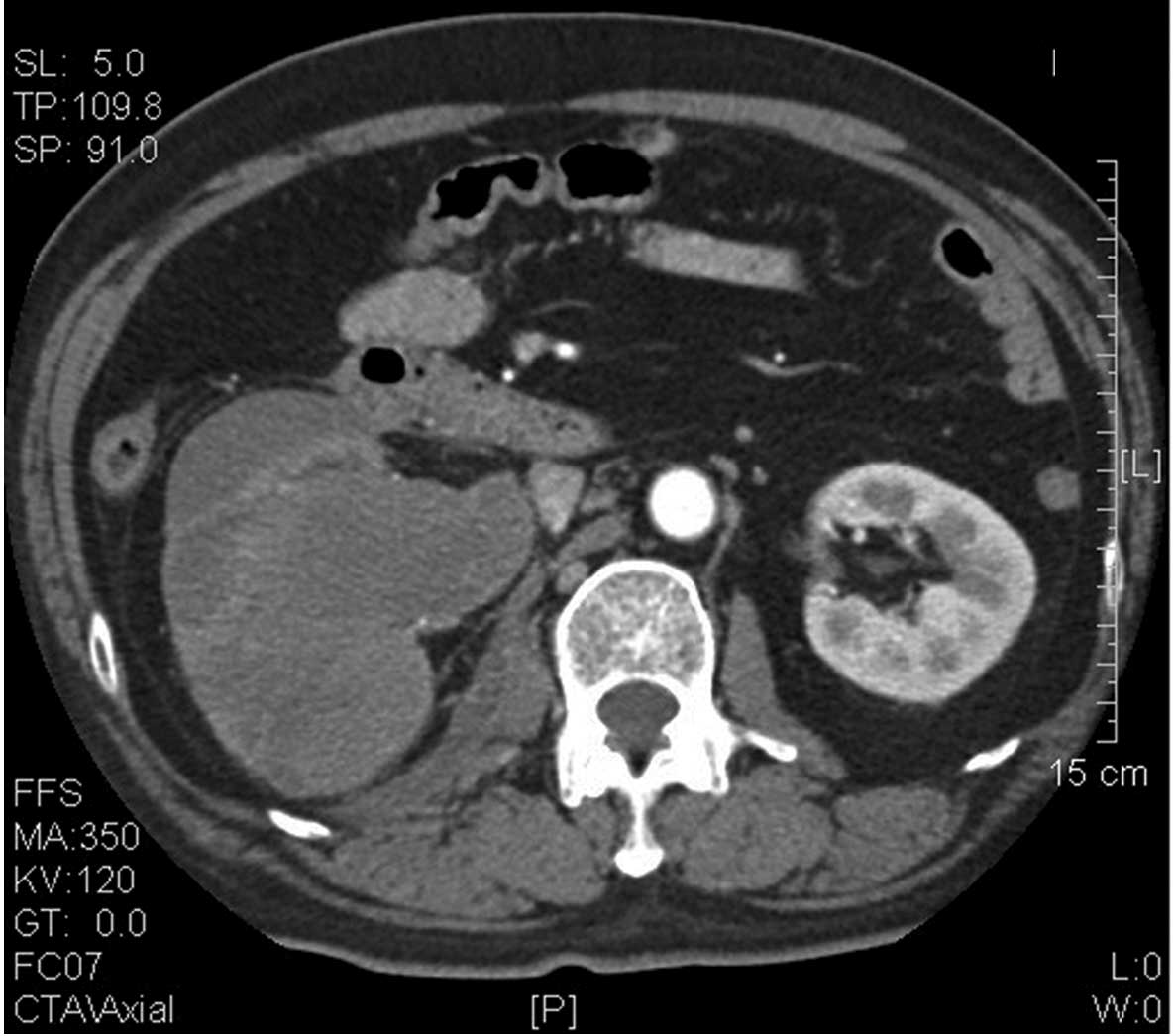
computed tomography (CT) indicated a right renal pelvic carcinoma
(Fig. 1), for which the patient
underwent a radical right nephrectomy on January 14, 2011.
Intraoperatively, the tumor measured 8.0×4.3×5.0 cm and had a hard
texture. Gerota’s fascia and lymph nodes that are adjacent to the
kidney and abdominal aorta were not involved. A postoperative test
showed no signs of hyperthyroidism. The patient had no personal or
family history of hyperthyroidism. Ultrasonography found no tumor
or other abnormal signs in the thyroid and other body parts.
Routine blood tests and thyroid-stimulating hormone (TSH),
triiodothyronine (T3) and thyroxine (T4) concentrations were within
normal limits. Following the surgery, the patient has shown no
signs of tumor recurrence or metastasis.
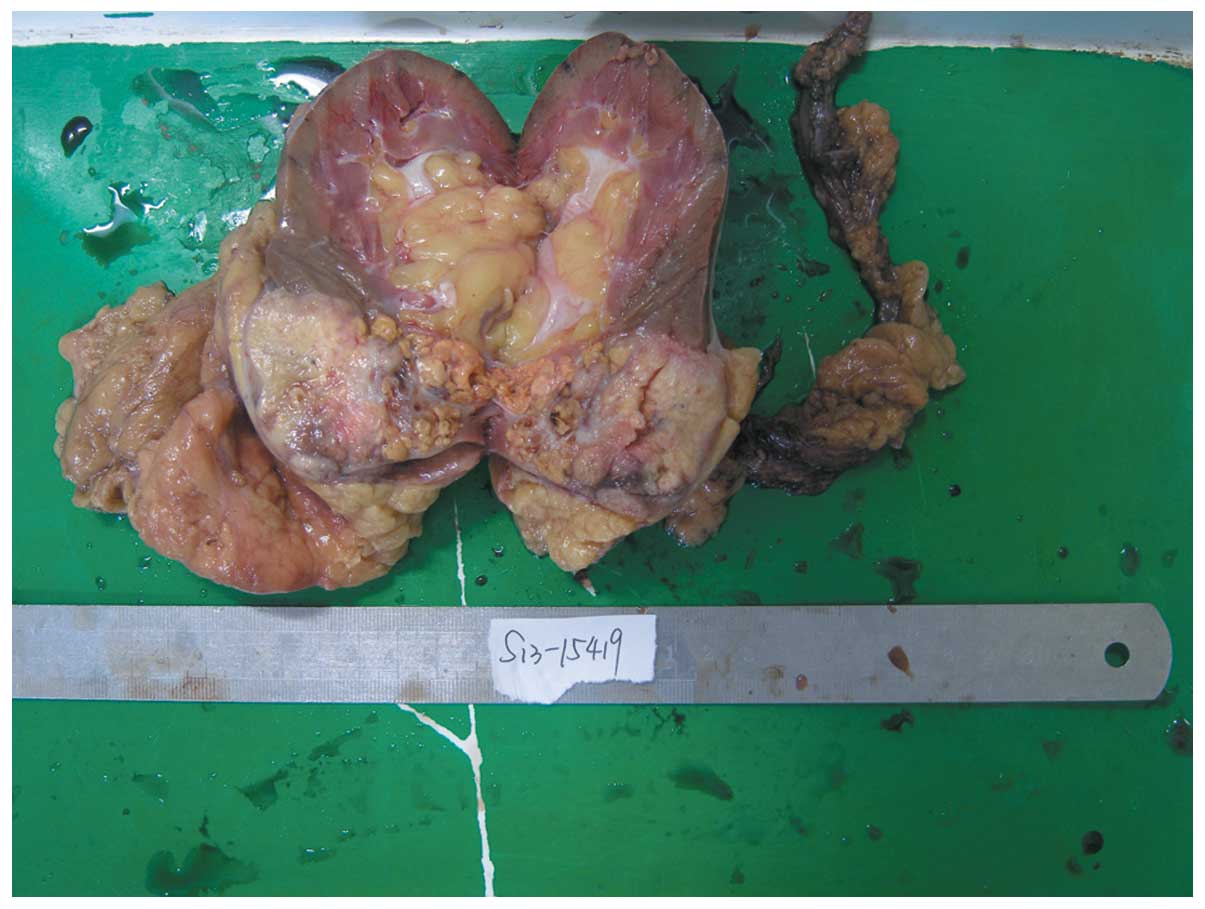
Macroscopically, the resected kidney measured
13.0×7.0×6.0 cm, with the tumor located in the renal parenchyma
measuring 8.0×4.3×5.0 cm. The cut surface revealed a
well-circumscribed gray-yellow solid tumor. Scattered gray-yellow
necrotic areas and gray-red hemorrhagic areas were observed with
small cystic cavities (Fig. 2).
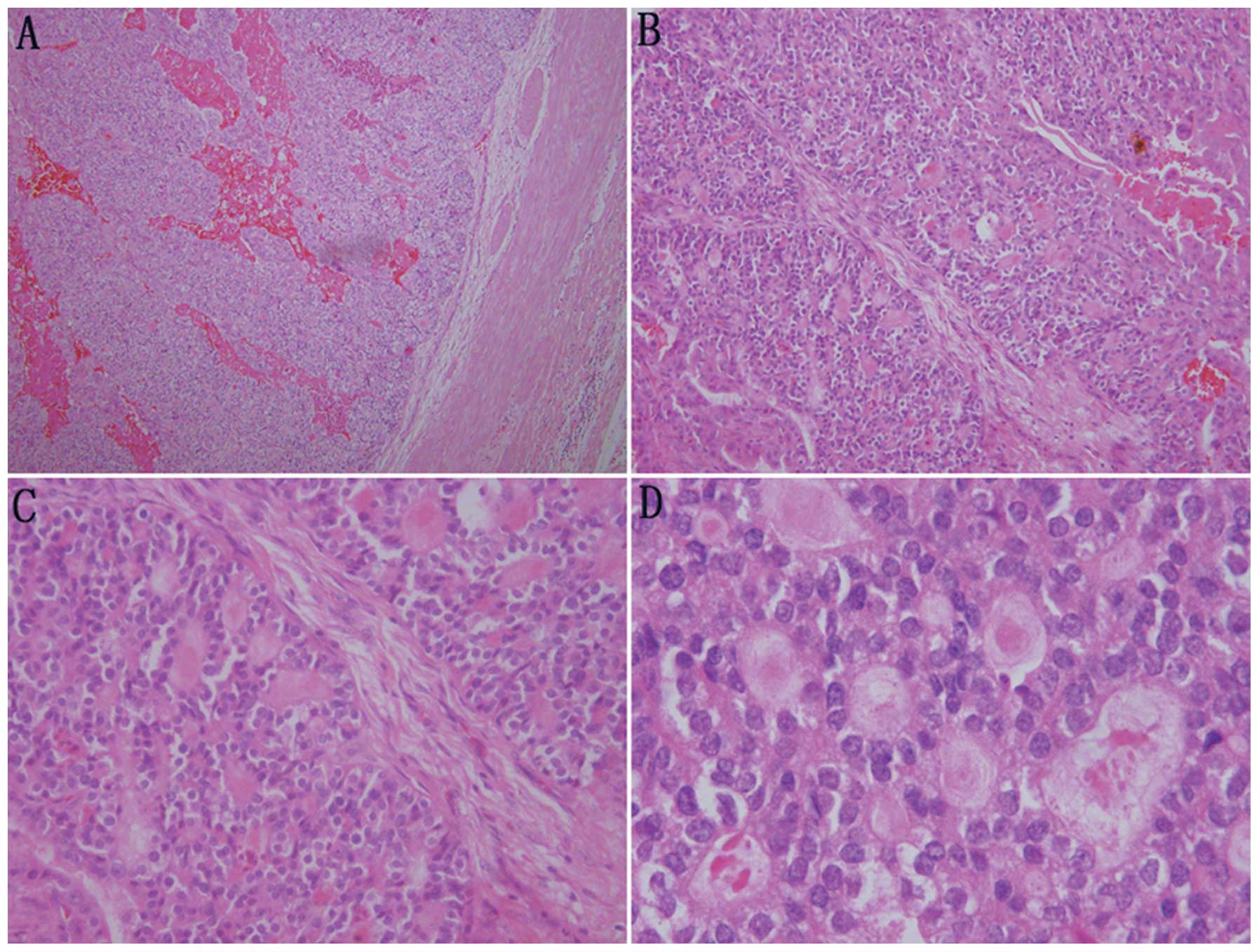
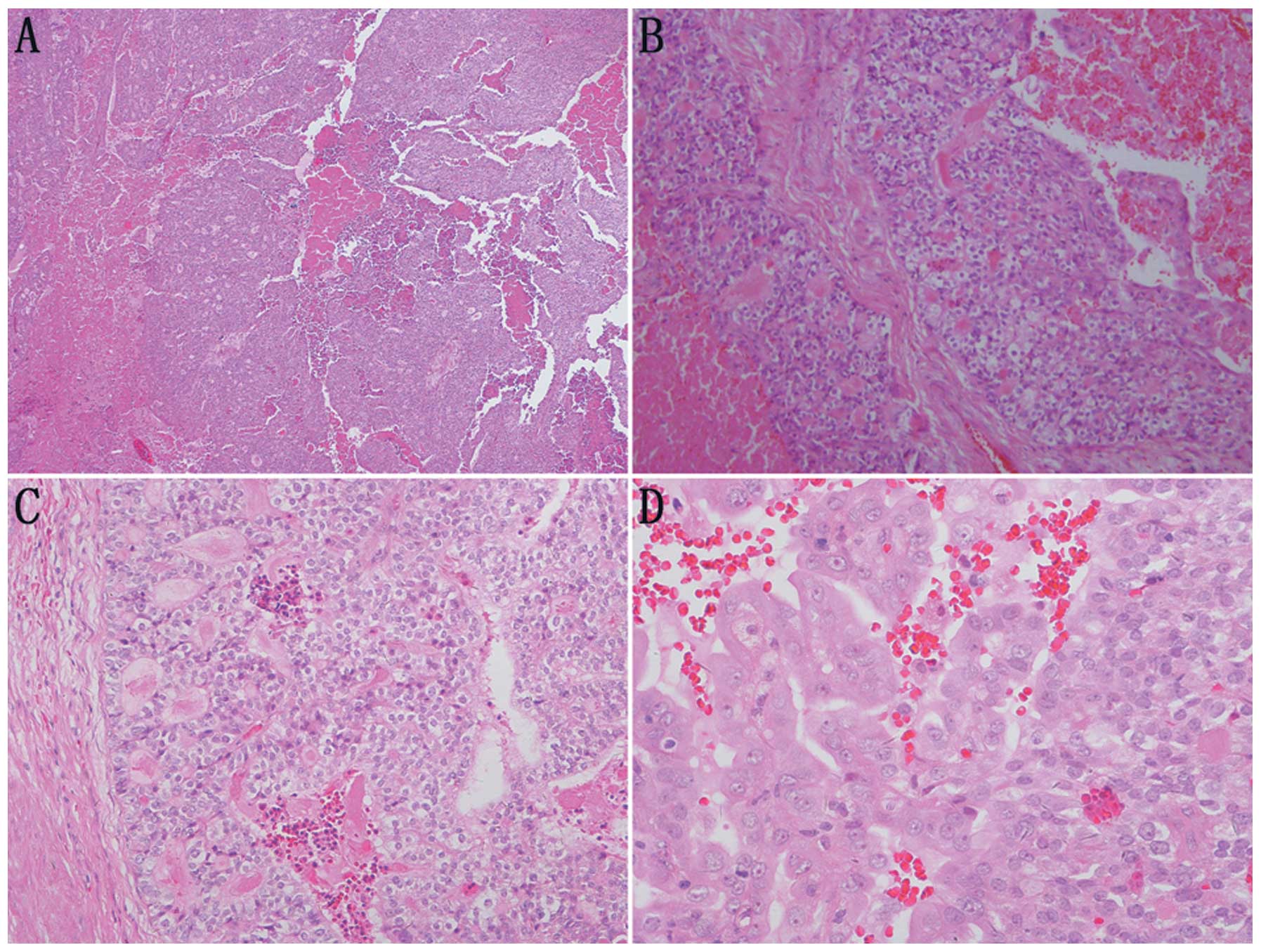
Microscopically, the tumor cells showed a morphology similar to
thyroid follicles or a sieve, occupying over 50% of the visual
field. The follicular lumens were filled with evenly red-stained
colloid-like material. Vacuoles were rarely observed around the
lumens. The colloid-like material broke a number of the lumens and
merged, resulting in the tumor cells exhibiting a morphology of
‘dried follicles’. The tumor cells showed indistinctive nuclear
heteromorphism, reduced transparent cytoplasm and unclear borders.
No clear cell type or other type of renal cell carcinoma was
identified (Fig. 3).
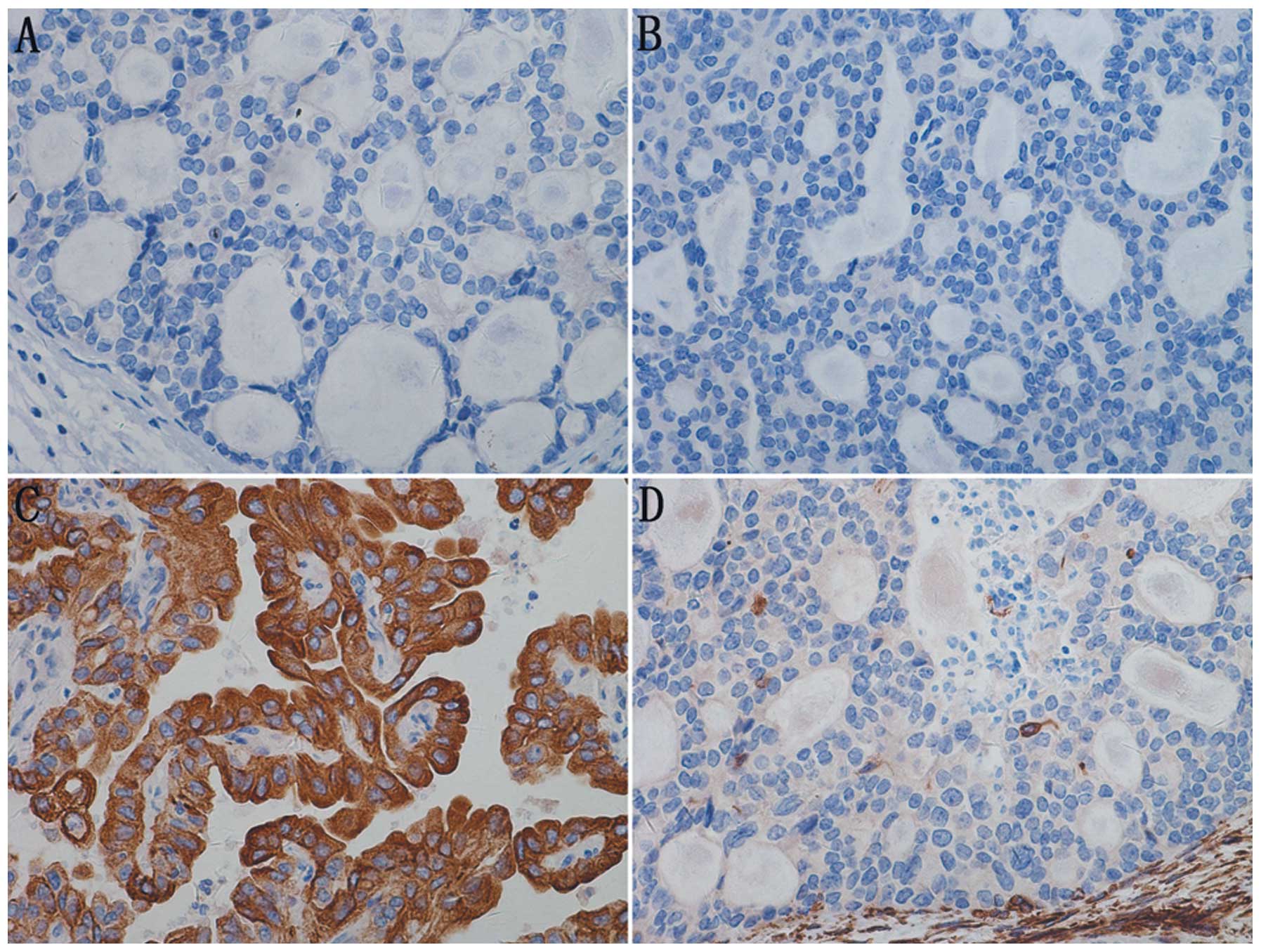
Immunohistochemically, the tumor was positive for
vimentin, epithelial membrane antigen (EMA), cytokeratin (CK), CK7
and neuron specific enolase (NSE); and negative for CK34BE12,
synapsin (Syn), CK20, cluster of differentiation 56 (CD56), CD10,
Wilm’s tumor-1 (WT-1), CD34, CD57, P53, CD99, thyroid transcription
factor-1 (TTF-1), CD15 and thyroglobulin (TG); and had a Ki-67
labeling index (LI) of 30%. The pathological diagnosis was TLFCK
(Fig. 4).
Patient 2
A 59-year-old man was found to have a mass in the
right kidney during a routine health examination and was admitted.
The patient had no personal or family history of thyroid
dysfunction. A physical examination showed normal vital signs,
heart, lungs, liver and spleen, and no percussion tenderness over
the right kidney region. Color Doppler ultrasonography revealed a
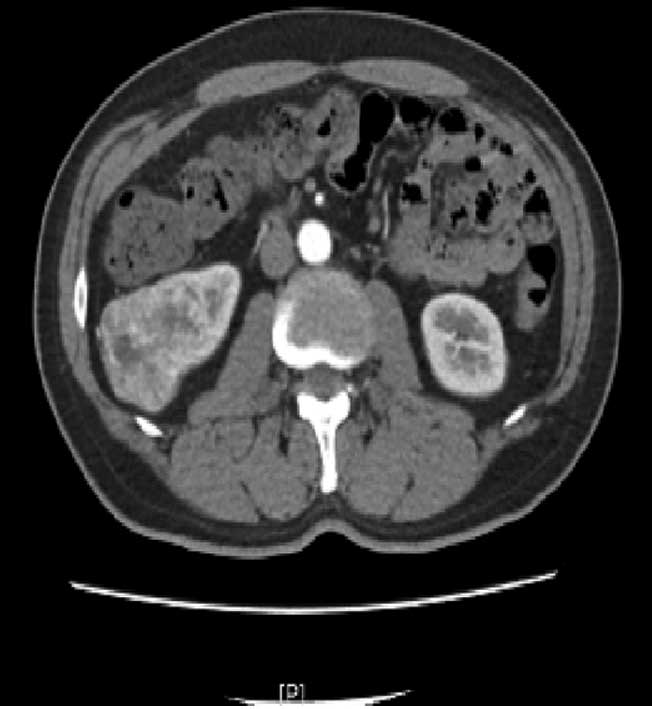
mass in the right kidney, which was confirmed by enhanced CT
(Fig. 5). Owing to the possibility
of malignancy, the patient was initially diagnosed with renal
carcinoma, for which radical right nephrectomy was performed on May
3, 2013. During the surgery, the tumor was found to be located in
the middle-lower pole of the right kidney. It measured 6.0×4.5×5.0
cm and had a hard texture. The tumor was well-circumscribed with a
mild adhesion to the adjacent tissues. Gerota’s fascia and the
lymph nodes that are adjacent to the kidney and abdominal aorta
were not involved. Ultrasonography found no tumor or other abnormal
signs in the thyroid and other body parts. Routine blood tests,
TSH, T3 and T4 were within normal limits. Following the surgery
there has been no evidence of tumor recurrence or metastasis.
The resected kidney measured 14.0×7.0×7.0 cm, with
the tumor measuring 6.0×5.0×5.4 cm, located in the middle-lower
pole. The cut surface revealed a solid, gray-white,
well-circumscribed tumor with invasion of the surrounding renal
tissues and capsules. Scattered gray-yellow ischemic areas and
small cystic cavities were also observed (Fig. 6).
Microscopically, the tumor was covered by a fibrous
pseudocapsule. The majority of the tumor cells were arranged as
thyroid follicles or a sieve, with certain cells appearing in
patches or fibrous septae. The follicular lumens were filled with
evenly red-stained colloid-like material. Vacuoles were rarely
observed around the lumens. The colloid-like material broke a few
of the lumens and merged, resulting in the tumor cells exhibiting a
morphology of ‘dried follicles’. The tumor cells showed a reduced
transparent cytoplasm and unclear borders. The nuclei were enlarged
and overlapped in round, oval or spindle shapes with a fine
chromatin pattern and one to two inconspicuous nucleoli per
nucleus. The nuclear groove was unremarkable and mitosis was rarely
observed. No clear cell type or other type of renal cell carcinoma
was identified (Fig. 7).
Immunohistochemically, the tumor cells were positive
for vimentin, EMA, CK7 and CK20; and negative for CD56, CD10, WT-1,
CD34, CD57, P53, CD117, TTF-1, CD15, CD99, TG, chromogranin A (CgA)
and Syn; and had a Ki-67 LI of 20% (Fig. 8).
Discussion
The first case of TLFCK was described in 2006; all
cases of TLFCK reported are summarized in Table I. The locations and clinical
manifestations of TLFCK do not distinguish these tumors from other
types of renal carcinoma. The majority of patients with TLFCK are
adults, aged 22 to 83 years, with a female to male predominance
(8:5, including the two patients in the present study) (1–5). The
majority of tumors involve the right kidney more than the left, as
is the case in the present study.
 | Table IClinicopathological features of
primary TLFCK reported between 2006 and 2012. |
Table I
Clinicopathological features of
primary TLFCK reported between 2006 and 2012.
| First author/s
(ref) | Age, year | Gender | Presentation | Tumor location | Tumor size, cm
(location) | TNM stage | Treatment | Disease-free
survival |
|---|
| Amin et al
(2) | 53 | Female | Incidental | Right kidney (middle
pole) | 2.1 | pT1aNx | RN | 4 years and 6
months |
| 29 | Female | Incidental | Right kidney (upper
pole) | 1.9 | pT1aNx | RN | 7 years |
| 45 | Male | Incidental | Right kidney (lower
pole) | 3.5 | pT1aN1 | RN | 1 year and 5
months |
| 83 | Male | Incidental | Left kidney (lower
pole) | 2.1 | pT1aNx | RN | 4 years |
| 35 | Male | Incidental | Right kidney (middle
pole) | 3.0 | pT1aNx | RN | 1 year and 8
months |
| 50 | Female | Incidental | Right kidney (middle
pole) | 4.0 | pT1aN0 | RN | 7 months |
| Jung et al
(1) | 32 | Female | Incidental | Right kidney (middle
and lower poles) | 11.8 | pT2Nx | RN | 6 months |
| Xu and Zang(3) | 36 | Female | Hematuria of the
middle course of urination with blood clots | Left kidney
(middle-lower pole) | 10.0 | pT2Nx | RN | 1 year |
| He et al
(4) | 22 | Female | Painless
hematuria | Left kidney | 8.0 | pT1aN0 | RN | No data |
| Sterlacci et
al (5) | 29 | Female | Incidental | Left kidney (middle
pole) | 4.3 | / | RN | Left lung lower lobe
metastasis at 2 months, disease-free survival for 5 years |
| Dhillon et al
(6) | 34 | Female | Hematuria and right
back pain | Right kidney (middle
pole) and bilateral lungs | 6.3 (right
kidney)
2.1 (left lung)
3.4 (right lung) | pT2N0M1 | RN | 1 year |
| Present study | 65 | Male | Hematuria and right
back pain | Right kidney
(middle-lower pole) | 8.0 | pT2N0 | RN | 15 months |
| 59 | Male | Incidental | Right kidney
(middle-lower pole) | 5.2 | pT1aN0 | RN | 1 month |
The tumors in the present study were pathologically
confirmed as having originated in the renal parenchyma, with
well-circumscribed pseudo-capsules. The cut surfaces of the tumor
were yellow-white or gray-white with focal necrosis. There was no
evidence of morphology that is typical of clear cell type renal
carcinoma, consistent with previous reports (2–4). The
sizes of TLFCK have been reported to range from 1.9–11.8 cm
(1–2). The most significant microscopic
feature of TLFCK is the striking resemblance to the
well-differentiated follicular carcinoma of the thyroid gland, with
follicular structures and colloid-like material. By contrast, no
TLFCK has shown morphological features similar to clear or other
types of renal carcinoma (2–4).
The tumors in the present study were negative for
TTF-1 and TG, ruling out the possibility of metastasis from thyroid
tumors and supporting a diagnosis of TLFCK (1–5).
WT-1 expression was found to be
immunohistochemically positive in TLFCK tumor cell nuclei,
indicating that these tumors originate in the kidneys (3). Thyroid follicle-like structures have
been observed in patients with chronic pyelonephritis and end-stage
renal disease, indicating that TLFCKs may originate from renal
tubular epithelial cells (2).
The immune phenotypes of reported TLFCK are listed
in Table II. Immunostaining
results for epithelial markers have differed among studies;
therefore, a combination of epithelial cell and renal tubular
epithelial cell markers have been used in the majority of studies
(2–4). Assays of primary and metastasized
tumors of TLFCK have found that paired box gene 2 (PAX2) and PAX8
are expressed, whereas TG and TTF-1 are not, supporting the renal
origin of this malignancy (6).
| Table IIImmune phenotypes of primary
TLFCK. |
Table II
Immune phenotypes of primary
TLFCK.
| First author/s
(ref) | CK7 | CK19 | CK20 | CK10 | EMA | LCK | HCK | PCK | TTF-1 | TG | PAX2 | PAX8 |
|---|
| Sterlacci et
al (5) | + | NA | + | − | NA | NA | NA | NA | − | − | NA | NA |
| He et al
(4) | Weak+ | + | Weak+ | Weak+ | Weak+ | NA | NA | + | − | − | NA | NA |
| Xu and Zang (3) | + | NA | NA |
Focal+ | + | NA | NA | NA | − | − | NA | NA |
| Dhillon et al
(6) | + | NA | + | + | + | NA | NA | NA | − | − | + | + |
| Case 1 |
Focal+ | + | − | − | + | NA | NA | NA | − | − | NA | NA |
| Case 2 | + | NA | + | NA | + | NA | NA | NA | − | − | NA | NA |
Chromosomal analysis has shown gains of chromosomes
7q36, 8q24, 12, 16, 17p11-q11, 17q24, 19q, 20q13, 21q22.3 and Xp
(1), and losses of chromosomes
1p36, 3 and 9q21–33 (1) and 1, 3,
7, 9p21, 12, 17 and X (5) in TLFCK.
TLFCKs were shown to be positive for the expression of 135 genes
but negative for an additional 46 genes (2). A 2.5-fold increase in the expression
of mixed lineage leukemia gene has been observed; this gene encodes
a transcription factor that is involved in the development of
several types of hematological malignancies, such as acute leukemia
(7).
TLFCK should be differentiated from renal metastases
from thyroid follicle carcinoma, however, only 10 cases of the
latter have been reported (8,9).
Thyroid follicle carcinomas tend to metastasize to lymph nodes,
lung and bone. Renal metastasis of thyroid follicle carcinoma
usually occurs following multiple systemic metastases. The majority
of thyroid carcinomas are markedly positive for TTF-1 and TG,
although these two proteins may not be expressed by certain poorly
differentiated or sarcoma-like thyroid carcinomas (10). Therefore, further examination is
required to diagnose TLFCK. TLFCK should also be differentiated
from malignant ovarian teratoma with thyroid tissue as the sole
component. Renal metastasis from these malignancies can be ruled
out by imaging of the ovaries (11,12).
In addition, the absence of expression of TTF-1 and TG can be
diagnostic of TLFCK, and TLFCK should be differentiated from other
renal carcinomas. Rare renal carcinoid tumors can have a follicular
structure and red-stained colloid material, but these tumors have
smaller nuclei, finer chromatin and no evidence of necrosis.
Neuroendocrine carcinomas are characterized by nuclear
heteromorphism and frequent mitosis, and a clear positivity for
NSE, CD56, CgA and synaptophysin. Epithelial-type nephroblastomas
usually occur in children with undifferentiated embryo, epithelial
and mesenchymal tissues. This malignancy usually shows epithelial
rosettes, but no follicular structure or colloid-like material has
been reported.
Radical nephrectomy is the major treatment method
for TLFCK and can achieve good prognosis (1,2,6).
Patient 1 has remained disease-free for over two years, whereas
patient 2 has shown no evidence of tumor relapse one month
following the surgery. Follow-up of six TLFCK patients for a mean
of 47.3 months identified that five were disease-free and one
showed metastasis to renal hilar lymph nodes, indicating that these
tumors have a low malignancy (2).
One patient was found to develop metastasis to the left lower lung
two months following surgery (5);
the metastasis was surgically removed and the patient has shown no
evidence of recurrence or metastasis during a follow-up period of
five years. TLFCK is regarded as having medium invasiveness, but
long-term survival can be achieved by radical resection of the
tumor (6).
References
|
1
|
Jung SJ, Chung JI, Park SH, Ayala AG and
Ro JY: Thyroid follicular carcinoma-like tumor of kidney: a case
report with morphologic, immunohistochemical, and genetic analysis.
Am J Surg Pathol. 30:411–415. 2006.PubMed/NCBI
|
|
2
|
Amin MB, Gupta R, Ondrej H, et al: Primary
thyroid-like follicular carcinoma of the kidney: report of 6 cases
of a histologically distinctive adult renal epithelial neoplasm. Am
J Surg Pathol. 33:393–400. 2009. View Article : Google Scholar
|
|
3
|
Xu H and Zang WY: Clinicopathological
features of thyroid follicular carcinoma-like renal cell carcinoma.
Zhen Duan Bing Li Xue Za Zhi. 17:46–49. 2010.
|
|
4
|
He CN, Li P, Zhao HF, Zhai JP, Liu YQ and
Ma LN: Thyroid follicular carcinoma-like tumor of kidney: report of
a case. Zhonghua Bing Li Xue Za Zhi. 37:428–430. 2008.(In
Chinese).
|
|
5
|
Sterlacci W, Verdorfer I, Gabriel M and
Mikuz G: Thyroid follicular carcinoma-like renal tumor: a case
report with morphologic, immunophenotypic, cytogenetic, and
scintigraphic studies. Virchows Arch. 452:91–95. 2008. View Article : Google Scholar
|
|
6
|
Dhillon J, Tannir NM, Matin SF, Tamboli P,
Czerniak BA and Guo CC: Thyroid-like follicular carcinoma of the
kidney with metastases to the lungs and retroperitoneal lymph
nodes. Hum Pathol. 42:146–150. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Slany RK: The molecular biology of mixed
lineage leukemia. Haematologica. 94:984–993. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Angell SK, Pruthi R and Freiha FS: Primary
thyroidlike carcinoma of the kidney. Urology. 48:632–635. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Garcia-Sanchis L, Lopez-Aznar D, Oltra A,
et al: Metastatic follicular thyroid carcinoma to the kidney: a
case report. Clin Nucl Med. 24:48–50. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Miettinen M and Franssila KO: Variable
expression of keratins and nearly uniform lack of thyroid
transcription factor 1 in thyroid anaplastic carcinoma. Hum Pathol.
31:1139–1145. 2000. View Article : Google Scholar
|
|
11
|
Devaney K, Snyder R, Norris HJ and
Tavassoli FA: Proliferative and histologically malignant struma
ovarii: a clinicopathologic study of 54 cases. Int J Gynecol
Pathol. 12:333–343. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nieminen U, Vonnumers C and Widholm O:
Struma ovarii. Acta Obstet Gynecol Scand. 42:399–424. 1964.
View Article : Google Scholar : PubMed/NCBI
|