Introduction
Cholangiocarcinoma (CCA), a highly aggressive
malignancy with a growth pattern characterized by periductal
extension and infiltration (1),
accounts for ~3% of all gastrointestinal malignancies (2). A recent study suggested that the
overall incidence and mortality of CCA appears to have increased
worldwide over the past decades (3). The prognosis of CCA is poor since
patients with CCA are usually at an advanced stage at the time of
diagnosis. Complete resection with negative margins is the only
treatment with the potential for cure. Although the surgical
outcomes and survival rates have gradually improved with the
advancement in diagnostic and surgical techniques over the past
decades (4), less than one-third of
patients present with resectable tumors at diagnosis (5–15).
Other treatment options for CCA include adjuvant radiotherapy and
chemotherapy and liver transplantation, while none of these
approaches have been shown to substantially improve the survival of
patients with resected or unresected CCA (16). Thus, novel therapeutic strategies
must be developed for the successful treatment of CCA.
Change in DNA methylation represents an important
epigenetic alteration during the multistep process of
carcinogenesis. DNA hypomethylation leads to genomic instability.
Notably, CpG islands in the promoters of tumor suppressor genes are
frequently hypermethylated, resulting in inactivation of the
corresponding tumor suppressors. Genes that are commonly silenced
by promoter hypermethylation are those regulating cell cycle
progression, DNA repair, apoptosis and metastasis (17). DNA methylation often occurs at the
C5 position of cytosine in a CpG dinucleotide context and is
catalyzed by the DNA methyltransferases (DNMTs) (18). DNMT3a and DNMT3b are mainly
responsible for de novo DNA methylation. DNMT1 maintains DNA
methylation by methylating the newly synthesized DNA strand
following DNA replication. Unlike genetic mutations, DNA
methylation may be reversed by inhibitors of DNMTs (DNMTIs). DNMTIs
are therefore emerging as powerful new tools in the epigenetic
therapy field. Decitabine (DAC; 5-aza-2′-deoxyazacytidine), one of
the well-characterized DNMTIs, functions as a cytosine analog and
induces cell death via several mechanisms, including obstruction of
DNA synthesis, induction of DNA structural instability and
degradation of DNMTs (19). DAC was
approved by the USA Food and Drug Administration in 2006 as the
standard care for myelodysplastic syndromes (20). DAC may also modulate the response of
cancer cells to chemo- and radiotherapy (21). In addition, increasing preclinical
and clinical studies have demonstrated a promising application of
DAC for the treatment of solid tumors.
To explore the effects of DAC on CCA, the current
study used CCA cell lines TFK-1 and QBC939 as models, and
investigated the cell proliferation, cell cycle arrest, apoptosis
and autophagy following DAC treatment in vitro. In addition,
an athymic nude mouse model bearing xenografts of TFK-1 cells was
examined to test whether DAC inhibits the growth of CCA
xenografts.
Materials and methods
Cell culture and reagents
The TFK-1 cell line was purchased from DSMZ
(Braunschweig, Germany) and the QBC939 cell line was provided by
the Third Military Medical University (Chongqing, China). TFK-1 and
QBC939 cells were cultured and maintained in a humidified
atmosphere containing 5% CO2 at 37°C, in RPMI-1640
supplemented with 10% fetal bovine serum and 1%
antibiotic-antimycotic (all Gibco-BRL, Carlsbad, CA, USA). DAC was
purchased from Sigma-Aldrich (St. Louis, MO, USA).
Measurement of cell viability
The growth of TFK-1 and QBC939 cells was evaluated
by Cell Counting Kit-8 assay (CCK-8; Dojindo, Kunamoto, Japan),
according to the manufacturer’s instructions. Briefly, TFK-1
(1×104) and QBC939 (5×103) cells were seeded
in 96-well plates. Following incubation with various concentrations
of DAC for 24–120 h, CCK-8 solution was added to each well to a
final concentration of 10 μl/100 μl medium and incubated for an
additional 2 h at 37°C. The absorbance was measured at 450 nm with
a reference wavelength of 630 nm by microplate reader (Thermo
Multiscan GO, Thermo Fisher Scientific, Waltham, MA, USA).
Clonogenic survival assay
TFK-1 cells were treated with 0.5, 5.0 or 50.0 μM
DAC for five days with culture media changed daily. The cells were
then trypsinized, counted and reseeded for clonogenic survival
assay on petri dishes at 200 cells per dish. Following incubation
at 37°C for three weeks, the cells were fixed with 50% ethanol in
ice-cold phosphate-buffered saline (PBS) and stained with 5%
crystal violet. The colonies with >50 cells were counted under a
microscope (Primo Star, Carl Zeiss, Oberkochen, Germany).
Cell cycle analysis
Following treatment with various concentrations of
DAC for 24, 72 and 120 h, cells were collected and fixed overnight
with 70% ethanol (−20°C). At the time of analysis, cells were
incubated with 50 mg/ml RNase A for 30 min at 37°C. Following
incubation, propidium iodide (PI) was added in the dark to a final
concentration of 50 μg/ml. Subsequently, the cell population was
analyzed by flow cytometry (BD-LSR; BD Biosciences, Franklin Lakes,
NJ, USA).
Apoptosis detection
Apoptosis was determined by flow cytometry-based
assay. Briefly, TFK-1 cells were exposed to DAC at the desired
concentration for 24, 48, 72, 96 or 120 h. Apoptosis was evaluated
using the Annexin V-FITC apoptosis detection kit (Nanjing KeyGen
Biotech., Co., Ltd., Nanjing, China) according to manufacturer’s
instructions.
Hoechst 33342/PI staining
Following incubation with DAC at 25 μM for 120 h,
TFK-1 cells were harvested and fixed in methanol for 10 min at room
temperature. Following washing with PBS, cells were incubated with
Hoechst 33342 (10 μg/ml; Nanjing KeyGen Biotech., Co., Ltd.) and PI
(2.5 μg/ml; Nanjing KeyGen Biotech., Co., Ltd.) for 10 min at room
temperature. The morphology of the apoptotic cells was observed
under a fluorescence microscope (Axio Zoom.V16, Carl Zeiss) and
recorded.
Detection of autophagy with green
fluorescent protein (GFP)-tagged MAP-LC3
TFK-1 and QBC939 cells were incubated with DAC for
three days and transfected with GFP-tagged MAP-LC3 (GFP-LC3)
plasmid. After 24 h, the cells were fixed in 4% paraformaldehyde
for 30 min and mounted for confocal microscopy (Leica, Buffalo
Grove, IL, USA). GFP fluorescence was observed under a confocal
microscope (TCS SP8, Leica). Autophagic cells that showed GFP-LC3
staining were counted.
Tumor growth and treatment in nude
mice
DAC-induced effects in vivo with xenografts
of TFK-1 cell lines in six-week-old male Balb-c nu/nu mice with a
median weight of 14–16 g were evaluated. All animal experiments
were performed according to the instructions approved by the
Experimental Animal Center of Huazhong University of Science and
Technology (Wuhan, China). A total of 10 mice were divided into two
groups. All mice were transplanted subcutaneously into the upper
right flank with 2×106 TFK-1 cells. Following the
detection of a measurable tumor, animals were treated with 0.8
mg/kg DAC or vehicle alone (4% dimethylsulfoxide) by
intraperitoneal injection daily for 14 consecutive days. Tumor
volumes were calculated every two days using the following formula:
Tumor volume (mm3)= π/(6xDxd2), where ‘D’ is
the largest diameter (in mm) and ‘d’ is the smallest diameter (in
mm). Mice were monitored daily for treatment-related morbidity and
mortality.
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 5 (GraphPad Software, San Diego, CA, USA). All in
vitro and in vivo experiments were repeated
independently in triplicate. The Mann-Whitney U test was performed
to determine the level of significance for the in vitro
studies. For in vivo studies, the statistical significance
was analyzed using the long-rank test. Data are expressed as the
mean ± standard deviation, accompanied by the number of tests.
P<0.05 was considered to indicate a statistically significant
difference.
Results
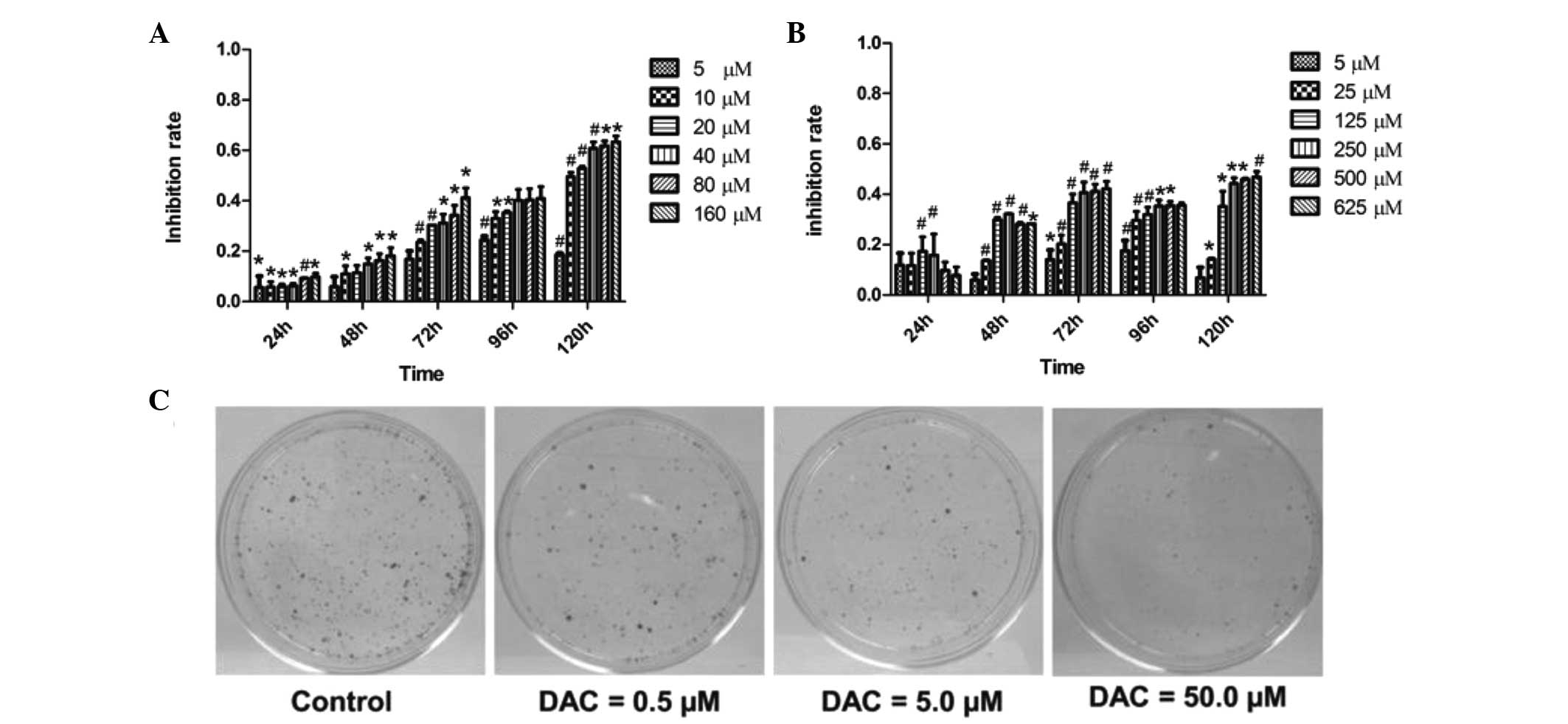
DAC inhibits the growth of CCA cells
To investigate the antiproliferative effects of DAC
on CCA cells, the viability of TFK-1 and QBC939 cells treated with
various concentrations of DAC was assessed for 24–120 h and the
cell viability was determined using CCK-8 assay. DAC was observed
to inhibit the proliferation of the two cell lines in a time- and
dose-dependent manner (P<0.05; Fig.
1). In TFK-1 cells, treatment with 10 μM DAC for 120 h resulted
in 50% suppression of cell proliferation (Fig. 1A). In QBC939 cells, DAC also
significantly inhibited the cell growth, but to a lesser extent
than in TFK-1 cells (Fig. 1B),
indicating that TFK-1 cells are more sensitive to DAC than QBC939
cells. The long-term effect of DAC on CCA cells was assessed by
clonogenic assay. Treatment of TFK-1 cells with DAC for five days
led to a loss of clonogenicity in a dose-dependent manner (Fig. 1C). As shown in Fig. 1C, the number of tumor clones in the
0.5 μM DAC-treated group was markedly greater than that in the
50.0-μM group. The results demonstrated that DAC may reduce the
proliferation of CCA cells.
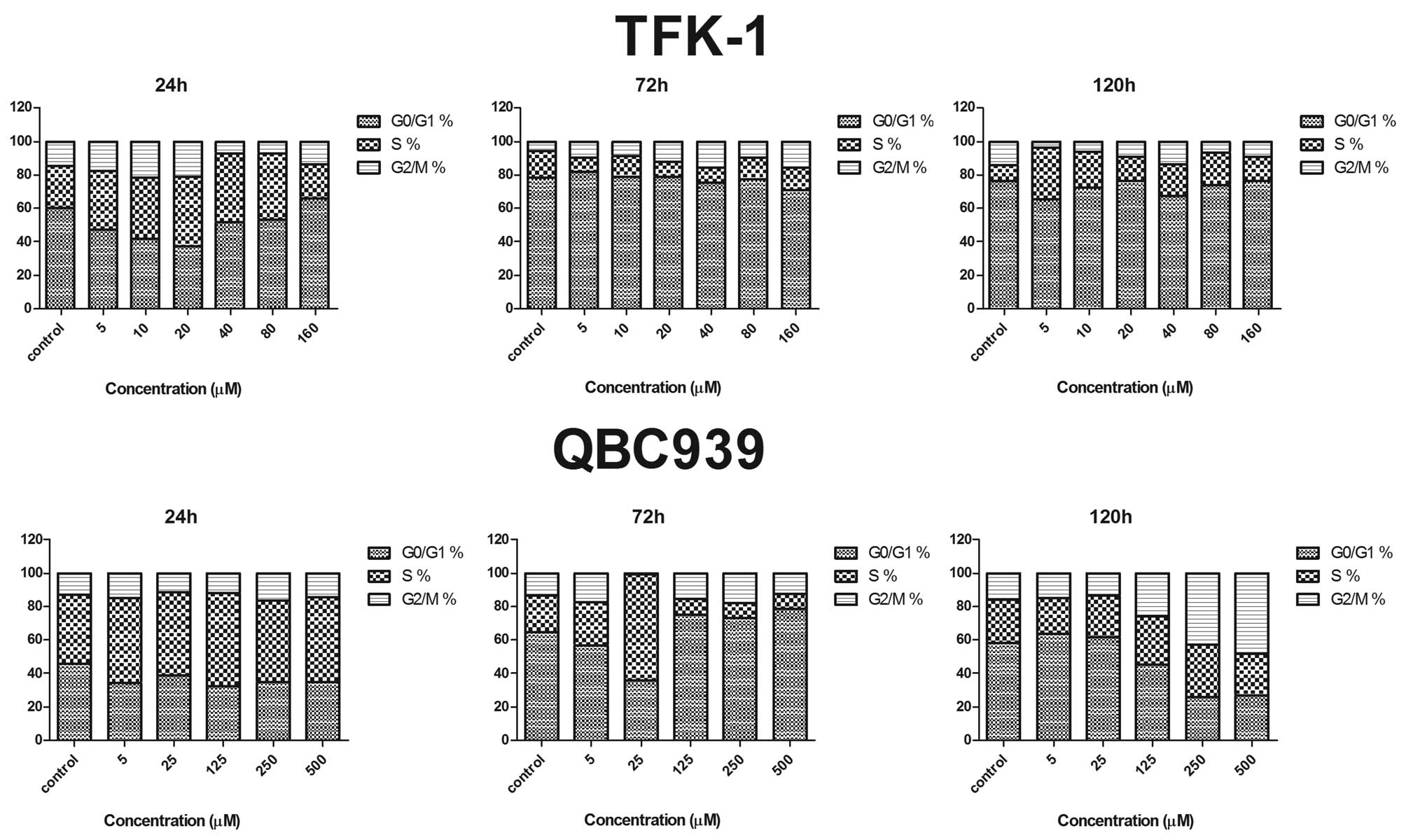
DAC induces cell cycle arrest in CCA
cells
To determine the mechanism by which DAC inhibits the
proliferation of CCA cells, the cell cycle distribution of TFK-1
and QBC939 cells treated with DAC for 24, 72 and 120 h was
determined. The percentage of cells in G0/G1, S and G2/M phases are
shown in Fig. 2. TFK-1 cells were
arrested slightly in G2/M phase in a dose-dependent manner when the
DAC concentration was at 40 μM. The cell number in G2/M phase
decreased rapidly when the concentration of DAC reached 80 μM, but
the cell number in G2/M phase increased slightly when the DAC
concentration exceeded 80 μM. Compared with the untreated TFK-1
cells, the accumulation of the cell population in G2/M phase was
accompanied by a concomitant decrease in the cell population in
G0/G1 phase. By contrast, no apparent alteration of cell cycle
distribution was identified in QBC939 cells following DAC treatment
for 24 and 72 h. However, following 120 h, an apparent increase in
G2/M and decrease in G0/G1 cells was observed following DAC
treatment (0–500 μM) in a dosage-dependent manner (Fig. 2; lower panel).
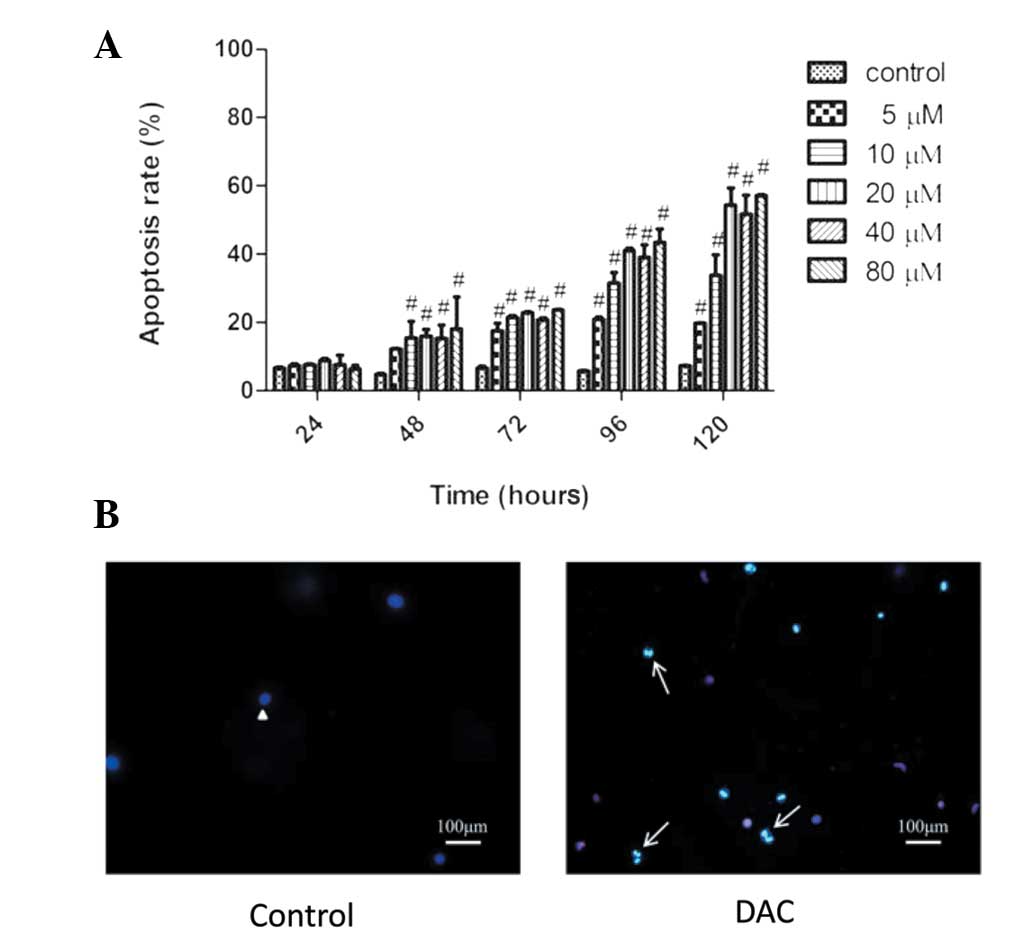
Inductive effect of DAC on TFK-1 cells
apoptosis
The effect of apoptosis in CCA cells following DAC
treatment was analyzed. TFK-1 cells were incubated with 0–80 μM DAC
for 24–120 h and then stained with Annexin V and PI. DAC
significantly induced apoptosis in a time- and dose-dependent
manner (Fig. 3A). With the increase
of concentration at 120 h, the percentage of apoptotic cells
increased from 10.8% in the control group to 57.03% in the 80 μM
DAC group. In addition, with the time of incubation, the percentage
of apoptotic cells varied from 9.17% at 24 h to 41.59% at 120 h in
the 20 μM-treated group. To further support the observed apoptosis
by DAC, the apoptotic morphological changes were determined under
the fluorescence microscope using Hoechst 33342/PI staining. As
shown in Fig. 3B, compared with the
control, TFK-1 cells exhibited typical apoptotic features following
DAC treatment, including cellular morphological change, apoptotic
bodies and condensation of chromatin (indicated by bright blue
staining).
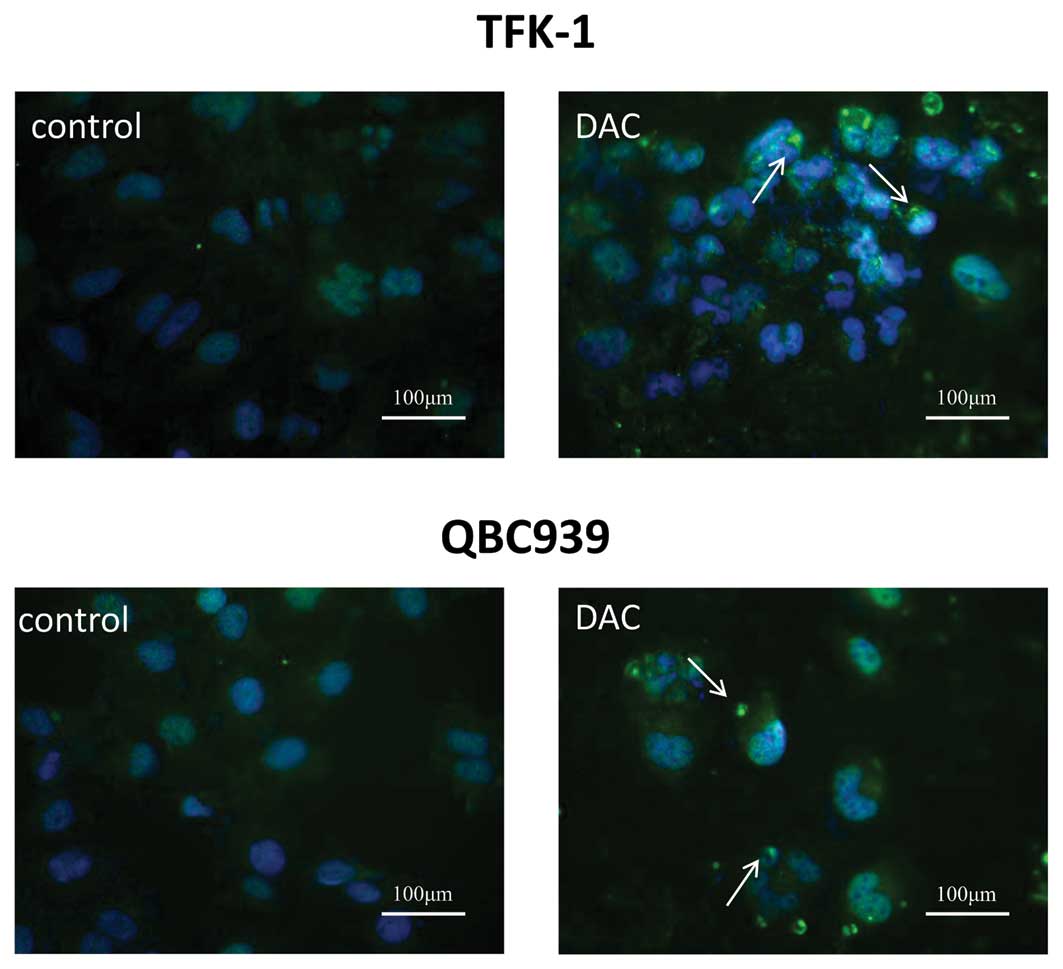
DAC induces autophagy of CCA cells
To assess whether a third possible mechanism may
contribute to the DAC-induced growth inhibition, autophagic cell
death in TFK-1 and QBC939 cells transiently transfected with a
GFP-LC3 plasmid was tested. Following treatment with DAC for three
days, GFP-LC3 puncta were examined under a confocal fluorescence
microscope. In TFK-1 cells, the number of puncta increased from 13
puncta per 100 cells for the control cells to 98 puncta per 100
cells for cells treated with DAC. Similarly, 77 puncta per 100
DAC-treated cells were identified, in comparison with eight puncta
per 100 control cells in QBC939 cells (Fig. 4). The results suggested that DAC may
induce autophagic cell death in CCA cells.
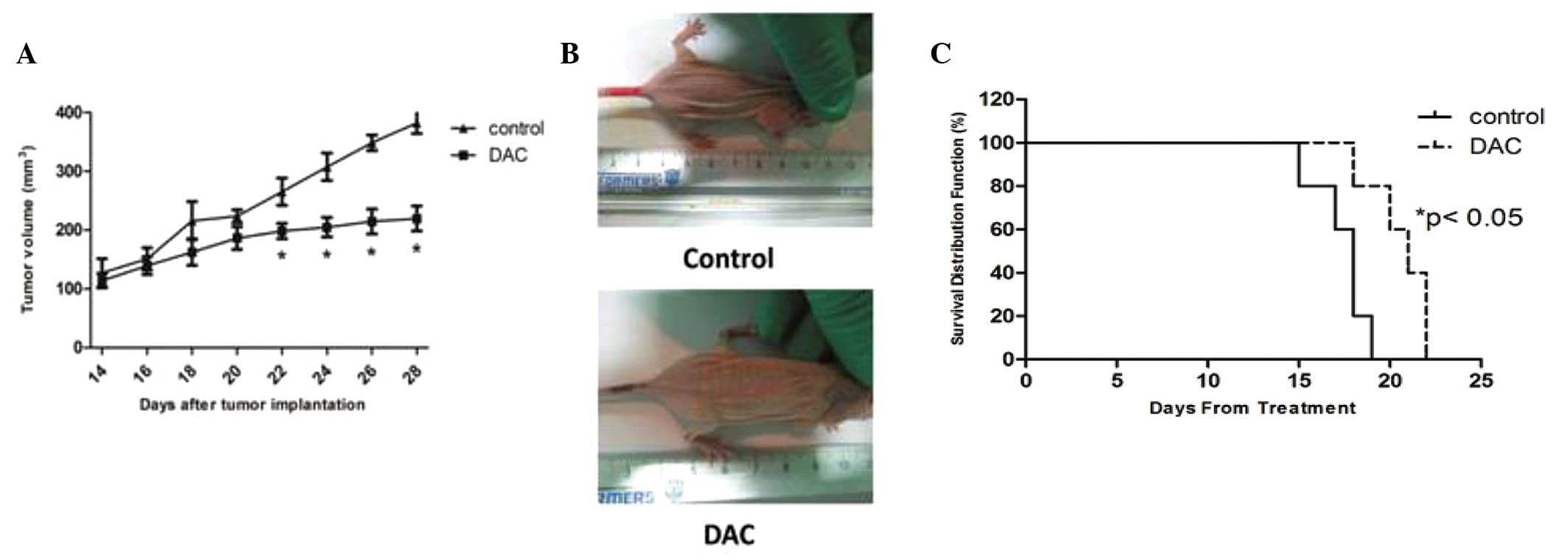
DAC reduces the growth of CCA
xenografts
To evaluate the value of DAC therapy in vivo,
a CCA xenograft mouse model generated by subcutaneous injection of
TFK-1 cells into nu/nu mice was used. As shown in Figs. 5A and B, daily administration of DAC
(0.8 mg/kg) for a two-week time period was able to reduce tumor
growth by ~42.5%. Furthermore, tumor growth inhibition was
associated with a significant increase in the survival of
DAC-treated animals. The Kaplan-Meier survival curves for each of
the two treatment groups are shown in Fig. 5C and the average survival rate of
DAC-treated animals was significantly increased. Thus, this clearly
demonstrated that DAC exerts a significant antitumor activity
against human CCA in vivo.
Discussion
DAC has been widely used as a DNA demethylating
agent to reactivate tumor suppressor genes silenced by aberrant
promoter hypermethylation. Following phosphorylation by
deoxycytidine kinase, DAC is incorporated into DNA. Once in the
DNA, DAC is recognized as a target cytosine by the DNMT enzyme. DAC
catalyzes the same reaction as normal cytosines with the formation
of a covalent intermediate between the catalytic cysteine of the
enzyme and 6-position of cytosine analogues (22). DNMT is thereby trapped on the DNA by
the suicide inhibitor, triggering DNA repair and degradation of the
enzyme. Previously, it has been well documented that DAC exhibits
potent antitumor activity, particularly in hematological
malignancies. Its therapeutic potential is currently under
investigation for treating various types of solid tumor. The
majority of previous trials have been designed to determine DAC
efficacy on solid tumors in combination with histone deacetylase
inhibitors, chemotherapy agents or even stimulators of the immune
system (23). In the present study,
DAC inhibited the growth of CCA cells in cultured cells and mouse
xenografts. CCK-8 assays showed that CCA TFK-1 and QBC939 cells
treated with DAC evidently lost cell viability, particularly with
the elongated incubation time and increased concentration. However,
the TFK-1 cells were more sensitive to DAC than QBC939 cells,
suggesting that the inhibitory effect of DAC may be cell
type-dependent. Consistently, the colony formation assay showed
that DAC could significantly decrease the clonogenic survival of
CCA cells, indicating that DAC treatment also produces long-term
effects on CCA cell growth.
One of the mechanisms by which antineoplastic agents
retard tumor growth is by arresting cell cycle progression. The
results of the present study showed that DAC was capable of
inducing a G2/M cell cycle arrest in CCA cell lines to a certain
extent. Thus, these experimental results indicated that the
antitumor effect of DAC on TFK-1 and QBC939 cells is associated
with cell cycle arrest.
Additionally, consistent with other solid tumors
(23), the results of the current
study showed that DAC induces apparent apoptosis in CCA cell lines.
TFK-1 cells showed shrinkage and condensation of the nuclear
chromatin and cytoplasm. The results of morphological changes were
consistent with those of flow percentage of apoptotic cells
measured by flow cytometry following Annexin V/PI staining.
Furthermore, Schnekenburger et al previously
reported that autophagy may be involved in DAC-induced cytotoxicity
in human chronic myelogenous leukemias (24). In the present study, compared with
the control group, treatment with DAC enhanced the formation of
autophagosomes. Therefore, DAC-mediated growth inhibition of CCA
cells may also be via induction of autophagy.
In the current study, the effects of DAC on CCA
tumor cell lines were also evaluated in vivo using a CCA
xenograft model. A total of 0.8 mg/kg DAC for two weeks
significantly reduced the growth of xenografted TFK-1 cells by
42.5%. In addition, the mice treated with DAC suffered from a
comparatively decreased tumor burden and exhibited prolonged
survival than that of the control groups. The results are
consistent with those from the Lu Z group (25). However, Yi et al previously
reported that the treatment of endometrial tumors with DAC at a
dose of 15 mg intraperitoneally injected thrice weekly for six
consecutive weeks was unable to significantly suppress the tumor
growth, with the exception of treatment with a combination of DAC
and valproic acid (26). The
results suggested that different tumor types require different DAC
regimens.
In summary, the present study demonstrated that DAC
is capable of suppressing the growth of CCA cells in vitro
and in vivo, suggesting a promising therapeutic development
of DAC for treating CCA.
Acknowledgements
The study was supported by the Development of Novel
Nano-Drug Delivery System Loaded with Traditional Chinese
Anticancer Medicine for the Targeted Therapy of Malignant Tumors
grant, which was issued by the Chinese Ministry of Science and
Technology (no. 2010DFA31870).
References
|
1
|
de Groen PC, Gores GJ, LaRusso NF,
Gunderson LL and Nagorney DM: Biliary tract cancers. N Engl J Med.
341:1368–1378. 1999.
|
|
2
|
Shaib Y and El-Serag HB: The epidemiology
of cholangiocarcinoma. Semin Liver Dis. 24:115–125. 2004.
|
|
3
|
Khan SA, Emadossadaty S, Ladep NG, et al:
Rising trends in cholangiocarcinoma: is the ICD classification
system misleading us? J Hepatol. 56:848–854. 2012.
|
|
4
|
Nagino M, Ebata T, Yokoyama Y, et al:
Evolution of surgical treatment for perihilar cholangiocarcinoma: a
single-center 34-year review of 574 consecutive resections. Ann
Surg. 258:129–140. 2013.
|
|
5
|
Kitiyakara T and Chapman RW:
Chemoprevention and screening in primary sclerosing cholangitis.
Postgrad Med J. 84:228–237. 2008.
|
|
6
|
Wiencke K and Boberg KM: Current consensus
on the management of primary sclerosing cholangitis. Clin Res
Hepatol Gastroenterol. 35:786–791. 2011.
|
|
7
|
Beuers U: EASL Recognition Awardee 2009:
Prof. Raoul Poupon J Hepatol. 51:617–619. 2009.
|
|
8
|
Chapman R, Fevery J, Kalloo A, et al:
Diagnosis and management of primary sclerosing cholangitis.
Hepatology. 51:660–678. 2010.
|
|
9
|
Sano T, Shimada K, Sakamoto Y, Yamamoto J,
Yamasaki S and Kosuge T: One hundred two consecutive hepatobiliary
resections for perihilar cholangiocarcinoma with zero mortality.
Ann Surg. 244:240–247. 2006.
|
|
10
|
Shaib YH, Davila JA, Henderson L, McGlynn
KA and El-Serag HB: Endoscopic and surgical therapy for
intrahepatic cholangiocarcinoma in the united states: a
population-based study. J Clin Gastroenterol. 41:911–917. 2007.
|
|
11
|
Kozarek RA: Inflammation and
carcinogenesis of the biliary tract: update on endoscopic
treatment. Clin Gastroenterol Hepatol. 7(11 Suppl): S89–S94.
2009.
|
|
12
|
Blechacz B and Gores GJ:
Cholangiocarcinoma: advances in pathogenesis, diagnosis, and
treatment. Hepatology. 48:308–321. 2008.
|
|
13
|
Ustundag Y and Bayraktar Y:
Cholangiocarcinoma: a compact review of the literature. World J
Gastroenterol. 14:6458–6466. 2008.
|
|
14
|
Ramacciato G, Nigri G, Bellagamba R, et
al: Univariate and multivariate analysis of prognostic factors in
the surgical treatment of hilar cholangiocarcinoma. Am Surg.
76:1260–1268. 2010.
|
|
15
|
Nuzzo G, Giuliante F, Ardito F, et al:
Intrahepatic cholangiocarcinoma: prognostic factors after liver
resection. Updates Surg. 62:11–19. 2010.
|
|
16
|
Anderson CD, Pinson CW, Berlin J and Chari
RS: Diagnosis and treatment of cholangiocarcinoma. Oncologist.
9:43–57. 2004.
|
|
17
|
Cheung HH, Lee TL, Davis AJ, Taft DH,
Rennert OM and Chan WY: Genome-wide DNA methylation profiling
reveals novel epigenetically regulated genes and non-coding RNAs in
human testicular cancer. Br J Cancer. 102:419–427. 2010.
|
|
18
|
Jurkowska RZ, Jurkowski TP and Jeltsch A:
Structure and function of mammalian DNA methyltransferases.
Chembiochem. 12:206–222. 2011.
|
|
19
|
Oki Y, Aoki E and Issa JP: Decitabine -
bedside to bench. Crit Rev Oncol Hematol. 61:140–152. 2007.
|
|
20
|
Griffiths EA and Gore SD: DNA
methyltransferase and histone deacetylase inhibitors in the
treatment of myelodysplastic syndromes. Semin Hematol. 45:23–30.
2008.
|
|
21
|
Gravina GL, Festuccia C, Marampon F, et
al: Biological rationale for the use of DNA methyltransferase
inhibitors as new strategy for modulation of tumor response to
chemotherapy and radiation. Mol Cancer. 9:3052010.
|
|
22
|
Damaraju VL, Mowles D, Yao S, et al: Role
of human nucleoside transporters in the uptake and cytotoxicity of
azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids.
31:236–255. 2012.
|
|
23
|
Yang D, Torres CM, Bardhan K, Zimmerman M,
McGaha TL and Liu K: Decitabine and vorinostat cooperate to
sensitize colon carcinoma cells to Fas ligand-induced apoptosis in
vitro and tumor suppression in vivo. J Immunol. 188:4441–4449.
2012.
|
|
24
|
Schnekenburger M, Grandjenette C, Ghelfi
J, et al: Sustained exposure to the DNA demethylating agent,
2′-deoxy-5-azacytidine, leads to apoptotic cell death in chronic
myeloid leukemia by promoting differentiation, senescence, and
autophagy. Biochem Pharmacol. 81:364–378. 2011.
|
|
25
|
Chen MY, Liao WS, Lu Z, et al: Decitabine
and suberoylanilide hydroxamic acid (SAHA) inhibit growth of
ovarian cancer cell lines and xenografts while inducing expression
of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and
autophagy. Cancer. 117:4424–4438. 2011.
|
|
26
|
Yi TZ, Li J, Han X, et al: DNMT inhibitors
and HDAC inhibitors regulate E-cadherin and Bcl-2 expression in
endometrial carcinoma in vitro and in vivo. Chemotherapy. 58:19–29.
2012.
|