Introduction
Juvenile papillomatosis (JP), also termed Swiss
cheese disease, of the breast is a rare and benign disease, which
predominantly occurs in females aged <30 years. JP is often
preoperatively diagnosed as fibroadenoma due to similarities in the
clinical manifestations. However, ductal papillomatosis and cysts
are dominant microscopic features that are distinct to JP (1–3). Since
JP was first presented by Rosen et al (1) in 1980, ~400 cases have been reported,
with the majority observed in Caucasian patients and rare cases
occurring in patients of Asian origin (4). A family history of breast cancer (in
first- or second-degree relatives) has been identified in 26–58% of
JP patients (5–8) and breast carcinomas coexisting with JP
lesions have been observed in certain cases (1,5,7). A
long-term follow-up study demonstrated that certain patients
developed breast cancer eight-nine years subsequent to the
diagnosis of JP (8); therefore, JP
patients appear to have an increased risk of developing breast
cancer. The current report presents a case of bifocal JP in an
11-year-old Chinese female and describes a review of the
literature. Furthermore, the diagnosis and treatment of JP, as well
as the association between JP and breast carcinoma are presented.
Written informed consent was obtained from the patient’s
family.
Case report
In January 2010, an 11-year-old Chinese female was
admitted to the Second Department of Breast Cancer, Tianjin Medical
University Cancer Institute and Hospital (Tianjin, China)presenting
with a tender mass in the left breast, which had initially been
identified three months previously and had gradually enlarged. The
patient had not experienced menarche. The patient’s grandmother was
diagnosed with breast cancer at 52 years of age and the patient’s
mother had required a lumpectomy for a breast fibroadenoma at 16
years of age, and both individuals have been cured. No history of
hormonal or other teratogenic agent use was recorded in the patient
or in the mother during pregnancy. Upon physical examination the
patient appeared healthy with bilateral normal development of the
breasts. A firm, mobile, poorly-circumscribed, tender mass was
identified in the upper outer quadrant of the left breast, 1 cm
from the areola (size, 3×3×2 cm). The nipple and areola were normal
with no nipple discharge and no swollen axillary lymph nodes were
observed.
Ultrasonography of the breasts revealed that the
palpable mass was poorly defined, irregularly-shaped and
inhomogeneous with blood-flow signals. Furthermore, two additional,
smaller, impalpable lumps were identified. The first lesion was
located in the lower outer quadrant of the left breast (size,
0.9×0.5×1.0 cm) and did not exhibit subcutaneous association with
the primary lesion. The second lesion was identified in the upper
outer quadrant of the right breast (size, 0.6×0.4×0.4 cm). The two
masses were well-circumscribed, regularly shaped and hypoechoic.
Ultrasonography determined the three masses as fibroadenomas. A
mammography was not performed due to the young age of the patient
and no abnormalities were identified during blood tests.
The patient and the patient’s parents selected
medical follow-up examinations for the mass in the right breast,
rather than surgery or a core needle biopsy; however, the two
lesions in the left breast were completely excised. One tumor mass
measured 2.4×1.5×1.3 cm and the other tumor mass measured
1.1×0.7×0.6 cm. The two lesions were gray, well-demarcated, firm
and exhibited no visible cysts, which was consistent with the
diagnosis of fibroadenoma, as determined by ultrasound.
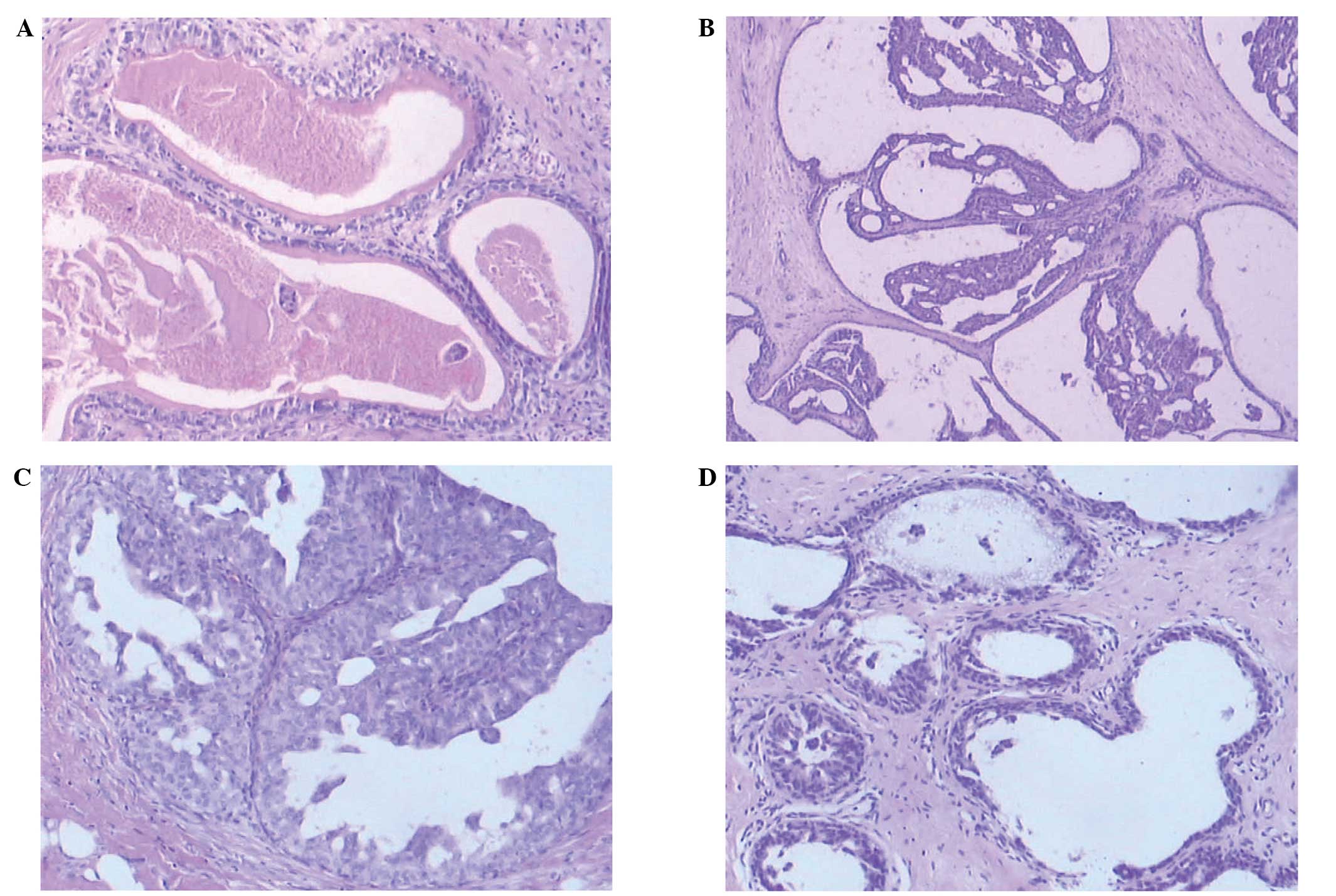
Furthermore, upon microscopic examination, the two lesions
demonstrated similar features. However, histopathology revealed
multiple dilated ducts containing inspissated secretions and foamy
cells, as well as intracystic papillary epithelial proliferation
with apocrine metaplasia (Fig. 1).
Thus, the diagnosis of JP of the breast was established. The JP was
bifocal as the two lesions were in different quadrants of the
breast and were not subcutaneously associated.
Regular patient follow-up, by physical examination
and ultrasonography of the breasts, is ongoing, and to date has
been conducted for 48 months. No local recurrence or malignant
change occurred and ultrasonography of the mass in the right breast
exhibited no change. Further follow-up did not indicate the
development of new breast disorders among the patient’s
relatives.
Discussion
Papillomatosis has an average of age of onset of 40
years and is associated with a relative risk for breast cancer
development in adults (3). In 1980,
Rosen et al (1) reported 37
cases of papillomatosis in young females with a mean age of 19
years (range, 10–44 years), and defined this novel disease as JP
due to its clinical and microscopic features. Thus far, ~400 cases
of JP have been reported, the majority in Caucasian females aged
<30 years at the time of diagnosis. The present study identified
10 cases of JP in males to date by conducting a search of the
English literature using PubMed (http://www.ncbi.nlm.nih.gov/pubmed) (9–11).
Cases of JP in Asian individuals were rare and, thus, fewer reports
exist. Whether the difference in incidence between Caucasian and
Asian populations is due to genetic or environmental factors
remains unclear.
The typical manifestation of JP is a unifocal tumor,
commonly located in the upper outer quadrant or outer half of the
breast, and is firm, well-circumscribed, mobile, painless and
generally measures <3 cm in diameter (5). Reports of bloody nipple discharge were
unusual (3,5,12).
When the clinical diagnosis was determined prior to surgery, it was
typically fibroadenoma. Mammography is not routinely recommended
for diagnosis or follow-up in females <35 years; however, the
few reported mammographic findings regarding JP revealed a
well-circumscribed homogeneous opacity, which is similar to that
observed in fibroadenomas and cysts (13). Ultrasonography is the preferred
imaging technique for JP patients as it facilitates with the
differentiation between JP and similar cystic lesions,
fibroadenomas, phyllodes tumors, intracystic papillomas and breast
cancer (14). Sonographically, the
JP lesion presented as a poorly-defined heterogeneous mass with
various small, round, echo-free areas, predominantly observed close
to the border of the lesion (15).
Microscopically, the typical histopathological features are duct
papillomatosis with or without epithelial atypia, apocrine and
non-apocrine cysts, duct stasis and sclerosing adenosis (1). Papillomatosis and cysts are the
dominant diagnostic criteria of JP. A case report describing the
fine-needle aspiration cytology of JP (16) revealed the tumor to be comprised of
sheets of hyperplastic breast epithelium with areas resembling
fibroadenoma, and containing macrophages and apocrine cells.
Although it is difficult to diagnose JP solely by its cytology, a
combination of clinical and cytological findings may facilitate
with the diagnosis of JP. There is no evidence to associate
hormonal agent use or reproductive history with the occurrence of
JP in young individuals, nor to associate JP with the maternal use
of teratogenic agents during pregnancy (5).
Various breast disorders in children and young
adults must be distinguished from JP. Rosen (6) described the rare types of papillary
duct hyperplasia, which are observed in adolescence, including
papilloma, papillomatosis and sclerosing papillomatosis. The most
common symptom of papillary duct hyperplasia was the presence of a
mass, although certain cases also exhibited nipple discharge, or
presented with nipple discharge alone; however, all of these
lesions lacked the cystic component that is characteristic of JP
(6,17). Breast cancer is rare in children,
however, when it does occur it most commonly takes the form of a
secretory carcinoma and presents as a long-standing breast mass,
which is occasionally painful (18). Nipple discharge was rarely
identified. Secretory breast cancer is characterized by the
presence of abundant intracellular and extracellular secretions,
and intracytoplasmic vacuoles (19). Furthermore, immunoperoxidase
staining for α-lactalbumin is typically positive in secretory
carcinoma, but negative in JP (18). However, Rosen et al (5) reported a case of secretory carcinoma
arising from JP and a case of JP with contralateral secretory
carcinoma, therefore, the association between JP and secretory
breast carcinoma appears likely and requires investigation.
Previous studies have demonstrated a relatively
strong association between JP and breast cancer, particularly when
there is a family history of breast cancer. Rosen et al
(5) reviewed 84 cases of JP in 1982
and identified that 26% of the patients had a family history of
breast cancer in at least one female relative. The majority of
breast cancer cases were observed in older, secondary relatives
(for example grandmothers or great aunts), although instances of
maternal breast carcinoma were also reported. This may have been
due to the young age of the JP patients and, therefore, the
patients’ mothers or young female relatives may not have reached
the peak age of breast cancer incidence. This is supported by a
follow-up study of JP patients by Rosen and Kimmel in 1990
(8), in which 58% of cases had a
family history of breast cancer, with mothers and maternal aunts
exhibiting the highest risk. Additionally, Bazzocchi et al
(7) observed that 33% of JP
patients had a family history of breast cancer. These findings
indicate that JP may be a marker of breast cancer in the family of
the JP patients, thus, a thorough medical follow-up is recommended
for JP patients and their families. Furthermore, microscopic
evaluation revealed that breast carcinoma coexisted with JP in
certain cases. Bazzocchi et al (7) identified that 15% of the JP patients
presented with a coexisting carcinoma and Rosen et al
(5) described three cases of other
types of cancer coexisting with JP (n=84). Two of the patients
exhibited secretory carcinoma (one arising from JP and another with
contralateral secretory cancer) and the two patients had a maternal
history of breast cancer. In addition, although the follow-up data
was insufficient, the JP patients appeared to be at an increased
risk of developing breast cancer. A previous study of 41 patients
with a median follow-up period of 14 years demonstrated a 10%
incidence of subsequent breast carcinoma in patients with JP
(8). Although the risk of breast
cancer should not be exaggerated, patients exhibiting any one of
the following characteristics should be closely monitored for the
subsequent development of breast cancer: i) Positive family history
of breast cancer; ii) atypical proliferative lesions; iii)
bilateral lesions; iv) multifocal lesions; or v) recurrence of JP.
Patients with a positive family history of breast cancer and
recurrent bilateral JP are considered to be at the greatest risk.
However, due to the young age at which JP was diagnosed, the
majority of the patients identified in the current study had not
reached the peak age of incidence of breast cancer (range, 50–70
years). Further studies with a longer follow-up period are required
to determine the incidence of breast cancer in patients with
JP.
In conclusion, the recommended treatment strategy
for JP is complete excision of the cancerous lesion to reduce local
recurrence. On consideration of the current literature, it is
prudent to advise an annual clinical follow-up, including a
physical examination and/or ultrasonography of the breasts for JP
patients, and for the patients’ female relatives, particularly
those with a family history of breast cancer and with recurrent or
bilateral JP.
References
|
1
|
Rosen PP, Cantrell B, Mullen DL and DePalo
A: Juvenile papillomatosis (Swiss cheese disease) of the breast. Am
J Surg Pathol. 4:3–12. 1980.
|
|
2
|
Ibarra JA: Papillary lesions of the
breast. Breast J. 12:237–251. 2006.
|
|
3
|
Gill J and Greenall M: Juvenile
papillomatosis and breast cancer. J Surg Educ. 64:234–236.
2007.
|
|
4
|
Hsieh SC, Chen KC, Chu CC and Chou JM:
Juvenile papillomatosis of the breast in a 9-year-old girl. Pediatr
Surg Int. 17:206–208. 2001.
|
|
5
|
Rosen PP, Lyngholm B, Kinne DW and Beattie
EJ Jr: Juvenile papillomatosis of the breast and family history of
breast carcinoma. Cancer. 49:2591–2595. 1982.
|
|
6
|
Rosen PP: Papillary duct hyperplasia of
the breast in children and young adults. Cancer. 56:1611–1617.
1985.
|
|
7
|
Bazzocchi F, Santini D, Martinelli G,
Piccaluga A, Taffurelli M, Grassigli A and Marrano D: Juvenile
papillomatosis (epitheliosis) of the breast. A clinical and
pathologic study of 13 cases. Am J Clin Pathol. 86:745–748.
1986.
|
|
8
|
Rosen PP and Kimmel M: Juvenile
papillomatosis of the breast. A follow-up study of 41 patients
having biopsies before 1979. Am J Clin Pathol. 93:599–603.
1990.
|
|
9
|
Sund BS, Topstad TK and Nesland JM: A case
of juvenile papillomatosis of the male breast. Cancer. 70:126–128.
1992.
|
|
10
|
Pacilli M, Sebire NJ, Thambapillai E and
Pierro A: Juvenile papillomatosis of the breast in a male infant
with Noonan Syndrome, café au lait spots, and family history of
breast carcinoma. Pediatr Blood Cancer. 45:991–993. 2005.
|
|
11
|
Tan TY, Amor DJ and Chow CW: Juvenile
papillomatosis of the breast associated with neurofibromatosis 1.
Pediatr Blood Cancer. 49:363–364. 2007.
|
|
12
|
Sanguinetti A, Fioriti L, Brugia M, Roila
F, Farabi R, Sidoni A and Avenia N: Juvenile papillomatosis of the
breast in young male: a case report. G Chir. 32:374–375. 2011.
|
|
13
|
Taffurelli M, Santini D, Martinelli G, et
al: Juvenile papillomatosis of the breast: A multidisciplinary
study. Pathol Annu. 26:25–35. 1991.
|
|
14
|
Ohlinger R, Schwesinger G, Schimming A,
Köhler G and Frese H: Juvenile papillomatosis (JP) of the female
breast (Swiss Cheese Disease) - role of breast ultrasonography.
Ultraschall Med. 26:42–45. 2005.
|
|
15
|
Kersschot EA, Hermans ME, Pauwels C, et
al: Juvenile papillomatosis of the breast: sonographic appearance.
Radiology. 169:631–633. 1988.
|
|
16
|
Ostrzega N: Fine-needle aspiration
cytology of juvenile papillomatosis of breast: a case report. Diagn
Cytopathol. 9:457–460. 1993.
|
|
17
|
Batchelor JS, Farah G and Fisher C:
Multiple breast papillomas in adolescence. J Surg Oncol. 54:64–66.
1993.
|
|
18
|
Buchino JJ, Moore GD and Bond SJ:
Secretory carcinoma in a 9-year-old girl. Diagn Cytopathol.
31:430–431. 2004.
|
|
19
|
Serour F, Gilad A, Kopolovic J and Krispin
M: Secretory breast cancer in childhood and adolescence: report of
a case and review of the literature. Med Pediatr Oncol. 20:341–344.
1992.
|