Introduction
Primitive neuroectodermal tumor (PNET) is included
in the Ewing family of tumors (ESFTs) as they exhibit identical
cytogenetic changes. The specific translocation t(11;22)(q24;q12)
was identified in Ewing’s sarcoma and PNET, however, various other
translocation patterns may also be involved (1). ESFTs, which also include extraosseous
Ewing’s sarcoma and Askin’s tumor (Ewing’s sarcoma of the chest
wall) are aggressive malignant primary tumors of the bone, which
are the second most common bone tumor in children and adolescents.
PNET is divided into central PNET and peripheral PNET according to
their location or origin. There are various symptoms in peripheral
PNET which can occur in almost all areas of the body except for the
central nerve system (2,3). A common feature of image findings of
peripheral PNET is a large and infiltrative soft tissue mass with
an ill-defined, necrotic region and heterogeneous enhancement
(2,4). However, it is not easy to diagnose
peripheral PNET by imaging alone due to insufficient radiological
studies (2). Combination therapy
including chemotherapy, radiation therapy and surgery are chosen
for treatment against peripheral PNET (5), however the patient prognosis is poor
(6). Recently, a few cases of PNET
occurring in the urologic region such as bladder, kidney and
adrenal glands have been described (7,8). PNET
of the prostate, which was also classified as peripheral PNET, was
first reported in 2003 (9). To the
best of our knowledge, the present study reports the ninth case of
PNET of the prostate gland thus far. Written informed consent was
obtained from the family of the patient.
Case report
A 23-year-old male presented to Oita Medical Center
(Oita, Japan) with the complaint of dysuria and anal pain. A
computed tomography (CT) scan and magnetic resonance imaging (MRI)
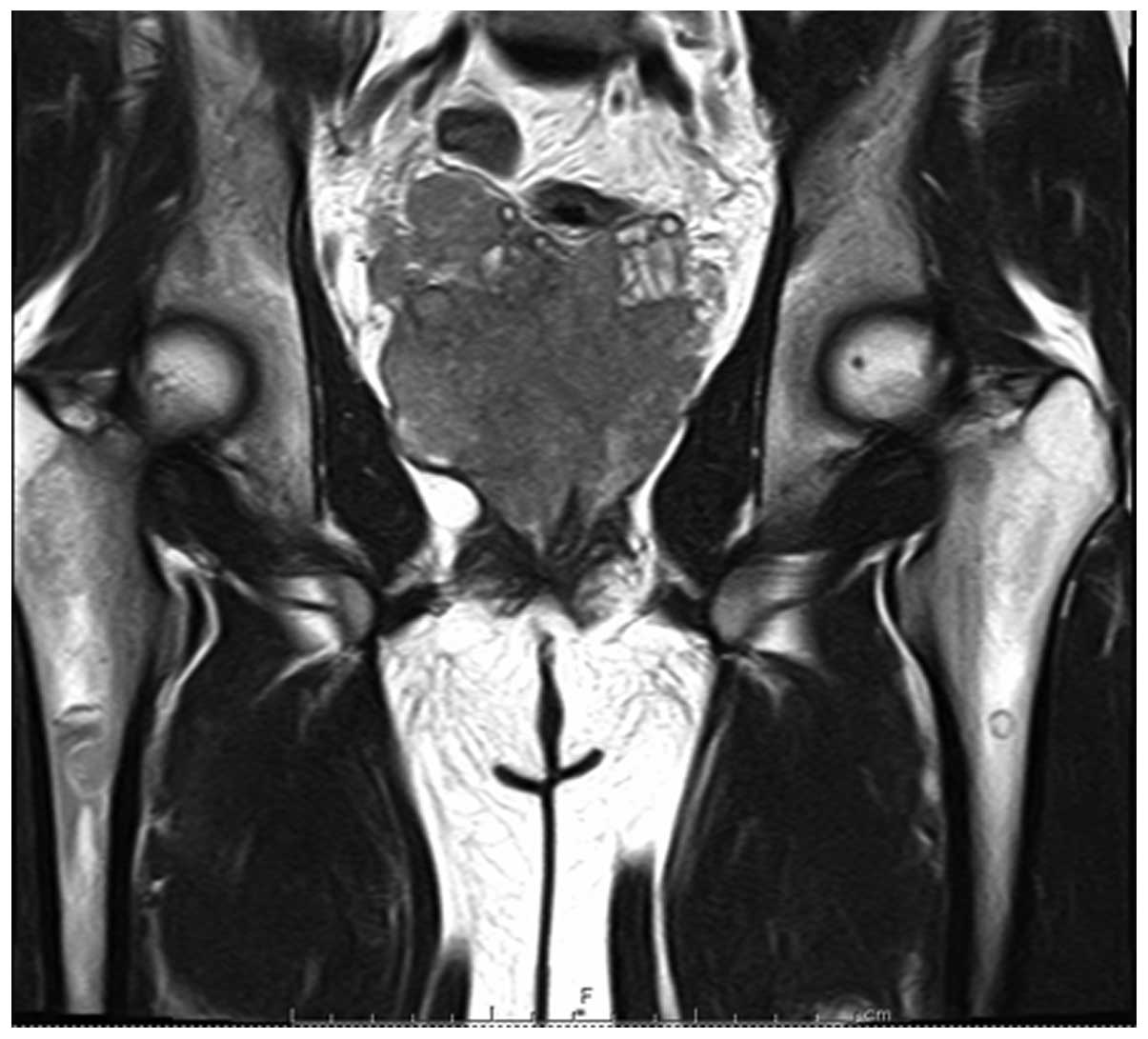
revealed a large solid tumor in the pelvic region replacing the
prostate (Fig. 1) and multiple
swollen lymph nodes. Cystostomy and biopsy of the prostate gland
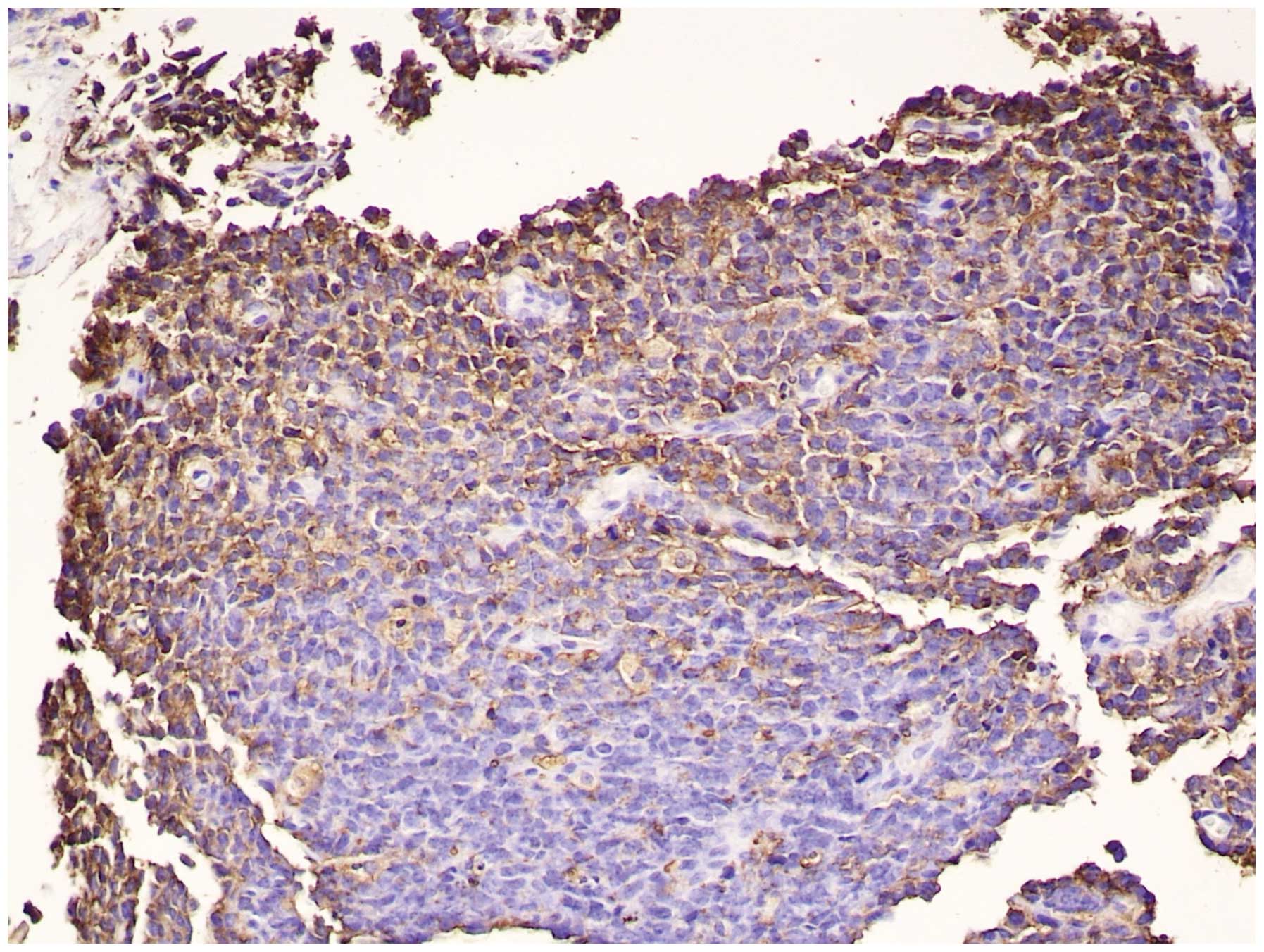
were performed. Histopathological analysis demonstrated small round
tumor cells, indicating a PNET, however, no rosette structure was
observed. The tumor cells were positive for MIC-2 (Fig. 2), cytokeratin, vimentin and neural
cell adhesion molecule, but negative for the other immunostains,
chromogranin A, neurofilament, neuron-specific enolase, leukocyte
common antigen, desmin, HHF35, sarcomeric actin, myogenic
differentiation 1 and myoglobin. The histopathological results
indicated a diagnosis of PNET of the prostate, and the patient was
admitted to Oita University Hospital (Yufu, Japan) for treatment.
Two days after admission, the patient complained of lower back
pain. Re-examination by CT scan and MRI revealed multiple bone
metastases to the dorsal and lumbar vertebrae, pelvic bone and
bilateral thighbones, multiple lung metastases and a fracture of
the fourth lumbar vertebra caused by metastasis. An additional
genetic examination was performed; the Ewing’s sarcoma-friend
leukemia virus integration 1 fusion gene was not detected by
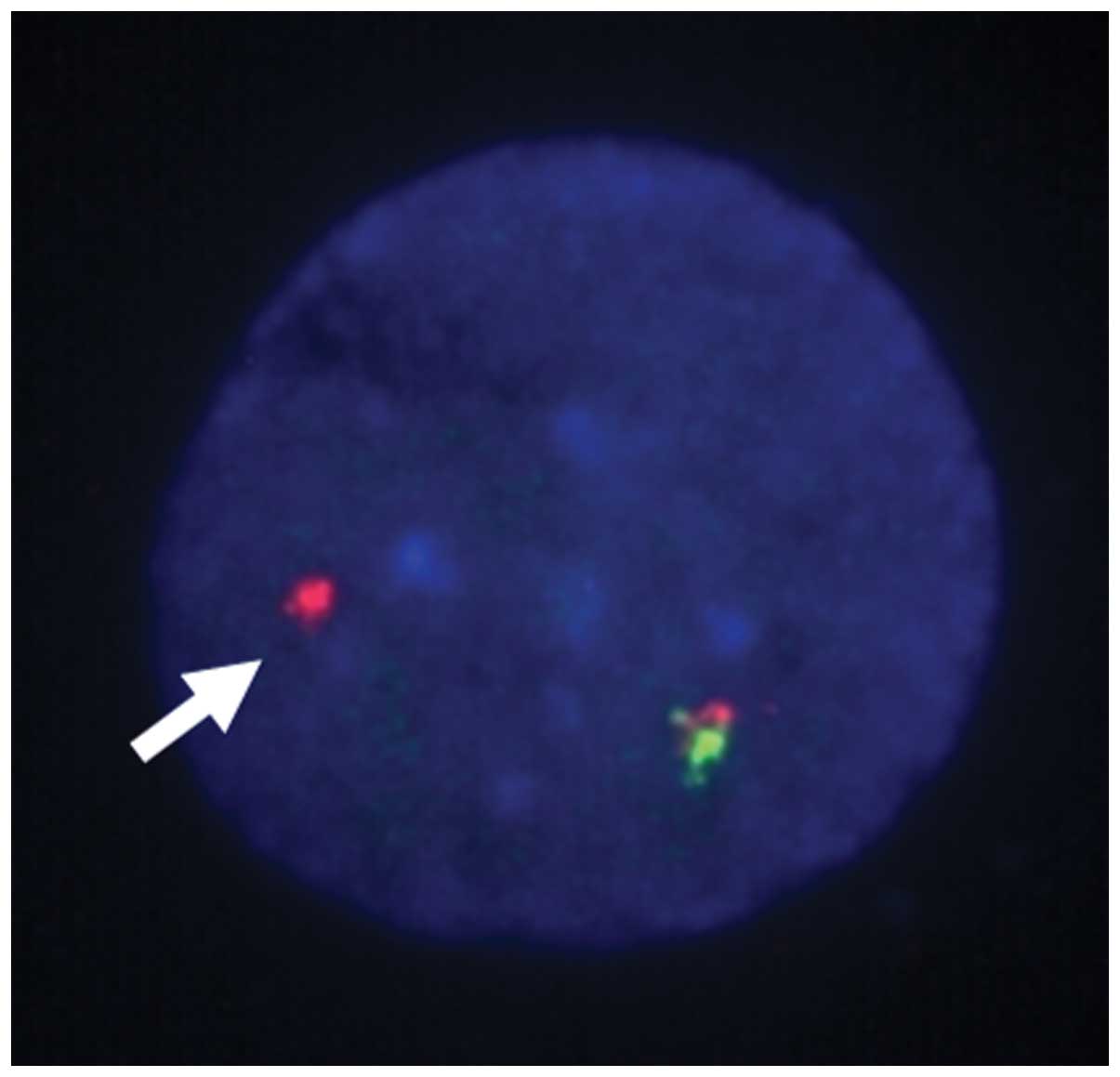
quantitative polymerase chain reaction, however, split signals were
observed in fluorescence in situ hybridization (FISH)
analysis using a Ewing sarcoma breakpoint region 1 (EWSR1) probe
(SRL Laboratories, Inc., Tokyo, Japan; Fig. 3). A diagnosis of PNET of the
prostate was established, and treatment with systemic chemotherapy
commenced. Following one cycle of chemotherapy (ifosfamide, 2
mg/m2/week) the patient exhibited various side-effects,
including dizziness, headache and nausea. MRI revealed multiple
metastases to the intracranial meninges and a CT scan indicated a
poor chemotherapy response. Therefore, best supportive care was
administered and the patient succumbed approximately four months
after the initial onset of symptoms.
Discussion
A multimodal therapeutic regimen that includes a
combination of chemotherapy, surgery and radiotherapy is the
current treatment strategy for the Ewing sarcoma family of tumors.
Additionally, PNET of the prostate is commonly treated by
combination therapy (Table I)
(9,12–18).
In all but one of the reported cases of PNET of the prostate, in
which the treatment strategy was not detailed, neoadjuvant or
adjuvant chemotherapy were effectively administered. Two cases in
the literature exhibited a reduction in the size of the primary
tumor following neoadjuvant chemotherapy (12,15)
and four cases underwent radical prostatectomy (9,12,15,18).
Chemotherapeutic agents, including ifosfamide, etoposide,
vincristine, Adriamycin and doxorubicin, were used as single or
combination therapies. Although a standard treatment has not yet
been established, chemotherapy is a potentially effective treatment
for localized PNET of the prostate (1). In the present study, however, multiple
metastases in various regions inhibited the effectiveness of the
chemotherapy. Previously, the presence of metastatic disease was
reported as the most unfavorable prognostic factor for Ewing
sarcoma family tumors (19). In
particular, patients exhibiting extrapulmonary metastatic disease
have demonstrated a worse prognosis compared with individuals
exhibiting solitary pulmonary metastatic disease. Furthermore,
tumor size (>100 ml), location within the pelvis, older age
(>14 years) and insufficient response to initial therapy are
reported as poor prognostic factors (20–22).
In the current study, the presence of a number of these poor
prognostic factors resulted in unsatisfactory progress. Thus,
treatment for high-risk patients may require the development of a
novel therapeutic regimen distinct from those used to treat local
cases of Ewing’s sarcoma, for example gemcitabine, docetaxel or
targeted molecular agents.
 | Table IClinical characteristics of reported
primitive neuroectodermal tumor of prostate cases. |
Table I
Clinical characteristics of reported
primitive neuroectodermal tumor of prostate cases.
| Case, n | First author
(reference) | Age, years | Metastases | Chemotherapeutic
agents | Radiation | Surgery | Survival, months |
|---|
| 1 | Colecchia et
al (9) | 31 | None | Ifosfamide,
etoposide, vincristine, Adriamycin | Yes | Yes | N/A |
| 2 | Peyromaure et
al (12) | 27 | None | Ifosfamide,
etoposide, vincristine, doxorubicin | Yes | Yes | 2 |
| 3 | Thete et al
(13) | 26 | None | N/A | N/A | N/A | N/A |
| 4 | Kumar et al
(14) | 25 | None | Ifosfamide,
vincristine, actinomycin D, Adriamycin | N/A | N/A | N/A |
| 5 | Funahashi et
al (15) | 20 | Lung | Ifosfamidenone | No | Yes | 10 |
| 6 | Mohsin et al
(16) | 29 | Lymph node
Lung | First-line;
vincristine, doxorubicin
Second-line; docetaxel, gemzar
Third-line; ifosfamide | No | No | N/A |
| 7 | Al Haddabi et
al (17) | 24 | None | Ifosfamide,
doxorubiin, vincristine, etoposide | No | No | N/A |
| 8 | Wu et al
(18) | 29 | Lung | Multiagent (detail
N/A) | No | Yes | 12 |
The present case lacked the specific Ewing’s
sarcoma/PNET translocation t(11;22)(q24;q12), however, EWSR1 gene
rearrangement on chromosome 22 was detected by performing FISH
analysis. Overall, ~15% of histopathologically defined
MIC-2-positive Ewing sarcomas lack the classical specific
translocation. However, in the majority of the remaining cases,
variant translocations involve chromosome 22q12. Chromosome 21q22
(10% of Ewing sarcoma), and chromosomes 7q22, 17q12 and 2q36
(<1% of Ewing sarcoma) were reported as translocation partners
(5), however, no translocation
partner could be detected in the present case. Identifying the
translocation partner may be important as the translocation type
may contribute to an unfavorable prognostic outcome.
In all reported cases of PNET of the prostate gland,
a large-sized primary tumor replaced the prostate at the diagnosis.
Despite the large size of the tumor in the pelvic area, the
predominant complaint of the patients was the common symptom of
dysuria. Thus, PNET of the prostate gland should be considered when
young males (20–30 years old) present to hospital with the
complaint of dysuria, to improve the rate of early diagnosis.
Non-invasive examinations, including ultrasound or digital rectal
examination, are sufficient to identify the presence of this
disease in the prostatic region.
References
|
1
|
Balamuth NJ and Womer RB: Ewing’s sarcoma.
Lancet Oncol. 11:184–192. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gupta P, Hari S and Thulkar S: Imaging
spectrum of peripheral primitive neuroendocrine tumours. Singapore
Med J. 54:463–472. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Khong PL, Chan GC, Shek TW, Tam PK and
Chan FL: Imaging of peripheral PNET: common and uncommon locations.
Clin Radiol. 57:272–277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Qian X, Kai X, Shaodong L, Gaohong C, Hong
M and Jingjing L: Radiological and clinicopathological features of
pPNET. Eur J Radiol. 82:e888–e893. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bemstein M, Kovar H, Paulussen M, et al:
Ewing’s sarcoma family of tumors: current management. Oncologist.
11:503–519. 2006. View Article : Google Scholar
|
|
6
|
Ibarburen C, Haberman JJ and Zerhouni EA:
Peripheral primitive neuroectodermal tumors. CT and MRI evaluation.
Eur J Radiol. 21:225–232. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ellinger J, Bastian PJ, Hauser S, Biermann
K and Müller SC: Primitive neuroectodermal tumor: rare, highly
aggressive differential diagnosis in urologic malignancies.
Urology. 68:257–262. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tsang YP, Lang BH, Tam SC and Wong KP:
Primitive neuroectodermal adrenal gland tumour. Hong Kong Med J.
20:444–446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Colecchia M, Degrada G, Poliani PL,
Messina A and Pilotti S: Primary primitive peripheral
neuroectodermal tumor of the prostate. Immunophenotypic and
molecular study of a case. Arch Pathol Lab Med. 127:e190–e193.
2003.PubMed/NCBI
|
|
10
|
Scotlandi K, Benini S, Manara MC, et al:
Murine model for skeletal metastases of Ewing’s sarcoma. J Orthop
Res. 18:959–966. 2000. View Article : Google Scholar
|
|
11
|
de Alava E and Gerald WL: Molecular
biology of the Ewing’s sarcoma/primitive neuroectodermal tumor
family. J Clin Oncol. 18:204–213. 2000.PubMed/NCBI
|
|
12
|
Peyromaure M, Vieillefond A, Boucher E, et
al: Primitive neuroectodermal tumor of the prostate. J Urol.
170:182–183. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thete N, Rastogi D, Arya S, et al:
Primitive neuroectodermal tumour of the prostate gland: ultrasound
and MRI findings. Br J Radiol. 80:e180–e183. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kumar V, Khurana N, Rathi AK, et al:
Primitive neuroectodermal tumor of prostate. Indian J Pathol
Microbiol. 51:386–388. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Funahashi Y, Yoshino Y and Hattori R:
Ewing’s sarcoma/primitive neuroectodermal tumor of the prostate.
Int J Urol. 16:7692009. View Article : Google Scholar
|
|
16
|
Mohsin R, Hashmi A, Mubarak M, et al:
Primitive neuroectodermal tumor/Ewing’s sarcoma in adult
uro-oncology: A case series from a developing country. Urol Ann.
3:103–107. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al Haddabi I, Al Bahri M and Burney I:
Cytokeratin-positive primitive neuroectodermal tumor of the
prostate: case report and review of literature. Indian J Pathol
Microbiol. 55:569–571. 2012. View Article : Google Scholar
|
|
18
|
Wu T, Jin T, Luo D, Chen L and Li X:
Ewing’s sarcoma/primitive neuroectodermal tumor of the prostate: A
case report and literature review. Can Urol Assoc J. 7:E458–E459.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Subbian V, Anderson P, Lazar AJ, et al:
Ewing’s sarcoma: standard experiental treatment options. Curr Treat
Options Oncol. 10:126–140. 2009. View Article : Google Scholar
|
|
20
|
Cotterill SJ, Ahrens S, Paulussen M, et
al: Prognostic factors in Ewing’s tumor of bone: analysis of 975
patients from the European Intergroup Cooperative Ewing’s Sarcoma
Study Group. J Clin Oncol. 18:3108–3114. 2000.PubMed/NCBI
|
|
21
|
Paulussen M, Ahrens S, Dunst J, et al:
Localized Ewing tumor of bone: final results of the cooperative
Ewing’s Sarcoma Study CESS 86. J Clin Oncol. 19:1818–1829.
2001.PubMed/NCBI
|
|
22
|
Seddon BM and Whelan JS: Emerging
chemotherapeutic strategies and the role of treatment
stratification in Ewing sarcoma. Paediatr Drugs. 10:93–105. 2008.
View Article : Google Scholar : PubMed/NCBI
|