Introduction
Inflammatory myofibroblastic tumor (IMT) is an
uncommon neoplasm initially described in the lungs by Brunn
(1) and was originally denoted as
inflammatory pseudotumor until the World Health Organization
officially named it IMT. In addition to pulmonary, there are many
IMTs that have been described in other organs throughout the body,
including the heart, liver and retroperitoneum (2–4). From
the literature IMT appears to present most commonly in children and
young adults, however, the etiology and pathogenesis of IMT remain
ambiguous as infection, surgery, autoimmunity and chromosomal
variation have all been hypothesized to contribute to IMT
development, however, the exact mechanism remains unclear.
Infection, surgery, autoimmunity and chromosomal variation are all
hypothesized to contribute to the development of IMT (5). Although it is described as a benign
neoplasm, some IMTs have the capacity for distant metastases
(6). To the best of our knowledge,
there has been no previously reported case of gastric IMT in an
adult that invaded the stomach and spleen. The current study
reports the case of an extremely large IMT that invaded the stomach
and spleen in a 50-year-old female with left-sided abdominal
distension for three months. Written informed consent was obtained
from the patient.
Case report
A 50-year-old female presented to the Department of
General Surgery, The Second Affiliated Hospital (Hangzhou,
China)with a three-month history of left-sided abdominal distension
and a lump, without abdominal pain, fever, nausea or vomiting. No
symptoms of reflux, melena, hematochezia or change in bowel habits
were reported. Urination became more frequent, however, no
hematuresis or odynuria were present. On examination, a smooth hard
lump with limited mobility was identified in the left
hypochondrium. The patient’s medical history was unremarkable and
did not include any autoimmune disease or surgery.
Laboratory analyses indicated a microcytic
hypochromic anemia with a hemoglobin level of 82 g/l (normal range,
113–151 g/l); hematocrit, 25.9% (normal range, 33.5–45.0%); mean
corpuscular volume, 73.3 fl (normal range. 84.0–94.0 fl) and mean
corpuscular hemoglobin, 23.1 pg (normal range, 27.0–34.0 pg). The
platelet count was 129,000/mm3 (normal range,
100,000–300,000/mm3). The results for the tumor
biological markers including CEA, AFP, CA199, CA125, NSE, SCC and
β-hCG were all normal.
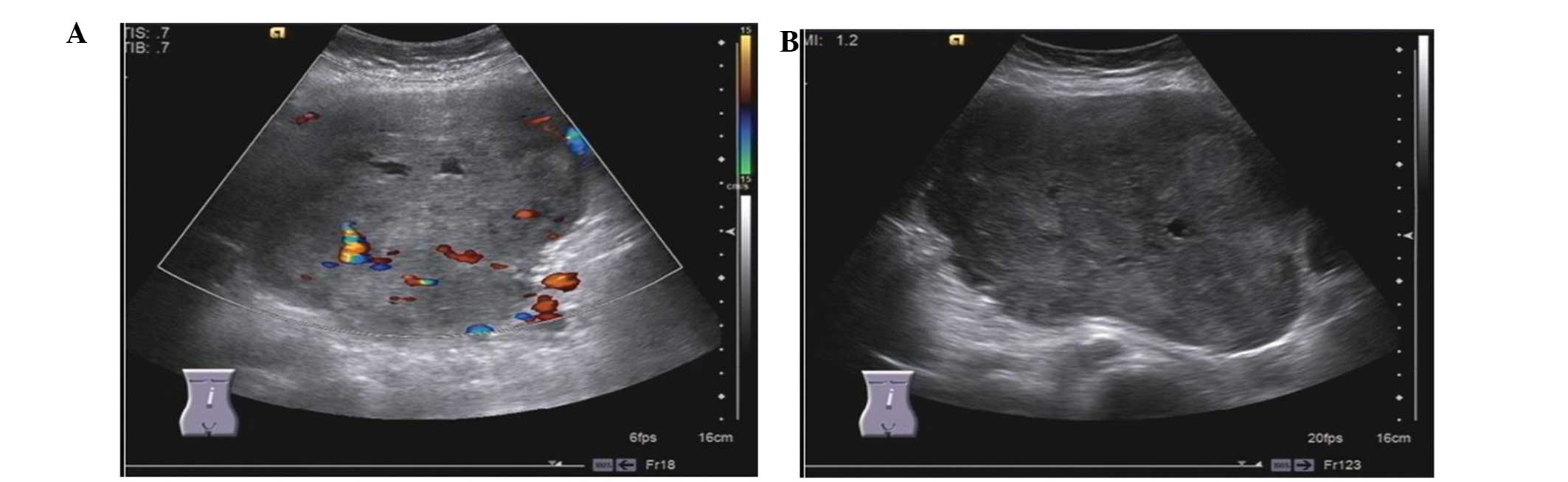
Ultrasound revealed a mass of 24.5×10.8 cm in size,
with its upper margin bound to the edge of liver and its lower
margin bound to the umbilical level in the left-sided abdomen
(Fig. 1). Abdominal computed
tomography revealed a mass of 22×14.2×11 cm in size between the
stomach and spleen, causing the surrounding tissues to alter their
locations (Fig. 2). Additionally, a
number of enlarged retroperitoneal lymph nodes and pelvic effusion
were revealed by enhanced computed tomography (Fig. 2).
Exploratory laparotomy showed a large solid mass and
numerous varicose vessels were observed on the surface. The mass
invaded the greater curvature of stomach and upper spleen with no
clear boundary. No liver, omentum or small bowel metastases were
identified. Therefore, the patient underwent a wide local excision
of the mass, accompanied by a total gastrectomy and
splenectomy.
On macroscopic examination, an extremely large mass,
measuring 22×13×8.5 cm, was tightly adhered to the stomach and
spleen. In the stomach lesion, tumor tissue invaded the entire
gastric wall and the overlying mucosa appeared ulcerated (7). Lymph nodes along the greater curvature
were excised and underwent biopsy, the result of which was negative
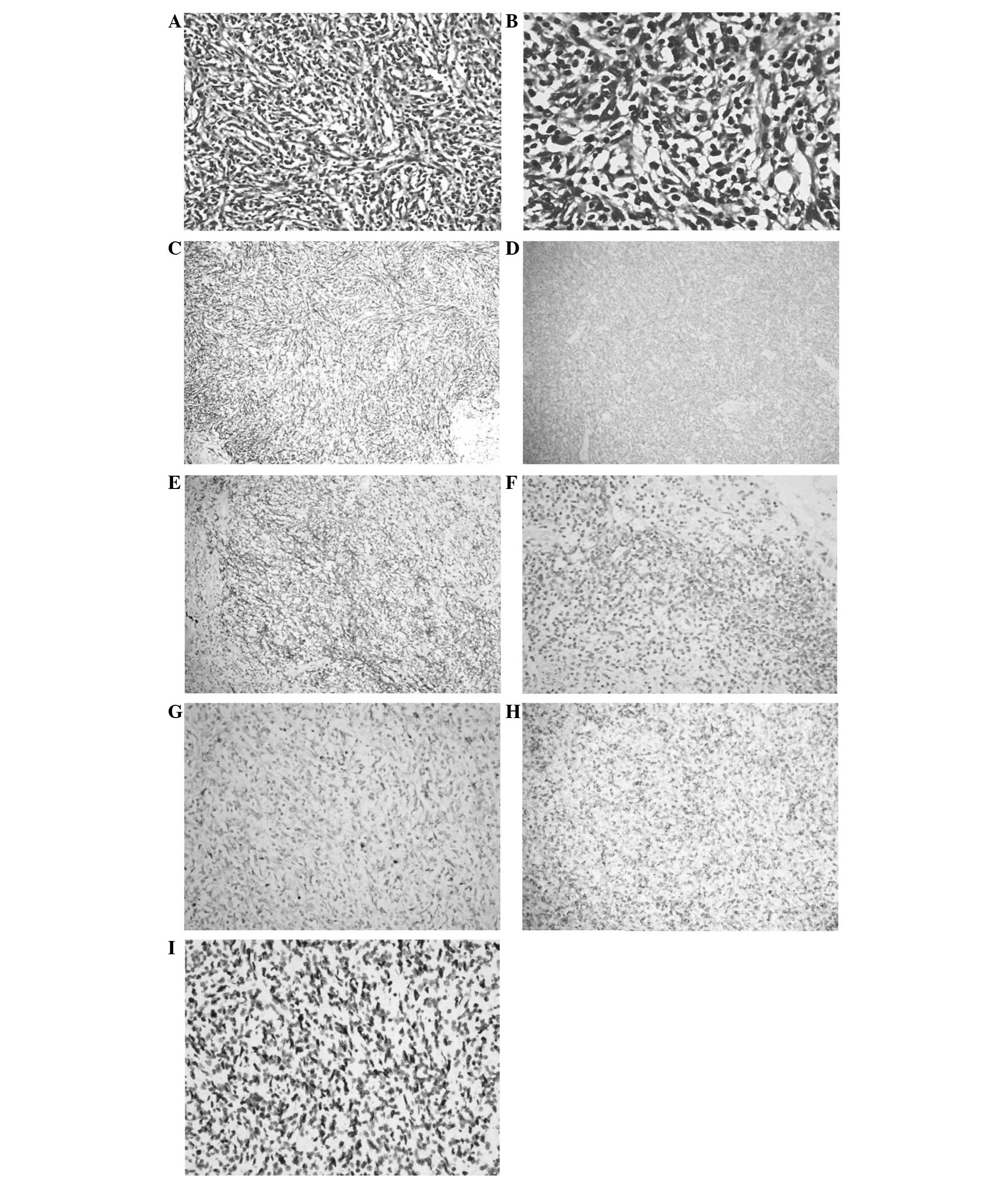
for tumor cells. On microscopic examination, a neoplasm composed of
spindle cells in inflammatory background with mixed lymphocytes,
plasma cells and eosinophils was observed (Fig. 3). Immunohistochemical staining was
positive for vimentin, smooth muscle actin (SMA), and negative for
CD30 and anaplastic lymphoma kinase (ALK). There were a number of
cells expressing CD68, but negative for LCA, excluding the
possibility that the tumor was derived from lymphatic and
hematopoietic system. Furthermore, for markers of dendritic cell
neoplasms, including CD21 and CD35, staining was negative.
Additionally, in situ hybridization revealed positive
results for EBV and EBVR. Based on these results, which were
confirmed by the Department of Pathology at the University of
California, Los Angeles (UCLA), a diagnosis of IMT was determined.
The patient experienced right-sided lower limb venous thrombosis,
however, four months following surgery, her recovery is
favorable.
Discussion
IMT (also known as inflammatory pseudotumor, plasma
cell granuloma, pseudosarcomatous myofibroblastic lesion, and
inflammatory myofibrohistiocytic lesion) was initially described in
the lungs (1). Following this, a
debate into whether IMT is a tumor or inflammation, and whether it
is benign or malignant arose, as it was considered to have the
potential of local recurrence and distant metastases; subsequently,
the World Health Organization classified IMT as a neoplasm of
intermediate biologic potential in 2002 (7,8). IMT
is histopathologically composed of myofibroblastic spindle cells
infiltrated with inflammatory cells, including plasma cells,
lymphocytes and eosinophiles (7).
Extrapulmonary IMTs occur in numerous sites throughout the body,
including the heart, liver, retroperitoneum, orbit and central
nervous system (2–5,9). The
first case with abdominal localization was described in the liver
by Pack and Baker (10). IMT is
most frequently observed in children and those <50 years old,
and primary gastric IMT in adults is rare (11).
Although it has been >70 years since the first
reported case of lung IMT (1), the
etiology of IMT is contentious. It has been proposed to be related
to infection, trauma, surgery, autoimmunity and chromosomal
variation, such as the abnormalities of ALK gene (12–15).
Viruses that were also hypothesized to affect the development of
IMTs include HIV, HHV-8 and EBV (16–18).
In the present case, while immunohistochemical analyses showed
negative staining for ALK, in situ hybridization revealed
infection of EBV, which may account for the occurrence of IMT. As
EBV infection is considered to be related to the neoplastic process
of IMTs, a longer follow-up is required for patients with
EBV-positive IMTs compared with those with EBV-negative IMTs
(17).
Even with a thorough diagnostic workup,
distinguishing IMT from other celiac malignancies is challenging.
Patients with IMTs most commonly present with symptoms of pain,
anemia, fatigue and weight loss, however, these symptoms are not
unique to IMTs. Therefore, pathological and immunocytochemical
analyses are the ‘gold standards’ for the diagnosis of IMTs. Coffin
et al reported three histological patterns of IMTs: A myxoid
vascular pattern, resembling nodular fasciitis; a compact spindle
cell pattern with a fascicular or storiform cellular arrangement;
and hypocellular collagenized pattern resembling a scar or desmoid
(7). In the present case,
histological examination revealed fascicles of spindle cells in a
mixed inflammatory background, with plasma cells.
Immunocytochemistry showed positive staining for vimentin, SMA, and
negative for ALK and CD30. Negative staining was also observed for
CD21 and CD35, eliminating the possibility of dendritic cell
neoplasm. Coffin et al (5)
also reported that almost 56% IMT cases exhibited diffuse
cytoplasmic ALK expression, however, a number of studies identified
IMTs in the spleen and lymph node that showed no ALK
overexpression, indicating that the splenic and nodal IMTs may
represent a discrete subset that are biologically distinct from
other IMTs (5,19,20).
In order to further confirm the diagnosis, the clinical information
and the results of immunohistochemical analyses were sent to UCLA;
the Department of General Surgery (The Second Affiliated Hospital)
and UCLA determined a diagnosis of IMT.
To date, the predominant therapy for the treatment
of IMTs remains complete resection of the tumor. In addition to
surgery, treatment with corticosteroids has also led to tumor
regression (21). For cases where
IMT cannot be completely resected or in the instance of metastatic
disease, chemotherapy is used despite the lack of definitive data
for the efficacy. The case presented in the current study showed a
rare mass of 22×14.2×11 cm in size that invaded the stomach and
spleen; this was marginally smaller than the largest reported IMT
described in liver, which measured 25 cm in diameter (22). Therefore, the most favorable
treatment option was the complete resection of the tumor with total
gastrectomy and splenectomy.
In recent years ALK-directed therapy, a novel
treatment option, has been developed, and has already been
demonstrated to have a partial therapeutic effect on IMT (23). The ALK fusion gene exists in ~56% of
IMTs and has been hypothesized to have significant effects in the
process of tumor development (5). A
recent phase I trial of crizotinib demonstrated a long-term partial
response in a patient with IMT carrying an ALK translocation, but
not in a patient with ALK-negative disease (23). However, the emergence of resistance
to crizotinib occurred approximately 5–8 months after the
initiation of therapy (23). The
development of more selective ALK inhibitors, which can overcome
emergent crizotinib resistance mutations, will be key to achieving
success in the future.
In conclusion, the current study reports the first
case of IMT that invaded the stomach and spleen, occurring in a 50
year-old female. In this case, it was important for the tumor to be
resected completely, as the volume of tumor was extremely large and
the efficacy of other treatment modalities are currently
unclear.
Acknowledgements
The authors would like to thank the Department of
Pathology of UCLA for providing clinicopathological analysis, which
contributed to the diagnosis and therapy of the patient.
Abbreviations:
|
IMT
|
Inflammatory myofibroblastic tumor
|
|
SMA
|
smooth muscle actin
|
|
ALK
|
anaplastic lymphoma kinase
|
References
|
1
|
Brunn H: Two interesting benign lung
tumors of contradictory histopathology. J Thorac Surg. 9:119–131.
1939.
|
|
2
|
Burke A, Li L, Kling E, Kutys R, Virmani R
and Miettinen M: Cardiac inflammatory myofibroblastic tumor: a
‘benign’ neoplasm that may result in syncope, myocardial
infarction, and sudden death. Am J Surg Pathol. 31:1115–1122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Díaz-Torné C, Narváez J, De Lama E, et al:
Inflammatory pseudotumor of the liver associated with rheumatoid
arthritis. Arthritis Rheum. 57:1102–1106. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Koirala R, Shakya VC, Agrawal CS, et al:
Retroperitoneal inflammatory myofibroblastic tumor. Am J Surg.
199:e17–e19. 2010. View Article : Google Scholar
|
|
5
|
Coffin CM, Hornick JL and Fletcher CD:
Inflammatory myofibroblastic tumor: comparison of
clinicopathologic, histologic, and immunohistochemical features
including ALK expression in atypical and aggressive cases. Am J
Surg Pathol. 31:509–520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gleason BC and Hornick JL: Inflammatory
myofibroblastic tumours: where are we now? J Clin Pathol.
61:428–437. 2008. View Article : Google Scholar
|
|
7
|
Coffin CM, Watterson J, Priest JR and
Dehner LP: Extrapulmonary inflammatory myofibroblastic tumor
(inflammatory pseudotumor). A clinicopathologic and
immunohistochemical study of 84 cases. Am J Surg Pathol.
19:859–872. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Coffin CM FJ: Inflammatory myofibroblastic
tumour. Fletcher CDM, Unni KK and Mertens F: Pathology and genetics
of tumours of soft tissue and bone. World Health Organization
classification of tumours Lyon: IARC Press; pp. 91–93. 2002
|
|
9
|
Sanahuja J, Ordoñez-Palau S, Begué R,
Brieva L and Boquet D: Primary Sjögren Syndrome with tumefactive
central nervous system involvement. AJNR Am J Neuroradiol.
29:1878–1879. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pack GT BH: Total right hepatic lobectomy;
report of a case. Ann Surg. 138:253–258. 1953. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi H, Wei L, Sun L and Guo A: Primary
gastric inflammatory myofibroblastic tumor: a clinicopathologic and
immunohistochemical study of 5 cases. Pathol Res Pract.
206:287–291. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Evans J, Chan C, Gluch L, Fielding I and
Eckstein R: Inflammatory pseudotumour secondary to actinomyces
infection. Aust N Z J Surg. 69:467–469. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rathinam S, Kuntz H, Panting J and Kalkat
MS: Inflammatory myofibroblastic tumour at the pacemaker site.
Interact Cardiovasc Thorac Surg. 10:443–445. 2010. View Article : Google Scholar
|
|
14
|
Griffin CA, Hawkins AL, Dvorak C, Henkle
C, Ellingham T and Perlman EJ: Recurrent involvement of 2p23 in
inflammatory myofibroblastic tumors. Cancer Res. 59:2776–2780.
1999.PubMed/NCBI
|
|
15
|
Perera MT, Wijesuriya SR, Kumarage SK,
Ariyaratne MH and Deen KI: Inflammatory pseudotumour of the liver
caused by a migrated fish bone. Ceylon Med J. 52:141–142. 2007.
|
|
16
|
Gonzalez-Duarte A, Sullivan S, Sips GJ, et
al: Inflammatory pseudotumor associated with HIV, JCV, and immune
reconstitution syndrome. Neurology. 72:289–290. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kiryu S, Takeuchi K, Shibahara J, et al:
Epstein-Barr virus-positive inflammatory pseudotumour and
inflammatory pseudotumour-like follicular dendritic cell tumour. Br
J Radiol. 82:e67–e71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gómez-Román JJ, Ocejo-Vinyals G,
Sánchez-Velasco P, Nieto EH, Leyva-Cobián F and Val-Bernal JF:
Presence of human herpesvirus-8 DNA sequences and overexpression of
human IL-6 and cyclin D1 in inflammatory myofibroblastic tumor
(inflammatory pseudotumor). Lab Invest. 80:1121–1126. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cheuk W, Chan JK, Shek TW, et al:
Inflammatory pseudotumor-like follicular dendritic cell tumor: a
distinctive low-grade malignant intra-abdominal neoplasm with
consistent Epstein-Barr virus association. Am J Surg Pathol.
25:721–731. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cook JR, Dehner LP, Collins MH, et al:
Anaplastic lymphoma kinase (ALK) expression in the inflammatory
myofibroblastic tumor: a comparative immunohistochemical study. Am
J Surg Pathol. 25:1364–1371. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li JY, Yong TY, Coleman M, et al:
Bilateral renal inflammatory pseudotumour effectively treated with
corticosteroid. Clin Exp Nephrol. 14:190–198. 2010. View Article : Google Scholar
|
|
22
|
Mathiak G, Meyer-Pannwitt U, Mathiak M,
Schröder S, Henne-Bruns D and Fröschle G: Inflammatory pseudotumor
of the liver-rare differential diagnosis of undetermined hepatic
space-occupying lesion. Case report and review of the literature.
Langenbecks Arch Chir. 381:309–317. 1996.(In German).
|
|
23
|
Butrynski JE, D’Adamo DR, Hornick JL, et
al: Crizotinib in ALK-rearranged inflammatory myofibroblastic
tumor. N Engl J Med. 363:1727–1733. 2010. View Article : Google Scholar : PubMed/NCBI
|