Introduction
Malignant triton tumor (MTT) is a malignant
peripheral nerve sheath tumor (MPNST) with rhabdomyoblastic
differentiation (1,2). The name ‘triton’ derived from an
experiment in which sciatic nerve tissue of the triton salamander
was stimulated to grow a neoplasm with skeletal muscle components
(3). It was first described as a
disease in 1932 (4) and first
reported as ‘malignant triton tumor’ in 1973 (5). Woodruff et al proposed three
criteria for its diagnosis (5): i)
arising along a peripheral nerve in ganglioneuroma or in a patient
with neurofibromatosis type 1 (NF-1) or representing a metastasis
from such a tumor; ii) having the growth characteristics of a
Schwann cell tumor; and iii) rhabdomyoblasts arise within the body
of the tumor. Daimaru et al considered that sporadic cases
fulfilling the latter two criteria could be diagnosed (6). Clinically, the morbidity of MTT is less
than 1 per 1,000,000 (7). As it is a
rare tumor, only limited cases have been reported so far, and no
large group studies exist. For further understanding of the
clinical features of MTT, we share our experiences of the treatment
of MTT in our hospital and conduct a literature analysis. Here, we
review two cases of MTT occurring in the head and neck, with no
evidence or symptoms of NF-1. Written informed consent was obtained
from both patients and the study was approved by the Research
Ethics Committee of Central South University (Changsha, China).
Case report
Case 1
A 27-year-old male was admitted to Xiangya Hospital,
Changsha, China, in March 2007, complaining of pain and a foreign
body sensation in the left side of the pharynx. Physical
examination revealed a cauliflower-like brown mass (30×20 mm)
covered with grayish membrane at the left tonsillar fossa. It
infiltrated into the surrounding tissues. The patient's mouth
opened freely and no lymphadenopathy was observed. Magnetic
resonance imaging (MRI) revealed isodense T1 and long T2 signals,
and there was no overt submaxillary bony destruction (Fig. 1A-C). On April 5, 2007, the patient
underwent ligation of the external carotid artery via a combined
jaw-and-neck approach and tumor resection en bloc via a
combined parapharyngeal space and intra-oral approach. The
surrounding tissues were invaded and the tumor had adhered too
tightly to be separated. The resected tissues included partial
mucosa of the posterior pharyngeal wall and left fauces, partial
medial and lateral pterygoid muscle and the left submandibular
gland en bloc. The histological diagnosis was MTT with
positive myogenin and S-100. The marginal mandibular branch was
preserved. Following an uneventful postoperative recovery, the
patient was discharged 7 days after surgery.
Following a course of radiotherapy at a total dose
of 52 Gy in May 2007, two courses of chemotherapy with combined
pirarubicin, ifosfamide and carboplatin were administered. The
patient underwent partial mandibulectomy for osteonecrosis in 2008
and was treated for granulocytopenia following chemotherapy.
Follow-up was carried out until April 2014. There was no evidence
of further disease.
Case 2
A 43-month-old boy presented with hoarseness for 10
days, failure to swallow saliva for 5 days and headache and
vomiting for 2 days. Clinical and radiological examinations
revealed a mass of ~2.2×2.8×2.5 cm in the right jugular foramen
area with destruction of the skull base. MRI demonstrated isodense
T1 and long T2 signals, while gadolinium-diethylenetriamine
pentaacetic acid-enhanced MRI indicated enhancement of the mass
with a distinct lobulated margin (Fig.
1D-F). Surgery revealed that the neoplasm was located behind
the temporomandibular joint and had destroyed the temporal bone and
skull base. With an invasion of the deep paroid, the neoplasm had a
tight connection with the internal jugular vein. Rapid
intraoperative pathological diagnosis suggested a spindle cell
sarcoma with an inclination of MPNST. At the request of his
parents, surgery was not performed and no treatment was offered.
Hematoxylin and eosin staining and immunohistochemistry results
suggested MTT with Bcl-2(+), CD34(+), CD57(−), CD99(+), desmin(+),
HMB45(−), myogenin(++), NF(+), vimentin(+) and S-100(−) (Fig. 2). The child then survived only seven
months.
Literature review
Database analysis
Literature searches on PubMed were conducted with
the terms ‘malignant triton tumor’ and ‘malignant peripheral nerve
sheath tumors AND rhabdomyosarcoma’ for the period prior to April
2014. All MTT cases were extracted from the databases. The relevant
information included standard demographic information (age, gender
and year of diagnosis) as well as NF-1, onset location, surgical
approach, type of adjuvant therapy (radiotherapy and chemotherapy),
relapse and metastasis. Survival time and final status were also
recorded and the follow-up period was designated to be 60
months.
The onset location of MTT was mainly classified as
trunk, head/neck or limbs, according to the original location.
Where the tumor occurred in more than two places without a known
original location, it was defined as ‘multiple locations’. The
concept of complete resection refers to radical resection,
amputation or en bloc resection with microscopically
negative margins. The data on adjuvant radiotherapy and
chemotherapy were recorded as dichotomous variables. Local
recurrence was defined as any tumor with complete resection which
redeveloped in the surgical excision bed. Metastasis was defined as
any distant tumor spread which was confirmed via biopsy or
diagnosed with clinical experience, such as positron emission
tomography-computed tomography (PET-CT) or radionuclide bone
imaging. One patient who succumbed during surgery was also excluded
from our analysis. The data that was not available from the
literature was classified as ‘Unknown’.
All the data were imported into SPSS version 18.0
software (SPSS Inc, Chicago, IL, USA). Standard descriptive
statistics were computed for those selected variables. Kaplan-Meier
survival analysis was performed for the whole cohort. The potential
effects of clinical variables on survival were determined by the
Cox proportional hazards model. All variables were entered
simultaneously into a multivariate model. P<0.05 was considered
to indicate a statistically significant difference.
Demographic and clinical data of
MTT
A total of 198 cases were retrieved from 135 studies
related to MTT in the databases. Pooling the data of our cases, the
data of 200 MTT cases were collected in total. The male-to-female
ratio was 1.5 to 1. The median age at diagnosis was 29 years old.
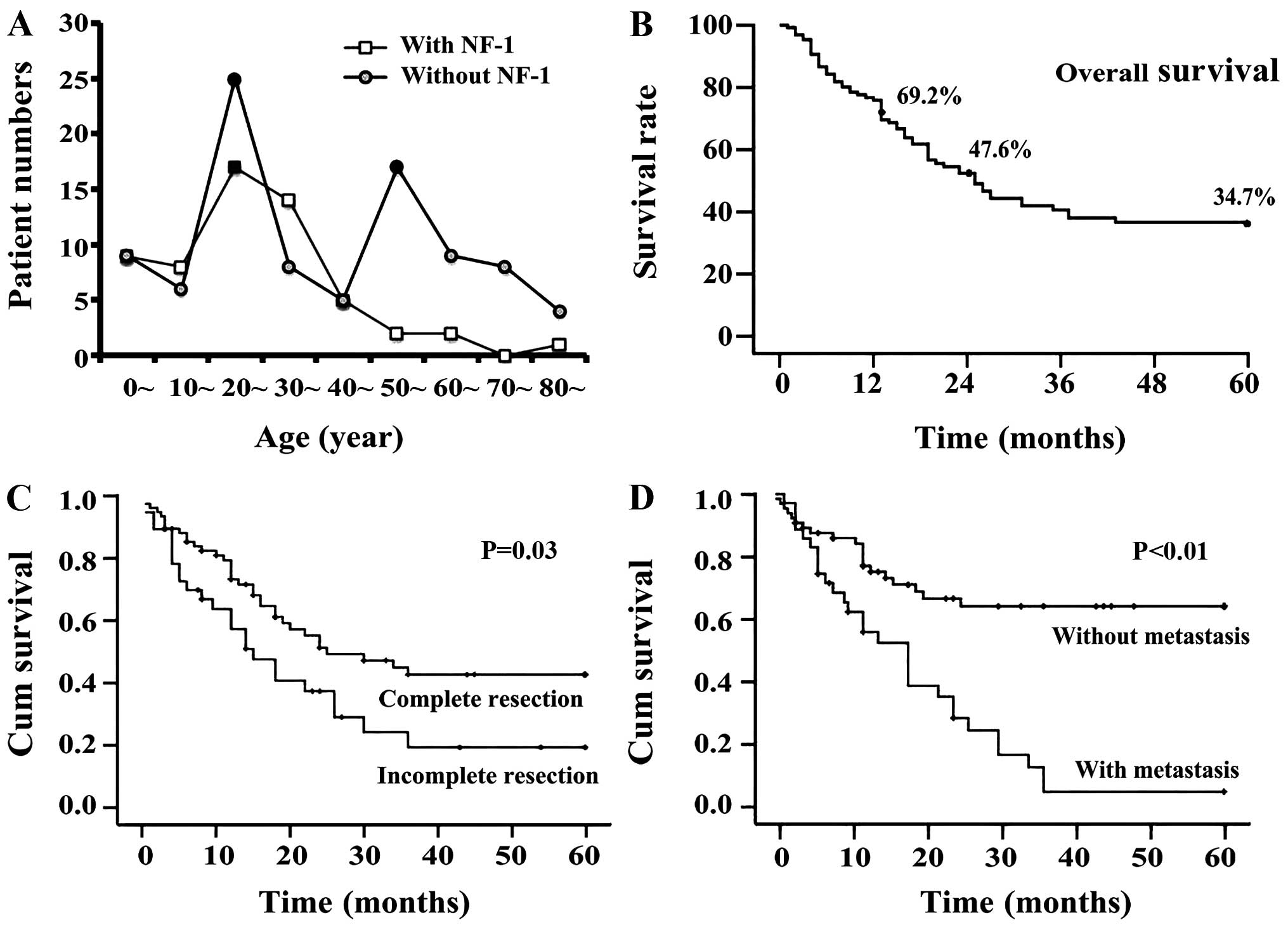
The age group distribution chart revealed that the patients with
NF-1 were most common in the 20–39 age groups while patients
without NF-1 had two peak age groups: the 20s and 50s (Fig. 3A). A total of 41.7% of patients had a
history of NF-1 and the others were sporadic. Among all patients,
the affected sites included the trunk (n=76, 38.0%), head and neck
(n=73, 36.5%) and extremities (n=44, 22.0%; Table I). The clinical treatment data
revealed that 68.7% of tumors were removed completely. Radiotherapy
and chemotherapy were administered to 54.0% and 35.3% of patients,
respectively. The rates of recurrence and metastasis were 42.6% and
34.4%, respectively.
 | Table I.Onset location of malignant triton
tumors. |
Table I.
Onset location of malignant triton
tumors.
| Onset location | Onset location in
detail | Cases, n (%) |
|---|
| Trunk | Pelvic cavity | 12 (6.0) |
|
| Mediastinum | 12 (6.0) |
|
| Spine | 10 (5.0) |
|
| Retroperitoneal
space | 9
(4.5) |
|
| Chest wall | 8
(4.0) |
|
| Buttocks | 5
(2.5) |
|
| Lung | 5
(2.5) |
|
| Back | 3
(1.5) |
|
| Other | 12 (6.0) |
|
| Total | 76
(38.0) |
| Head and neck | Nasal cavity and
sinus | 20
(10.0) |
|
| Neck | 19 (9.5) |
|
| Brain | 11 (5.5) |
|
| Oral cavity and
around | 9
(4.5) |
|
| Parapharyngeal
space | 4
(2.0) |
|
| Occiput | 3
(1.5) |
|
| Other | 7
(3.5) |
|
| Total | 73
(36.5) |
| Limbs | Legs | 35
(17.5) |
|
| Arm | 7
(3.5) |
|
| Palm | 1
(0.5) |
|
| Foot | 1
(0.5) |
|
| Total | 44
(22.0) |
| Multiple
locations | Total | 3
(1.5) |
| Unknown | Total | 4
(2.0) |
| Overall total |
| 200
(100.0) |
Analysis of prognostic factors of
MTT
A total of 133 cases with follow-up were selected
for the prognosis analysis. Life span analysis revealed that the
1-, 2- and 5-year survival rates were 69%, 48% and 35%,
respectively (Fig. 3B). The results
of Kaplan-Meier survival analysis demonstrated that five factors
had a statistically significant effect on the survival curves;
namely, age (P=0.013), presence of NF-1 (P<0.001), complete
resection (P=0.010, Fig. 3C),
radiotherapy (P=0.002) and presence of metastasis (P<0.001,
Fig. 3D). The factors of gender,
onset site, chemotherapy and relapse had no significant effect on
the survival curves (P>0.05, Table
II).
| Table II.Kaplan-Meier analysis and Cox
proportional hazards analyses of patients with malignant triton
tumor. |
Table II.
Kaplan-Meier analysis and Cox
proportional hazards analyses of patients with malignant triton
tumor.
|
| Kaplan-Meier
analysis | Cox proportional
hazards analyses |
|---|
|
|
|
|
|---|
| Category | Numbera | Mean time to death,
monthsb | P-value | HR | 95% CI for HR | P-value |
|---|
| Gender |
|
| 0.207 |
|
| 0.494 |
|
Male | 78 | 34.24 |
|
|
|
|
|
Female | 53 | 28.46 |
| 1.312 | 0.602–2.858 |
|
| Age,
yearsc |
|
|
|
|
|
|
|
≤32 | 68 | 25.68 | 0.013d |
|
| 0.423 |
|
>32 | 65 | 36.87 |
| 0.736 | 0.602–1.558 |
|
| Location |
|
| 0.638 |
|
|
|
| Head
and neck | 48 | 29.22 |
|
|
|
|
|
Trunk | 56 | 31.87 |
| 0.944 | 0.392–2.272 | 0.845 |
|
Limbs | 26 | 30.37 |
| 0.912 | 0.602–2.303 | 0.942 |
| NF-1 |
|
| 0.000d |
|
| 0.440 |
|
Yes | 72 | 17.80 |
| No | 40 | 36.59 |
| 1.312 | 0.628–2.914 |
|
| Complete
resection |
|
| 0.010d |
|
| 0.032d |
|
Yes | 76 | 34.23 |
|
|
|
|
| No | 39 | 22.50 |
| 0.396 | 0.170–0.925 |
|
| Radiotherapy |
|
| 0.002d |
|
| 0.131 |
|
Yes | 67 | 36.24 |
|
|
|
|
| No | 54 | 22.99 |
| 0.572 | 0.277–1.181 |
|
| Chemotherapy |
|
| 0.430 |
|
| 0.244 |
|
Yes | 41 | 31.97 |
|
|
|
|
| No | 80 | 29.15 |
| 0.594 | 0.248–1.426 |
|
| Recurrence |
|
| 0.129 |
|
| 0.511 |
|
Yes | 45 | 27.82 |
|
|
|
|
| No | 66 | 35.96 |
| 1.278 | 0.615–2.655 |
|
| Metastasis |
|
| 0.000d |
|
| 0.004d |
|
Yes | 37 | 18.12 |
|
|
|
|
| No | 71 | 39.80 |
| 3.188 | 1.450–7.008 |
|
On this basis, all factors in the Kaplan-Meier
survival analysis were entered simultaneously into the Cox
proportional hazards model for analysis. The result revealed that
the factors of complete resection and metastasis had a significant
effect on survival (P<0.05, Table
II). The hazard ratio (HR) for the former was 0.396. This
indicated that if tumor excision was performed en bloc,
there was a reduction in mortality risk. The HR of the latter was
3.188, implying that mortality risk increased more than three
times.
Discussion
Although the mechanism of MTT remains elusive, its
association with nerves or nervous lesions should be noted. The
literature revealed that a number of nerves could be involved,
including the cervical plexus (8),
brachial plexus (9), optic nerve
(10), sciatic nerve (11), cervical sympathetic nerve (12) and spinal nerve root (13). There was no conclusive evidence that
the masses in our cases came from the nerves; however, the onset
locations were observed to be in or around the parapharyngeal space
which contains the nerves. A total of 50–70% of cases were observed
to arise in patients with NF-1 (1,7), and this
rate was ~40% in our own data. Radiotherapy and repeated surgery on
the benign nerve fibroma may increase the risk of rhabdomyosarcoma
differentiation (11,14). At the sub-cellular level, aberrations
in chromosomes 1, 6, 7, 8, 9, 16, 17, 19, 20 and 22 were correlated
with the genesis and recurrence of MTT (15). The missense mutation and a loss of
heterozygosity were observed in the majority of MTT cases (16,17).
Nucleotide deletion in P53, amplification of C-myc and aberrations
in the hedgehog-patched pathway were also noted in MTT (18–20).
Clinically, the age at diagnosis age ranged from
newborn to over 80 years old, with a median age of 29–35 (1,21,22). More than 60% of MTT cases with NF-1
were diagnosed in patients between 20 and 50 years old, when the
benign masses were suspected to undergo a malignant transformation
(23). MTT was reported mainly in the
head, neck and trunk, and less frequently in the extremities
(1); our data yielded similar
results. In the early stage, patients often had no self-reported
symptoms, and only sought medical attention when there were such
symptoms as pain or dysfunction caused by the involvement with
adjacent tissues. The size of the abdominal mass increased due to
the lack of space constraints, and the maximal diameter was
reported at 82 cm (24).
MRI has been the most common method of identifying
the location, scale and shape of lesions. In most cases, the masses
demonstrated short or mixed isodensity T1 and long T2 signals. In
certain images, ring or linear forms were observed as a type of
isolation strip; these are likely to be due to necrosis and
hemorrhage in the mass as a result of a quick growth (25). Enhanced spiral CT scanning is capable
of visualizing bone erosion around the mass and providing
indications of its malignancy. 18FDG-PET-CT was also considered to
be a more effective method of estimating the prognosis and
metastasis (9). All of the above
methods are used for auxiliary examination only, and the final
diagnosis should be based upon pathology. Microscopically, MTT
always contains shuttle-like malignant peripheral nerve cells,
additional rhabdomyoblastic differentiation and zonal cytoplasm
(7). Immunohistochemical results
facilitated our diagnosis, particularly with positive desmin and
myoglobin (13). Here, additional
oncogene proteins including Bcl-2, CD34, CD99, HHF35, NF and
vimentin were also noted to be positive, which confirmed the
malignant features of this neoplasm.
Literature revealed that the 5-year survival rates
of patients with MPNST and MTT were 34–44% and 10–20%, respectively
(3,4).
Our data revealed 1-, 2- and 5-year survival rates of 69%, 48% and
35%, respectively. Theoretically, the actual survival rates may be
lower, since our censored proportion accounted for almost 20%. Our
analysis also revealed no survival difference among MTT of the
trunk, head and neck, or extremities. Terzic et al observed
that the 1-, 2- and 5-year survival rates of head and neck MTT were
76%, 61% and 49%, respectively, and that paranasal sinus MTT had an
even better prognosis (22). The poor
survival rate associated with distant metastasis was in keeping
with the findings made in previous studies (1,13).
In ~60% of the cases in the literature, tumors were
completely resected. Wide local excision, whole organ excision and
amputation were performed (7,22). In our analysis, Cox proportional
hazards analysis demonstrated that complete resection is an
independent prognostic factor, and decreased the mortality risk.
Thus, radical mass excision is key to prolonging postoperative
survival. The effectiveness of radiotherapy remains controversial.
The single factor analysis of radiotherapy revealed a statistically
significant difference; however, multi-factorial analysis revealed
no difference, which may be related to the limited number of cases.
Given our findings and experience, radiotherapy should be
considered as a routine for all MTT patients, whether the residual
mass remains or not. Ifosfamide, dactinomycin, carboplatin and
fluorouracil are the main drugs used in chemotherapy (4,22). One
study described a tumor expressing retinoic acid receptors-a and b,
and the treatment of isotretinoin and interferon-α was offered to
control the disease (26).
Kaplan-Meier analysis revealed no significant difference for the
patients with chemotherapy, which might be attributed to the reason
that there was no standard or guidelines. Simultaneously, the side
effects of radiotherapy and chemotherapy should be recorded and
managed promptly. Our patient (case 1) suffered osteonecrosis and
granulocytopenia following adjunctive therapies. Thus, prolonging
the survival time is not the only target for patients: improving
their quality of life is also considered to be a priority.
Acknowledgements
Grants were obtained by the National Natural Science
Foundation of China (nos. 81372426, 81372906, 81202128 and
81172558), the Research Fund for the Doctoral Program of Higher
Education of China (no. 20120162120049), the Graduate Student
Research Innovation Project of Hunan Province (no. CX2013B108), the
Freedom Explore Program of Central South University (no.
2012QNZT099), and the Open-End Fund for the Valuable and Precision
Instruments of Central South University (no. CSUZC2014048).
The authors are grateful for the statistical
guidance from Deng Jing at the Department of Statistics, School of
Public Health, Central South University, and Zhu Lei at the Library
of Medicine, Central South University, who offered assistance in
the literature searches.
References
|
1
|
McConnell YJ and Giacomantonio CA:
Malignant triton tumors - complete surgical resection and adjuvant
radiotherapy associated with improved survival. J Surg Oncol.
106:51–56. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Friesenbichler J, Leithner A, Maurer-Ertl
W, Szkandera J, Sadoghi P, Frings A, Maier A, Andreou D, Windhager
R and Tunn PU: Surgical therapy of primary malignant bone tumours
and soft tissue sarcomas of the chest wall: a two-institutional
experience. Int Orthop. 38:1235–1240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Locatelli P: Formation de Membres
Surnumeraires. C. R. Assoc. des Anatomistes (20e reunion Turin).
279–282. 1925.
|
|
4
|
Masson P: Recklinghausen's
neurofibromatosis, sensory neuromas and motor neuromas. Libman
Anniversary. 2:(New York, NY). International Press. 793–802.
1932.
|
|
5
|
Woodruff JM, Chernik NL, Smith MC, Millett
WB and Foote FW Jr: Peripheral nerve tumors with
rhabdomyosarcomatous differentiation (malignant ‘Triton’ tumors).
Cancer. 32:426–439. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Daimaru Y, Hashimoto H and Enjoji M:
Malignant ‘triton’ tumors: a clinicopathologic and
immunohistochemical study of nine cases. Hum Pathol. 15:768–778.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
John M: Anderson's Pathology.
Philadelphia: The C. V. Mosby Company. 1894–1896. 1990.
|
|
8
|
Yang BB, Jiang H and Chang HY: Malignant
triton tumour of the parapharyngeal space: a case arising from the
cervical sympathetic nerve. J Laryngol Otol. 122:531–534. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dartnell J, Pilling J, Ferner R, Cane P
and Lang-Lazdunski L: Malignant triton tumor of the brachial plexus
invading the left thoracic inlet: a rare differential diagnosis of
pancoast tumor. J Thorac Oncol. 4:135–137. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Han DH, Kim DG, Chi JG, Park SH, Jung HW
and Kim YG: Malignant triton tumor of the acoustic nerve. Case
report. J Neurosurg. 76:874–877. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tripathy K, Mallik R, Mishra A, Misra D,
Rout N, Nayak P, Samantray S and Rath J: A rare malignant triton
tumor. Case Rep Neurol. 2:69–73. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cano JR, Algar FJ, Alvarez A and
Salvatierra A: Triton tumor of the left sympathetic nerve. Interact
Cardiovasc Thorac Surg. 5:790–791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rekhi B, Jambhekar NA, Puri A, Agrawal M
and Chinoy RF: Clinicomorphologic features of a series of 10 cases
of malignant triton tumors diagnosed over 10 years at a tertiary
cancer hospital in Mumbai, India. Ann Diagn Pathol. 12:90–97. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ozer E, Erkilic S, Bayazit YA, Mumbuç S,
Aydin A and Kanlikama M: Malignant triton tumor of the
supraclavicular region arising after radiotherapy. Auris Nasus
Larynx. 29:405–407. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Koutsimpelas D, Brieger J, Heinrich U,
Torzewski M, Sommer C and Mann WJ: Cytogenetic analysis of a
malignant triton tumour by comparative genomic hybridization (CGH)
and review of the literature. Eur Arch Otorhinolaryngol.
268:1391–1396. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Strauss BL, Gutmann DH, Dehner LP, Hirbe
A, Zhu X, Marley EF and Liapis H: Molecular analysis of malignant
triton tumors. Hum Pathol. 30:984–988. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ferrari A, Bisogno G, Macaluso A, Casanova
M, D'Angelo P, Pierani P, Zanetti I, Alaggio R, Cecchetto G and
Carli M: Soft-tissue sarcomas in children and adolescents with
neurofibromatosis type 1. Cancer. 109:1406–1412. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chao MM, Levine JE, Ruiz RE, Kohlmann WK,
Bower MA, Petty EM and Mody RJ: Malignant triton tumor in a patient
with Li-Fraumeni syndrome and a novel TP53 mutation. Pediatr Blood
Cancer. 49:1000–1004. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Haddadin MH, Hawkins AL, Long P,
Morsberger LA, Depew D, Epstein JI and Griffin CA: Cytogenetic
study of malignant triton tumor: A case report. Cancer Genet
Cytogenet. 144:100–105. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bura M, Musani V, Cretnik M, Botica I and
Levanat S: Hedgehog-Patched pathway aberrations in a malignant
triton tumor case study. Oncol Rep. 20:347–352. 2008.PubMed/NCBI
|
|
21
|
Sönmez K, Türkyilmaz Z, Karabulut R,
Kapisiz A, Eser EP, Memiş L and Başaklar AC: A Triton tumor
mimicking sacrococcygeal teratoma. J Pediatr Surg. 44:e5–e8. 2009.
View Article : Google Scholar
|
|
22
|
Terzic A, Bode B, Gratz KW and Stoeckli
SJ: Prognostic factors for the malignant triton tumor of the head
and neck. Head Neck. 31:679–688. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Duat-Rodríguez A, Carceller Lechón F, Pino
López MÁ, Rodríguez Fernández C and González-Gutiérrez-Solana L:
Neurofibromatosis type 1 associated with moyamoya syndrome in
children. Pediatr Neurol. 50:96–98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Murtaza B, Gondal ZI, Mehmood A, Shah SS,
Abbasi MH, Tamimy MS and Kazmi ST: A huge malignant triton tumour.
J Coll Physicians Surg Pak. 15:728–730. 2005.PubMed/NCBI
|
|
25
|
Ghosh A, Sastri SB, Srinivas D, Mahadevan
A, Anandappa CB and Shankar SK: Malignant triton tumor of cervical
spine with hemorrhage. J Clin Neurosci. 18:721–723. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Köstler WJ, Amann G, Grunt TW, Singer CF,
Schneider SM, Brodowicz T, Tomek S and Zielinski CC: Recurrent
malignant Triton tumour: First report on a long time survivor.
Oncol Rep. 10:533–535. 2003.PubMed/NCBI
|