Introduction
Chronic myeloid leukemia (CML) is a
myeloproliferative disease characterized by the presence of the
Philadelphia (Ph) chromosome, which is formed by a t(9;22)
(q34;q11) balanced reciprocal translocation (1). The Ph chromosome translocation generates
the BCR-ABL oncogene that encodes for the BCR-ABL oncoprotein,
which exhibits constitutively active tyrosine kinase activity that
promotes the growth of leukemic cells (2). CML patients in the chronic phase (CP)
that are treated with tyrosine kinase inhibitors (TKIs) achieve a
significant effect (1). However, a
significant percentage of patients develop TKI resistance and
disease recurrence (3), which
involves a variety of cellular mechanisms. Therefore, identifying
molecules that are involved in the development and progression of
CML may provide novel therapeutic targets for CML treatment.
Programmed cell death 4 (PDCD4) is a novel tumor
suppressor that inhibits tumor growth via suppression of protein
translation by binding to eukaryotic initiation factor (EIF) 4A via
two MA-3 domains, which are highly homologous to eIF4G (4). PDCD4 also suppresses translation
elongation by combining directly with target gene coding regions,
such as c-myb (5) and A-myb (6). PDCD4-deficient mice exhibit a
significantly reduced life span and develop spontaneous lymphomas
with frequent metastasis to the liver and kidneys (7). PDCD4 transgenic mice exhibit resistance
to tumor promotion and progression in response to a multistage
carcinogenesis regimen (8).
Additionally, decreased or absent PDCD4 expression has been
identified in several types of human cancer, including lung
(9), hepatocellular (10), colon (11), glioma (12) and ovarian cancers (13). Overexpression of PDCD4 suppresses
tumor phenotypes by inhibiting activator protein-1 (AP-1)
transactivation in JB6 cells (14).
PDCD4 overexpression and has also been demonstrated to inhibit
invasive capacity in colon RKO cells (15), inhibit tumor cell intravasation
(16) and suppress malignant
phenotypes of human ovarian cancer (13). Furthermore, PDCD4 knockdown
significantly promotes invasion and activates both β-catenin/T cell
factor (Tcf) and AP-1-dependent transcription (17). Additionally, PDCD4 knockdown leads to
increased Snail expression and subsequent downregulation of
E-cadherin resulting in the activation of catenin/Tcf-dependent
transcription and the expression of c-Myc and urokinase receptor
(18).
Taken together, these findings indicate that PDCD4
presents a potential target in the diagnosis and treatment of
neoplasms. However, whether PDCD4 is involved in human hematologic
neoplasms remains unclear. In the present study, PDCD4 expression
levels in CML patients were evaluated using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analysis.
Materials and methods
Patients and samples
A total of 50 bone marrow aspirate samples were
obtained from patients diagnosed with CML, according to the World
Health Organization guidelines (19),
at Yuhuangding Hospital of Qingdao University (Yantai, China)
between June 2012 and September 2014. The CML patient sample
included 23 females and 27 males with a mean age (± standard
deviation) of 41.94 years (±14.13 years). Patients were divided
into the following three groups depending on their clinical and
laboratory data: into 30 CP (n=30), accelerated phase (AP; n=10)
and blast phase (BP; n=10). The criteria for diagnosis of CML-CP
was the presence of t (9;22) or the BCR-ABL fusion gene, <10%
bone marrow blasts, and does not satisfy the diagnostic criteria of
CML-AP or CML-BP. Peripheral blood samples were also obtained from
20 healthy individuals, which served as the control group. Patient
characteristics are listed in Table
I. The final protocol for the use of patient samples in the
present study was approved by the local Institutional Review Board
of Qingdao University and informed consent was obtained from all
patients and controls.
 | Table I.Clinical parameters and PDCD4
expression in 50 CML patients and 20 healthy individuals. |
Table I.
Clinical parameters and PDCD4
expression in 50 CML patients and 20 healthy individuals.
|
|
| CML patients |
|---|
|
|
|
|
|---|
| Parameter | Healthy controls | CML-CP | CML-AP | CML-BP |
|---|
| Gender, n |
|
|
|
|
| Male | 10 | 13 | 5 | 4 |
|
Female | 10 | 17 | 5 | 6 |
| Age, years |
|
|
|
|
|
Median | 36 | 49 | 49 | 51 |
|
Range | 24–59 | 15–71 | 32–63 | 39–76 |
| PDCD4/GAPDH
(±SEM) | 1.65±0.17 | 0.150±0.03 | 0.13±0.04 | 0.15±0.05 |
Cell lines and treatment
K562 cells were purchased from the Shanghai Cell
Bank of Chinese Academy of Sciences (Shanghai, China) and cultured
in RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Waltham, MA,
USA) supplemented with 10% heat-inactivated fetal bovine serum
(Gibco; Thermo Fisher Scientific,), 1% penicillin and streptomycin
at 37°C in a humidified incubator with 5% CO2. A total
of 2×105 cells/well were seeded in 6-well plates and
treated with 0.5 and 1 µM imatinib (LC Laboratories, Woburn, MA,
USA) for 6, 24 and 48 h. Next, cells were harvested and washed once
with phosphate-buffered saline and subjected to RT-PCR and
immunoblotting analyses. Experiments were performed in
triplicate.
MTT assay
A total of 8×103 cells/well were seeded
into 96-well plates and treated with 0.5 and 1 µM imatinib. After
6, 24 and 48 h of imatinib treatment, 20 µl MTT reagent (5 mg/ml;
Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) was added to
each well and incubated in the dark for 4 h at 37°C. After 4 h, 100
µl dimethyl sulfoxide was added to each well and the absorbance was
determined using a Benchmark Plus Microplate Spectrophotometer
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) at 570 nm. Cell
proliferation of treated cells was calculated from the average
optical density at 570 nm values compared to that of untreated
cells. Experiments were performed in triplicate.
RNA isolation and RT-qPCR
RNA was extracted from isolated peripheral blood
mononuclear cells using a modified TRIzol one-step extraction
method (Invitrogen; Thermo Fisher Scientific). RNA concentrations
were determined based on the absorbance at 260 nm. Total RNA (2 µg)
was reversely transcribed to cDNA using the Reverse-Transcribe Kit
(Promega Corp., Madison, WI, USA). Quantitative PCR was performed
using SYBR® Selected Master Mix (Applied Biosystems;
Thermo Fisher Scientific) and specific primer pairs (Invitrogen;
Thermo Fisher Scientific Inc.). The sequences of the sense and
antisense primers were as follows: Sense,
5′-TGTAAACCCTGCAGATCCTGATAA-3′ and antisense,
5′-TGGAGGATGCTGAAATCCAA-3′ for PDCD4; sense,
5′-GGAGCTGCAGATGCTGACCACC-3′ and antisense,
5′-TCAGACCCTGAGGCTCAAAGTC-3′ for BCR-ABL; sense,
5′-AACGGATTTGGTCGTATTGGG-3′ and antisense,
5′-CCTGGAAGATGGTGATGGGAT-3′ for glyceraldehyde 3-phosphate
dehydrogenase (GAPDH). The samples were denatured at 95°C for 10
min, followed by 39 cycles of 95°C for 30 sec and 60°C for 1 min
and 65°C for 30 sec to stop the reaction. Each experiment was
conducted in triplicate. Relative PDCD4 expression was calculated
using the ΔΔCq model (20). All
samples were normalized to GAPDH, which served as an endogenous
control.
SDS-PAGE and western blot
analysis
Cells were lysed in SDS sample buffer (Beyotime
Institute of Biotechnology, Shanghai, China) and the protein
concentration of the homogenized lysates was measured using a
bicinchoninic acid protein assay kit (Beyotime Institute of
Biotechnology) and equal amounts of protein were separated on 10%
SDS-PAGE (Beyotime Institute of Biotechnology) and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). Membranes were then blocked with 5% skimmed milk in
Tris-buffered saline containing 0.1% Tween-20 (Beyotime Institute
of Biotechnology) for 1 h and incubated overnight at 4°C with
rabbit monoclonal anti-PDCD4 (catalog no. 9535S; dilution, 1:1,000;
Cell Signaling Technology, Inc., Beverly, MA, USA) and human
polyclonal anti-β-actin (catalog no. sc-130656; dilution, 1:2,000;
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) antibodies.
The membranes were then incubated with peroxidase-conjugated goat
anti-rabbit immunoglobulin G secondary antibody (catalog no.
sc-2004; dilution, 1:5,000; Santa Cruz Biotechnology, Inc.) for 1 h
at room temperature. After washing with Tris-buffered saline with
0.1% Tween-20, signals were visualized with SuperSignal West Pico
Chemiluminescent Substrate (Pierce Biotechnology, Inc., Rockford,
IL, USA). Quantification of PDCD4 protein was normalized to β-actin
using Quantity One® Version 4.3.1 (Bio-Rad Laboratories,
Inc.). Western blot analysis was performed at least three times for
each sample.
Statistical analysis
Data were obtained from at least three independent
experiments and are presented as the mean ± standard error of the
mean. All statistical analysis was performed using SPSS 10.0
statistical software (SPSS, Inc., Chicago, IL, USA). Data was
analyzed using the Student's t-test or one-way analysis of
variance. Pearson's coefficient was calculated to analyze the
correlation between PDCD4 and BCR-ABL expression. P<0.05 was
considered to indicate a statistically significant difference.
Results
Decreased PDCD4 mRNA expression is
observed in CML patients
To investigate PDCD4 expression in primary CML,
PDCD4 mRNA expression was analyzed in 50 CML patients and 20
healthy controls by RT-qPCR. As shown in Fig. 1, high levels of PDCD4 mRNA expression
were observed in the 20 healthy control samples, however, all CML
patients exhibited extremely low PDCD4 expression. Furthermore, the
differences between PDCD4 expression in three phases of CML were
analyzed, however, no significant differences in PDCD4 mRNA
expression were identified in CML-CP, CML-AP and CML-BP patients
(Table I).
Decreased PDCD4 protein expression is
observed in CML patients
To evaluate PDCD4 protein expression levels in
primary CML, PDCD4 protein expression in 50 CML patients and 20
healthy controls was analyzed by western blot. The results
demonstrated that all healthy controls exhibited high PDCD4 protein
expression, however, PDCD4 protein expression was markedly
decreased or absent in the 50 CML samples, in accordance with that
of PDCD4 mRNA (Fig. 2A). PDCD4
protein expression in CML patients was significantly decreased when
compared with healthy controls (P<0.01; Fig. 2B).
PDCD4 expression is significantly
associated with BCR-ABL expression
BCR-ABL exhibits an important function in the
development of CML, therefore the association between PDCD4
expression and BCR-ABL expression was analyzed by Pearson
correlation analysis. As PDCD4 expression was markedly decreased or
absent in CML patients, 20 CML patients with relatively high levels
of PDCD4 expression (PDCD4/GAPDH>0.1) were selected for the
analysis. The results revealed that PDCD4 expression was negatively
correlated with BCR-ABL expression (n=20; r=−0.6716; P<0.001;
Fig. 3), which indicates that PDCD4
expression is associated with the development and progression of
CML.
Imatinib increases PDCD4 levels in the
K562 cell line
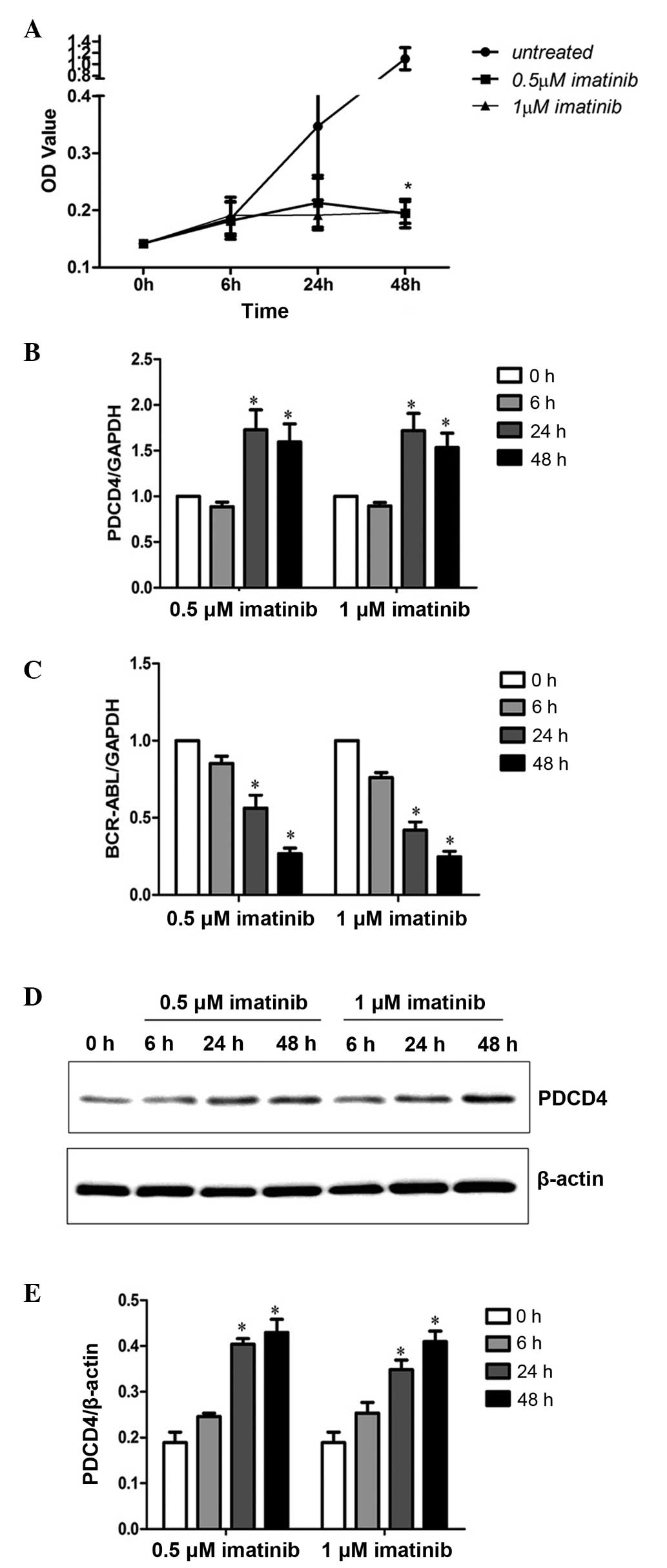
Since PDCD4 expression was negatively correlated
with BCR-ABL expression, PDCD4 expression in K562 cells after 6, 24
and 48 h treatment with 0.5 and 1 µM imatinib was analyzed by
RT-qPCR and western blot analysis. The results revealed that the
proliferation of K562 cells was significantly inhibited following
48 h of 0.5 and 1 µM imatinib treatment (P<0.05; Fig. 4A). Furthermore, PDCD4 mRNA expression
was significantly increased (P<0.05; Fig. 4B) and BCR-ABL expression (P<0.05;
Fig. 4C) was significantly decreased
after 24 and 48 h of treatment with 0.5 and 1 µM imatinib. Western
blot analysis of PDCD4 protein expression demonstrated the same
results (Fig. 4D and E). These
results indicated that decreased BCR-ABL expression may enhance
PDCD4 expression and inhibit malignant proliferation of K562
cells.
Discussion
CML is characterized by the presence of the Ph
chromosome, which results from a balanced reciprocal translocation
between the ABL gene on chromosome 9 and the BCR gene on chromosome
22 (1). A number of tumor suppressor
genes are inactivated or downregulated by BCR-ABL in Ph+
leukemia, including protein phosphatase 2 (21), p53 (22)
and phosphatase and tensin homolog (23). The results of the present study
revealed that the tumor suppressor, PDCD4, is also downregulated in
CML patients and is negatively correlated with BCR-ABL expression.
In addition, inhibition of BCR-ABL expression by imatinib induced
PDCD4 expression in the CML K562 cell line.
PDCD4 was recently identified as a tumor
suppressor gene and its expression is markedly decreased or absent
in a number of solid tumors (9–13).
However, the expression of PDCD4 in hematological neoplasms has
rarely been reported. Recently, it was reported that miR-21 is
frequently overexpressed in AML blasts, in association with a
marked PDCD4 protein downmodulation in nucleophosmin-mutant acute
myeloid leukemia (24). In the
present study, PDCD4 mRNA and protein expression were significantly
decreased in CML patients compared with healthy controls, which
suggests that PDCD4 is involved in normal hematopoietic
differentiation. PDCD4 is known to suppress protein translation by
directly interacting with eIF4A to inhibit the formation of the
translation-initiation complex (4) or
by interfering with translation elongation via an RNA-binding
domain (6), which indicates that
PDCD4 inhibits cell proliferation and is involved in tumor
development and progression. Furthermore, PDCD4 also suppresses
autophagy and promotes cell differentiation (25). In addition, it has been reported that
PDCD4 expression contributes to all-trans retinoic acid
(ATRA)-induced granulocytic but not monocytic/macrophagic
differentiation in acute myeloid leukemia, and ATRA induces PDCD4
expression via the inhibition of phosphoinositide 3-kinase
(PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR)
pathway (26). PDCD4 is also involved
in the germ line stem cell differentiation pathway: It is
hypothesized to relieve the inhibition of Bam by eIF4A (27). These findings suggest that PDCD4 may
contribute to cell differentiation of CML. However, in the present
study, no significant difference in PDCD4 expression was observed
between CML-CP, CML-AP and CML-BP patients, which indicates that
PDCD4 does not promote the transition of CML from CP to BP.
However, a recent study demonstrated that in the blast crisis of
CML, RBP2 expression was decreased; moreover, RBP2 could induce
cell differentiation and inhibit cell proliferation, which depend
on inhibiting miR-21 and increasing PDCD4 expression (28). All these findings suggest that PDCD4
may play a role in the blast crisis of CML.
The BCR-ABL oncoprotein constitutively activates
several downstream pathways (29).
One important target is the PI3K/AKT/mTor pathway, which is
constitutively activated in BCR-ABL-transformed cells and is
inhibited by imatinib mesylate (30).
In this study, the expression of PDCD4 and BCR-ABL in primary CML
patients was investigated, which revealed that PDCD4 expression was
negatively correlated with BCR-ABL expression. In addition,
following 24 and 48 h of imatinib treatment, PDCD4 expression was
significantly upregulated in K562 cells. These results suggest that
BCR-ABL negatively regulates PDCD4 expression in CML patients,
which is consistent with the results of previous studies, which
reported that inhibition of the BCR-ABL/mTOR/p70 S6K pathway
results in enhanced protein expression of PDCD4 in K562 cells
(31) and inhibition of p70 S6 kinase
activity by fluvastatin results in the upregulation of expression
of PDCD4 in renal cell carcinoma (32).
In conclusion, the results of the present study
indicate that the downregulation of PDCD4 expression is associated
with the development of human CML. Therefore, increased
understanding with regard to the involvement of PDCD4 in CML
progression may provide novel molecular targets for diagnosis and
therapy of human CML.
Acknowledgements
This study was supported by The National Natural
Science Foundation of China (grant nos. 81202069 and 81470403) and
The Yantai Science and Technology Development Plan (grant no.
2012071).
References
|
1
|
Quintás-Cardama A and Cortes JE: Chronic
myeloid leukemia: Diagnosis and treatment. Mayo Clin Proc.
81:973–988. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Quintás-Cardama A and Cortes JE: Molecular
biology of bcr-abl1-positive chronic myeloid leukemia. Blood.
113:1619–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Perrotti D, Jamieson C, Goldman J and
Skorski T: Chronic myeloid leukemia: Mechanisms of blastic
transformation. J Clin Invest. 120:2254–2264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang HS, Cho MH, Zakowicz H, Hegamyer G,
Sonenberg N and Colburn NH: A novel function of the MA-3 domains in
transformation and translation suppressor Pdcd4 is essential for
its binding to eukaryotic translation initiation factor 4A. Mol
Cell Biol. 24:3894–3906. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh P, Wedeken L, Waters LC, Carr MD and
Klempnauer KH: Pdcd4 directly binds the coding region of c-myb mRNA
and suppresses its translation. Oncogene. 30:4864–4873. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Biyanee A, Ohnheiser J, Singh P and
Klempnauer KH: A novel mechanism for the control of translation of
specific mRNAs by tumor suppressor protein Pdcd4: Inhibition of
translation elongation. Oncogene. 34:1384–1392. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hilliard A, Hilliard B, Zheng SJ, Sun H,
Miwa T, Song W, Göke R and Chen YH: Translational regulation of
autoimmune inflammation and lymphoma genesis by programmed cell
death 4. J Immunol. 177:8095–8102. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jansen AP, Camalier CE and Colburn NH:
Epidermal expression of the translation inhibitor programmed cell
death 4 suppresses tumorigenesis. Cancer Res. 65:6034–6041. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen Y, Knösel T, Kristiansen G, Pietas A,
Garber ME, Matsuhashi S, Ozaki I and Petersen I: Loss of PDCD4
expression in human lung cancer correlates with tumour progression
and prognosis. J Pathol. 200:640–646. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang H, Ozaki I, Mizuta T, Hamajima H,
Yasutake T, Eguchi Y, Ideguchi H, Yamamoto K and Matsuhashi S:
Involvement of programmed cell death 4 in transforming growth
factor-beta1-induced apoptosis in human hepatocellular carcinoma.
Oncogene. 25:6101–6112. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mudduluru G, Medved F, Grobholz R, Jost C,
Gruber A, Leupold JH, Post S, Jansen A, Colburn NH and Allgayer H:
Loss of programmed cell death 4 expression marks adenoma-carcinoma
transition, correlates inversely with phosphorylated protein kinase
B, and is an independent prognostic factor in resected colorectal
cancer. Cancer. 110:1697–1707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wei ZT, Zhang X, Wang XY, Gao F, Zhou CJ,
Zhu FL, Wang Q, Gao Q, Ma CH, Sun WS, et al: PDCD4 inhibits the
malignant phenotype of ovarian cancer cells. Cancer Sci.
100:1408–1413. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao F, Zhang P, Zhou C, Li J, Wang Q, Zhu
F, Ma C, Sun W and Zhang L: Frequent loss of PDCD4 expression in
human glioma: Possible role in the tumorigenesis of glioma. Oncol
Rep. 17:123–128. 2007.PubMed/NCBI
|
|
14
|
Yang HS, Knies JL, Stark C and Colburn NH:
Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1
transactivation. Oncogene. 22:3712–3720. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang HS, Matthews CP, Clair T, Wang Q,
Baker AR, Li CC, Tan TH and Colburn NH: Tumorigenesis suppressor
Pdcd4 down-regulates mitogen-activated protein kinase kinase kinase
kinase 1 expression to suppress colon carcinoma cell invasion. Mol
Cell Biol. 26:1297–1306. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Leupold JH, Yang HS, Colburn NH, Asangani
I, Post S and Allgayer H: Tumor suppressor Pdcd4 inhibits
invasion/intravasation and regulates urokinase receptor (u-PAR)
gene expression via Sp-transcription factors. Oncogene.
26:4550–4562. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Q, Sun Z and Yang HS: Downregulation
of tumor suppressor Pdcd4 promotes invasion and activates both
beta-catenin/Tcf and AP-1-dependent transcription in colon
carcinoma cells. Oncogene. 27:1527–1535. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang Q, Sun ZX, Allgayer H and Yang HS:
Downregulation of E-cadherin is an essential event in activating
beta-catenin/Tcf-dependent transcription and expression of its
target genes in Pdcd4 knockdown cells. Oncogene. 29:128–138. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Vardiman JW, Harris NL and Brunning RD:
The World Health Organization (WHO) classification of the myeloid
neoplasms. Blood. 100:2292–2302. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neviani P, Santhanam R, Trotta R, Notari
M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, et al:
The tumor suppressor PP2A is functionally inactivated in blast
crisis CML through the inhibitory activity of the BCR/ABL-regulated
SET protein. Cancer Cell. 8:355–368. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beck Z, Kiss A, Tóth FD, Szabó J, Bácsi A,
Balogh E, Borbély A, Telek B, Kovács E, Oláh E and Rak K:
Alterations of P53 and RB genes and the evolution of the
accelerated phase of chronic myeloid leukemia. Leuk Lymphoma.
38:587–597. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Peng C, Chen Y, Yang Z, Zhang H, Osterby
L, Rosmarin AG and Li S: PTEN is a tumor suppressor in CML stem
cells and BCR-ABL-induced leukemias in mice. Blood. 115:626–635.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Riccioni R, Lulli V, Castelli G, Biffoni
M, Tiberio R, Pelosi E, LoCoco F and Testa U: miR-21 is
overexpressed in NPM1-mutant acute myeloid leukemias. Leuk Res.
39:221–228. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Song X, Zhang X, Wang X, Zhu F, Guo C,
Wang Q, Shi Y, Wang J, Chen Y and Zhang L: Tumor suppressor gene
PDCD4 negatively regulates autophagy by inhibiting the expression
of autophagy-related gene ATG5. Autophagy. 9:743–755. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ozpolat B, Akar U, Steiner M,
ZorrillaCalancha I, TiradoGomez M, Colburn N, Danilenko M, Kornblau
S and Berestein GL: Programmed cell death-4 tumor suppressor
protein contributes to retinoic acid-induced terminal granulocytic
differentiation of human myeloid leukemia cells. Mol Cancer Res.
5:95–108. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cash AC and Andrews J: Fine scale analysis
of gene expression in Drosophila melanogaster gonads reveals
Programmed cell death 4 promotes the differentiation of female
germline stem cells. BMC Dev Biol. 12:42012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou M, Zeng J, Wang X, Wang X, Huang T,
Fu Y, Sun T, Jia J and Chen C: Histone demethylase RBP2 decreases
miR-21 in blast crisis of chronic myeloid leukemia. Oncotarget.
6:1249–1261. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cilloni D and Saglio G: Molecular
pathways: BCR-ABL. Clin Cancer Res. 18:930–937. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Parmar S, Smith J, Sassano A, Uddin S,
Katsoulidis E, Majchrzak B, Kambhampati S, Eklund EA, Tallman MS,
Fish EN and Platanias LC: Differential regulation of the p70 S6
kinase pathway by interferon alpha (IFNalpha) and imatinib mesylate
(STI571) in chronic myelogenous leukemia cells. Blood.
106:2436–2443. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carayol N, Katsoulidis E, Sassano A,
Altman JK, Druker BJ and Platanias LC: Suppression of programmed
cell death 4 (PDCD4) protein expression by BCR-ABL-regulated
engagement of themTOR/p70 S6 kinase pathway. J Biol Chem.
283:8601–8610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Woodard J, Sassano A, Hay N and Platanias
LC: Statin-dependent suppression of the Akt/mammalian target of
rapamycin signaling cascade and programmed cell death 4
up-regulation in renal cell carcinoma. Clin Cancer Res.
14:4640–4649. 2008. View Article : Google Scholar : PubMed/NCBI
|