Introduction
Central nervous system (CNS)-based cancers account
for approximately 1.4% of all cancers worldwide and account for
proportionally more deaths, i.e., 2.7% among cancer-related
mortalities (1). Estimates for
incidence of CNS cancer in 2016 were 6.4/100,000 individuals and
for deaths this was 4.3/100,000 (2).
Notably, of the many types of cells in CNS, most of the malignant
tumors in brain originate from astrocytes. There are four distinct
types of astrocytomas, classified on the basis of microscopic
characteristics, grades (I–IV). Only the relatively rare grade I
astrocytomas are curable while the other gliomas are incurable.
Although grade III and IV gliomas are considered high-grade
gliomas, only grade IV glioma is commonly known as glioblastoma
multiforme. Glioblastomas show a high mitotic/proliferation index,
diffused infiltration, angiogenesis, microvascular proliferation
and pleomorphic vessel resistance to apoptosis, nuclear atypia, and
pseudopalisading necrosis (3,4). Gliomas progress with time from the time
of diagnosis to aggressiveness and approximately 90% of astrocytoma
present de novo as a glioblastoma. From the stage of
glioblastoma occurrence, there is a poor survival rate, with a
median of 16 months and survival rate of approximately 3% (5). It is extremely difficult to eliminate
the glioblastoma even with total resection, as tumor cells persist
microscopically, with tumor recurrence occurring in 90% of the
patients at the original tumor location (6). The frequently seen extensive hypoxic
regions in glioblastomas contribute to the highly malignant
phenotype of these tumors, exacerbating the prognosis and clinical
outcomes of the patients. Hypoxic tumor cells are more resistant to
chemo- and radiation therapy (7,8) and are
protected by the vasculature that develops due to hypoxia-mediated
molecular processes (3). Hypoxia also
supports the survival of neural and glioma stem cells, which are
drug resistant and possess tumorigenicity potential (9,10).
Considering the significance of hypoxia in the growth and
aggressiveness of glioblastomas, targeting hypoxia potentially
improves the outcomes in patients with this lethal cancer type.
Significance of hypoxia in development of
glioblastoma
The pathognomonic feature of pseudopalisading
necrosis, which is the area of hypercellularity surrounding
necrotic regions, and vascular proliferation observed in
glioblastoma tumors is a manifestation of hypoxia. These
hypercellularity regions are highly hypoxic and represent tumor
cells migrating away from vasoocclusive, distorted and degenerating
blood vessels from the tumor center. Additionally, the cells have a
high expression of hypoxia-inducible factor-1α (HIF-1α) and
vascular endothelial growth factor (VEGF), which promote
angiogenesis (11). A subset of
growth factors including angiopoietins, fibroblast growth factors,
chemokines and matrix metalloproteinases, play an important role in
tumor angiogenesis (12). These new
vessels are deformed, leaky and have gaps between endothelial
cells, resulting in vascular stasis. Repeated cycle of events of
angiogenesis, vascular collapse due to deformation and cancer cell
migration, contribute to rapid tumor expansion in adjacent normal
tissue and invasion (13). Inasmuch
as hypoxia drives the progression and aggressiveness of
glioblastoma tumors, a strategy for the treatment of this type of
cancer has been developed by measuring tumor volume and the extent
of intratumoral hypoxia, using fluoromisonidazole probe-based
positron emission tomography, followed by appropriate targeting of
hypoxic cells to improve the treatment outcome (14). As mentioned above, tumor stem cells
residing in hypoxic pseudopalisading zones are protected from
chemoradiation because of vascular stasis and depletion of
molecular oxygen. In a prospective clinical trial testing
bevacizumab and irinotecan in glioblastoma patients, it was
observed that hypoxia-induced carbonic anhydrase (CA9) and HIF-2α
were major and significant predictors of treatment effectiveness
and overall survival (15).
Hypoxia, growth factors and tumor
angiogenesis
Hypoxic response is essentially mediated by HIF-1α,
which is induced under conditions of low oxygen, in a nuclear
factor-κB-dependent manner. HIF-1α is a transcription factor that
regulates the expression of approximately 60 genes involved in
several cell pathways such as glycolysis, angiogenesis, invasion
and epithelial-mesenchymal transition, which are critical for tumor
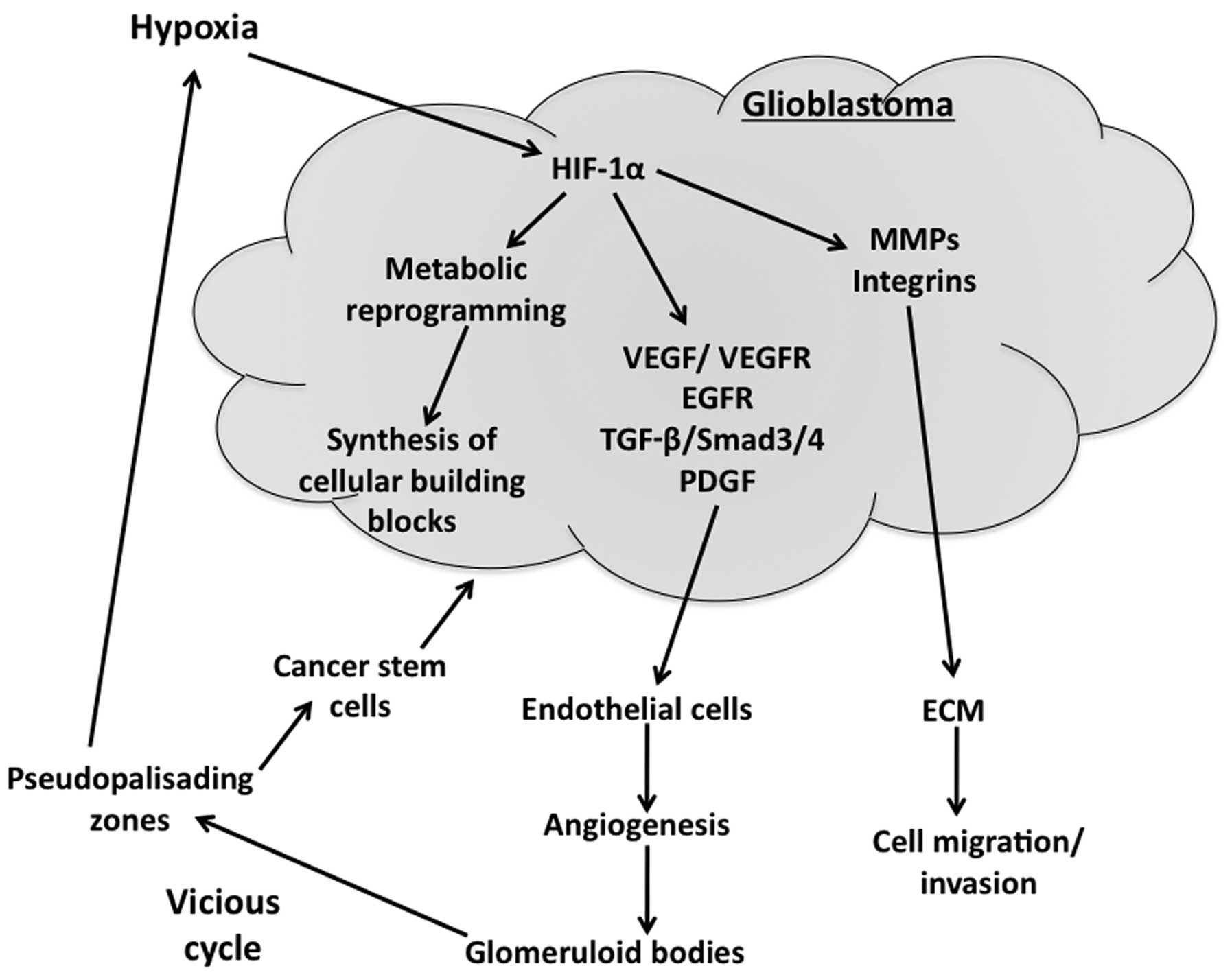
growth and proliferation (Fig. 1).
HIF-1α likely mediates the hypoxic response, partly through the
upregulation of glucose transporter 1 (Glut1) and glycolytic genes,
thereby promoting anerobic glycolysis and metabolism to survive the
unfavorable conditions encountered during hypoxia. Tumor initiating
cancer stem cells undergo HIF-1α-mediated adaptation to hypoxia and
thus have an elevated expression of various HIF-regulated genes
(15). It has been shown that hypoxic
area volume in the tumor is inversely proportional to the survival
of the patients and thus hypoxic volume ratios greater than the
median, survived only ~4 months as compared to >12 months in the
patients with less than median hypoxic burden (14).
Multiple pro-angiogenic factors, which are under the
regulation of HIF-1α and other transcription factors, are present
in glioblastoma and contribute to the formation of new vessels
(Fig. 1). VEGF, a prominent
pro-angiogenic factor and its receptor VEGF receptor 2 (VEGFR2),
are highly expressed in glioblastomas and tumor vasculature
(17,18). In addition to angiogenesis, VEGF
regulates vascular permeability and contributes to vasogenic edema.
Most of the approaches developed for glioblastoma therapy that
specifically address targeting angiogenesis, mainly focused on
blocking VEGF signaling pathways (19). Primary glioblastomas also exhibit a
high proportion of mutations and/or the overexpression of epidermal
growth factor receptor (EGFR) gene. The EGFR gene codes for a
170-kDa receptor, which is a tyrosine kinase receptor, and a
glycosylated plasma membrane. Altered EGFR function often leads to
oncogenesis in various cell types and contributes to glioblastoma
initiation, cancer cell proliferation and growth, invasion,
resistance to apoptosis, chemo- and radiotherapy and angiogenesis
(20,21). It has been observed that almost 40% of
glioblastomas have amplified EGFR gene expression with 50% of them
showing overexpression of the receptor protein (22). Even in less malignant astrocytomas and
oligodendrogliomas, there are elevated EGFR mRNA levels, suggesting
that other oncogenic events play a role in the amplification of the
EGFR gene (23).
Another important growth factor is transforming
growth factor-β (TGF-β), whose signaling controls different cell
functions such as proliferation, differentiation, and apoptosis and
thus plays an important role in cancer progression, remodeling of
the extracellular matrix and angiogenesis (24). The isoforms of TGF-β, TGF-β1/−β2 and
their complete cognate signaling machinery are known to be highly
expressed in glioblastomas, and contribute to enhanced
proliferation and invasion of tumor cells and tumor angiogenesis
(25). TGF-β2 has been shown to
contribute to aberrant vascular gene expression via Smad 2/4 and
Smad 3/4 signaling pathways in glioblastoma (26). Experimental studies indicated that
plate-derived growth factor (PDGF)-B overexpression in murine
neural and glial progenitor cells leads to the formation of
malignant gliomas (27), and it is
known that human glioblastomas show elevated expression of PDGF and
PDGF-receptor. PDGF-B, derived from tumor cells is shown to enhance
angiogenesis in endothelial cells via increased production of VEGF
(28). The net effect on overall
angiogenesis in glioblastoma tumors is dependent on the source and
availability of PDGF-B and the counteracting effects of
Wnt/β-catenin signaling on angiogenesis (29).
Significance of abnormal vessel formation in
glioblastoma
Glioblastomas are characterized by the presence of
morphologically abnormal and dysfunctional vasculature, also known
as glomeruloid bodies or vascular tufts (30). These glomeruloid bodies have multiple
layers of endothelial cells, pericytes and smooth muscle cells and
exhibit a thick basement membrane. Such morphology constitutes a
typical diagnosis for glioblastoma, albeit not for low-grade
gliomas (19). The origin of these
vessels is not clear and controversial. Previous in vitro
studies indicated that glioma stem-like cells can be induced to
differentiate to an endothelial cell type and that glioblastoma
stem-like cells contribute to vessel formation (Fig. 1) in experimental models (31,32).
Although the blood-brain barrier, which consists of endothelial
cells and pericytes astrocytes in normal brain vessels, is
disrupted in glioblastoma, with a resultant increase in vascular
permeability (33), this barrier
appears to remain intact at the invasive front of the tumor and the
normal cortex, which is invaded by migratory glioblastoma cells.
Thus, therapeutic agents that target glioblastoma to cross the
blood-brain barrier to attack the invasive glioblastoma cells. Of
note, the malformed blood vessels in the glioblastoma tumors in
turn contribute to increasing hypoxic conditions in the tumor, as
these vessels are poorly perfused (34). As mentioned earlier, hypoxia is a
potent inducer of tumor progression through metabolic
reprogramming, resistance to cell death, immunosuppression,
inflammation and epithelial-mesenchymal transition of cancer cells
(13). The stabilized and thus
elevated HIF-1α in glioma cells causes an increase in the
expression of VEGF and CXC chemokine ligand 12, both of which
promote angiogenesis through different mechanisms, eventually
leading to the formation of the malformed vessels, thus leading to
a vicious cycle that promotes glioblastoma tumor growth and
aggressiveness (Fig. 1) (35).
Hypoxia and glioblastoma stem cells
Cancer stem cells are more potent in inducing tumors
when injected into the brains of immunocompromised mice, as
compared to non-stem cells from tumors. Cancer stem cells have a
high level of resistance to radiation through activation of the DNA
damage checkpoint and to chemoresistance (36,37). An
important function of the tumor vasculature is to provide a supply
of glioblastoma stem-like cells (38), which are critical for the progression
of tumor growth and resistance to chemo- and radiotherapy. Notably,
hypoxia is shown to increase the expression of CD133, a stem cell
marker, in brain tumors and this increase is probably mediated by
the HIF family of transcription factors (36,39). Of
the HIF transcription factors, HIF-1α promotes proliferation and
survival of all cancer cells and is activated in normal neural
progenitors, thus limiting its value, in terms of therapeutic
targeting. HIF-2α, which is practically absent in non-glioma stem
cells, is specifically elevated in glioblastoma stem cells, even
under modest hypoxic conditions, thereby making HIF-2α an
interesting therapeutic target in glioblastomas (40). HIF-2α regulates chromatin structure by
activating proteins/processes that modify chromatin epigenetically,
such as the histone methyltransferase and mixed lineage leukemia 1
(41). Other hypoxia-induced genes,
that are expressed at elevated levels in glioblastoma stem cells,
include Oct4, Glut1, SerpinB9, and VEGF. In addition to promoting
angiogenesis by the glioma stem cells, hypoxia also increases other
microenvironmental interactions by these cancer stem cells
including the suppression of immune response. Hypoxia activates
pSTAT signaling, thereby increasing the secretion of
immunosuppressive cytokines such as CSF1 and CCL2, which are useful
in inhibiting T-cell proliferation and macrophage phagocytosis,
which in turn accelerate tumor progression (42).
Glioma stem cells show enhanced Notch signaling
pathways (43,44), which contribute to their
radioresistance (45). Thus, blocking
Notch signaling in human glioblastoma stem cells with high doses of
γ-secretase inhibitors reduces proliferation of these cancer stem
cells and tumor formation, and increases differentiation (46). Other enhanced signaling pathways
include PI3 kinase/Akt, Hedgehog and Stat3 (47–49).
Therapeutic implications
Inasmuch as hypoxia and hypoxia-induced factors
promote the growth of glioblastomas, therapeutic measures
addressing hypoxia have been developed. Thus, a monoclonal antibody
against VEGF, bevacizumab, has been developed and approved as a
second-line monotherapy of glioblastoma patients. However,
bevacizumab therapy achieved improvement in progression-free
survival but was not beneficial for overall survival (50). On the other hand, a subset of patients
who positively responded to cediranib, a pan-VEGF receptor tyrosine
kinase inhibitor, which increased blood perfusion, showed improved
survival (51). A better
understanding of the significance and pathophysiology of tumor
vasculature and associated factors is needed to develop effective
therapeutics that target these elements of glioblastomas.
An interesting direct approach using a prodrug is
via a hypoxia-activated TH-302, which is a nitroimidazole prodrug
of bromo-isophosphoramide mustard (Br-IPM), a cytotoxin. Under
hypoxic conditions, TH-302 is converted by intracellular reductases
to the alkylating agent Br-IPM, which functions as a DNA
cross-linking agent damaging the hypoxic tumor cells, in which it
is formed. Br-IPM may also diffuse into the extracellular matrix
and the nearby normoxic cells, showing its cytotoxic effects in
these cells as well. However, TH-302 per se is not effective under
normoxic conditions and requires hypoxic conditions to show its
cytotoxic effects (52). A recent
study showed that TH-302 is well tolerated when administered after
a 4-week period of postsurgical recovery, to glioblastoma patients,
in combination with bevacizumab (10 mg/kg), with a promising
clinical benefit rate of 62% (53).
A major advance in the treatment of glioblastoma
during the last decade is the concomitant chemoradiotherapy with
temozolomide. It has been shown that the median survival of
patients receiving radiation therapy alone is 12.1 months, whereas
a combination of radiation with temozolomide increased the median
survival to ≤14.6 months (54). One
of 5 glioblastoma patients survived in the temozolomide-treated
population, whereas essentially none of the patients without
temozolomide survived to 3 years, during the follow-up. The
survival promoting action of temozolomide is essentially due to the
epigenetic modification of the methyl guanine methyl transferase
(MGMT) promoter, in the glioblastoma cells, which leads to reduced
or no expression of this enzyme and thus the inability to repair
the guanine methylation induced by temozolomide. However, MGMT
silencing by promoter modification is seen only in 40% of the
glioblastoma cases. Therefore, the treatment benefit of
temozolomide is not evident in the remaining 60% of the
glioblastoma patients who harbor normal expression of the MGMT,
which is capable of repairing the temozolomide-mediated guanine
methylation (55).
Other agents that have been examined as therapeutic
agents targeting HIFs are amphotericin B, an anti-fungal drug,
which inhibits HIF-1α transcription (56); and 2-methoxyestradiol, an inhibitor of
HIF-1α, which has been tested in phase-I clinical trials with some
success showing stable disease in 38% of the enrolled patients
(57). Despite the significant leaps
in our understanding of the molecular events that underlie the
aggressiveness and progression of glioblastomas, much progress is
needed for effective therapeutic development that goes much further
than extending life by a short period of time.
Conclusions
Among the CNS cancer grade IV astrocytomas,
glioblasomas are highly aggressive. The extensive hypoxic regions
in glioblastomas contribute to the highly malignant phenotype of
these tumors, exacerbating the prognosis of the patients. Hypoxic
tumor cells are resistant to chemo- and radiation therapy and are
also protected by the malfunctional vasculature that developed due
to hypoxia. The abnormal and malfunctional vessels play a critical
role in generating pseudopalisading necrotic regions that are
hypoxic and protect cancer stem cells residing in that region from
therapeutic agents, which facilitates cancer stem cell
proliferation and tumor growth, thus causing a vicious cycle of
tumor growth. Therapeutic approaches that target hypoxia-induced
factors such as use of monoclonal antibody against VEGF,
bevacizumab, have been useful only in stabilizing the disease but
failed to increase the overall survival. Hypoxia-activated TH-302
appears to be more attractive due to its better beneficial effects
in glioblastoma patients. A better understanding of the hypoxia
mediated protection of the glioblastoma cells is needed in future
studies in order to develop more effective therapeutics.
References
|
1
|
Filippini G: Epidemiology of primary
central nervous system tumors. Handb Clin Neurol. 104:3–22. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Howlader N, Noone A, Krapcho M, Miller D,
Bishop K, Altekruse S, Kosary C, Yu M, Ruhl J, Tatalovich Z, et al:
Seer cancer statistics review, 1975-2013. National Cancer
Institute; Bethesda, MD: http://seer.Cancer.Gov/csr/1975_2013/Accessed.
April 15–2016
|
|
3
|
Louis DN, Ohgaki H, Wiestler OD, Cavenee
WK, Burger PC, Jouvet A, Scheithauer BW and Kleihues P: The 2007
WHO classification of tumours of the central nervous system. Acta
Neuropathol. 114:97–109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Furnari FB, Fenton T, Bachoo RM, Mukasa A,
Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, et al:
Malignant astrocytic glioma: Genetics, biology, and paths to
treatment. Genes Dev. 21:2683–2710. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ostrom QT, Gittleman H, Liao P, Rouse C,
Chen Y, Dowling J, Wolinsky Y, Kruchko C and Barnholtz-Sloan J:
CBTRUS statistical report: Primary brain and central nervous system
tumors diagnosed in the United States in 2007-2011. Neuro-oncol.
16(Suppl 4): iv1–iv63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hou LC, Veeravagu A, Hsu AR and Tse VC:
Recurrent glioblastoma multiforme: A review of natural history and
management options. Neurosurg Focus. 20:E52006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rong Y, Durden DL, Van Meir EG and Brat
DJ: ‘Pseudopalisading’ necrosis in glioblastoma: A familiar
morphologic feature that links vascular pathology, hypoxia, and
angiogenesis. J Neuropathol Exp Neurol. 65:529–539. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hsieh CH, Shyu WC, Chiang CY, Kuo JW, Shen
WC and Liu RS: NADPH oxidase subunit 4-mediated reactive oxygen
species contribute to cycling hypoxia-promoted tumor progression in
glioblastoma multiforme. PLoS One. 6:e239452011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bao S, Wu Q, McLendon RE, Hao Y, Shi Q,
Hjelmeland AB, Dewhirst MW, Bigner DD and Rich JN: Glioma stem
cells promote radioresistance by preferential activation of the DNA
damage response. Nature. 444:756–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brat DJ, CastellanoSanchez AA, Hunter SB,
Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B and Van Meir EG:
Pseudopalisades in glioblastoma are hypoxic, express extracellular
matrix proteases, and are formed by an actively migrating cell
population. Cancer Res. 64:920–927. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jain RK, di Tomaso E, Duda DG, Loeffler
JS, Sorensen AG and Batchelor TT: Angiogenesis in brain tumours.
Nat Rev Neurosci. 8:610–622. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jain RK: Normalizing tumor
microenvironment to treat cancer: Bench to bedside to biomarkers. J
Clin Oncol. 31:2205–2218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Spence AM, Muzi M, Swanson KR, O'Sullivan
F, Rockhill JK, Rajendran JG, Adamsen TC, Link JM, Swanson PE,
Yagle KJ, et al: Regional hypoxia in glioblastoma multiforme
quantified with [18F]fluoromisonidazole positron emission
tomography before radiotherapy: Correlation with time to
progression and survival. Clin Cancer Res. 14:2623–2630. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sathornsumetee S, Cao Y, Marcello JE,
Herndon JE II, McLendon RE, Desjardins A, Friedman HS, Dewhirst MW,
Vredenburgh JJ and Rich JN: Tumor angiogenic and hypoxic profiles
predict radiographic response and survival in malignant astrocytoma
patients treated with bevacizumab and irinotecan. J Clin Oncol.
26:271–278. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mannino M and Chalmers AJ: Radioresistance
of glioma stem cells: Intrinsic characteristic or property of the
‘microenvironment-stem cell unit’? Mol Oncol. 5:374–386. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Plate KH, Breier G, Weich HA and Risau W:
Vascular endothelial growth factor is a potential tumour
angiogenesis factor in human gliomas in vivo. Nature. 359:845–848.
1992. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Olsson AK, Dimberg A, Kreuger J and
Claesson-Welsh L: VEGF receptor signalling - in control of vascular
function. Nat Rev Mol Cell Biol. 7:359–371. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dimberg A: The glioblastoma vasculature as
a target for cancer therapy. Biochem Soc Trans. 42:1647–1652. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
KarpelMassler G, Schmidt U, Unterberg A
and Halatsch ME: Therapeutic inhibition of the epidermal growth
factor receptor in high-grade gliomas: Where do we stand? Mol
Cancer Res. 7:1000–1012. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Squatrito M and Holland EC: DNA damage
response and growth factor signaling pathways in gliomagenesis and
therapeutic resistance. Cancer Res. 71:5945–5949. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Riddick G and Fine HA: Integration and
analysis of genome-scale data from gliomas. Nat Rev Neurol.
7:439–450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Reifenberger J, Reifenberger G, Ichimura
K, Schmidt EE, Wechsler W and Collins VP: Epidermal growth factor
receptor expression in oligodendroglial tumors. Am J Pathol.
149:29–35. 1996.PubMed/NCBI
|
|
24
|
Massagué J: TGFbeta in Cancer. Cell.
134:215–230. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bruna A, Darken RS, Rojo F, Ocaña A,
Peñuelas S, Arias A, Paris R, Tortosa A, Mora J, Baselga J, et al:
High TGFbeta-Smad activity confers poor prognosis in glioma
patients and promotes cell proliferation depending on the
methylation of the PDGF-B gene. Cancer Cell. 11:147–160. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Dieterich LC, Mellberg S, Langenkamp E,
Zhang L, Zieba A, Salomäki H, Teichert M, Huang H, Edqvist PH,
Kraus T, et al: Transcriptional profiling of human glioblastoma
vessels indicates a key role of VEGF-A and TGFβ2 in vascular
abnormalization. J Pathol. 228:378–390. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shih AH and Holland EC: Platelet-derived
growth factor (PDGF) and glial tumorigenesis. Cancer Lett.
232:139–147. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ,
Cavenee WK and Cheng SY: Platelet-derived growth factor-B enhances
glioma angiogenesis by stimulating vascular endothelial growth
factor expression in tumor endothelia and by promoting pericyte
recruitment. Am J Pathol. 162:1083–1093. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lindblom P, Gerhardt H, Liebner S,
Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S,
Landegren U, Nystrom HC, et al: Endothelial PDGF-B retention is
required for proper investment of pericytes in the microvessel
wall. Genes Dev. 17:1835–1840. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kanu OO, Mehta A, Di C, Lin N, Bortoff K,
Bigner DD, Yan H and Adamson DC: Glioblastoma multiforme: A review
of therapeutic targets. Expert Opin Ther Targets. 13:701–718. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
RicciVitiani L, Pallini R, Biffoni M,
Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G,
Larocca LM, et al: Tumour vascularization via endothelial
differentiation of glioblastoma stem-like cells. Nature.
468:824–828. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Soda Y, Marumoto T, FriedmannMorvinski D,
Soda M, Liu F, Michiue H, Pastorino S, Yang M, Hoffman RM, Kesari
S, et al: Transdifferentiation of glioblastoma cells into vascular
endothelial cells. Proc Natl Acad Sci USA. 108:4274–4280. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wolburg H, Noell S, FallierBecker P, Mack
AF and Wolburg-Buchholz K: The disturbed blood-brain barrier in
human glioblastoma. Mol Aspects Med. 33:579–589. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gmeiner M, Sonnberger M, Wurm G and Weis
S: Glioblastoma with the appearance of arteriovenous malformation:
Pitfalls in diagnosis. Clin Neurol Neurosurg. 115:501–506. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Würth R, Bajetto A, Harrison JK, Barbieri
F and Florio T: CXCL12 modulation of CXCR4 and CXCR7 activity in
human glioblastoma stem-like cells and regulation of the tumor
microenvironment. Front Cell Neurosci. 8:1442014.PubMed/NCBI
|
|
36
|
Blazek ER, Foutch JL and Maki G: Daoy
medulloblastoma cells that express CD133 are radioresistant
relative to CD133- cells, and the CD133+ sector is enlarged by
hypoxia. Int J Radiat Oncol Biol Phys. 67:1–5. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Todaro M, Alea MP, Di Stefano AB,
Cammareri P, Vermeulen L, Iovino F, Tripodo C, Russo A, Gulotta G,
Medema JP, et al: Colon cancer stem cells dictate tumor growth and
resist cell death by production of interleukin-4. Cell Stem Cell.
1:389–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Calabrese C, Poppleton H, Kocak M, Hogg
TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, et
al: A perivascular niche for brain tumor stem cells. Cancer Cell.
11:69–82. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Platet N, Liu SY, Atifi ME, Oliver L,
Vallette FM, Berger F and Wion D: Influence of oxygen tension on
CD133 phenotype in human glioma cell cultures. Cancer Lett.
258:286–290. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Li Z, Bao S, Wu Q, Wang H, Eyler C,
Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, et al:
Hypoxia-inducible factors regulate tumorigenic capacity of glioma
stem cells. Cancer Cell. 15:501–513. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Heddleston JM, Wu Q, Rivera M, Minhas S,
Lathia JD, Sloan AE, Iliopoulos O, Hjelmeland AB and Rich JN:
Hypoxia-induced mixed-lineage leukemia 1 regulates glioma stem cell
tumorigenic potential. Cell Death Differ. 19:428–439. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wei J, Wu A, Kong LY, Wang Y, Fuller G,
Fokt I, Melillo G, Priebe W and Heimberger AB: Hypoxia potentiates
glioma- mediated immunosuppression. PLoS One. 6:e161952011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kanamori M, Kawaguchi T, Nigro JM,
Feuerstein BG, Berger MS, Miele L and Pieper RO: Contribution of
Notch signaling activation to human glioblastoma multiforme. J
Neurosurg. 106:417–427. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lino MM, Merlo A and Boulay JL: Notch
signaling in glioblastoma: A developmental drug target? BMC Med.
8:722010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wang J, Wakeman TP, Lathia JD, Hjelmeland
AB, Wang XF, White RR, Rich JN and Sullenger BA: Notch promotes
radioresistance of glioma stem cells. Stem Cells. 28:17–28.
2010.PubMed/NCBI
|
|
46
|
Fan X, Khaki L, Zhu TS, Soules ME, Talsma
CE, Gul N, Koh C, Zhang J, Li YM, Maciaczyk J, et al: NOTCH pathway
blockade depletes CD133-positive glioblastoma cells and inhibits
growth of tumor neurospheres and xenografts. Stem Cells. 28:5–16.
2010.PubMed/NCBI
|
|
47
|
Wei Y, Jiang Y, Zou F, Liu Y, Wang S, Xu
N, Xu W, Cui C, Xing Y, Liu Y, Cao B, Liu C, Wu G, Ao H, Zhang X
and Jiang: Activation of PI3K/Akt pathway by CD133-p85 interaction
promotes tumorigenic capacity of glioma stem cells. Proc Natl Acad
Sci USA. 110:6829–6834. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Morgenroth A, Vogg AT, Ermert K,
Zlatopolskiy B and Mottaghy FM: Hedgehog signaling sensitizes
glioma stem cells to endogenous nano-irradiation. Oncotarget.
5:5483–5493. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Liu M, Inoue K, Leng T, Guo S and Xiong
ZG: TRPM7 channels regulate glioma stem cell through STAT3 and
Notch signaling pathways. Cell Signal. 26:2773–2781. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Keunen O, Johansson M, Oudin A, Sanzey M,
Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R, et al:
Anti-VEGF treatment reduces blood supply and increases tumor cell
invasion in glioblastoma. Proc Natl Acad Sci USA. 108:3749–3754.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Sorensen AG, Emblem KE, Polaskova P,
Jennings D, Kim H, Ancukiewicz M, Wang M, Wen PY, Ivy P, Batchelor
TT, et al: Increased survival of glioblastoma patients who respond
to antiangiogenic therapy with elevated blood perfusion. Cancer
Res. 72:402–407. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Meng F, Evans JW, Bhupathi D, Banica M,
Lan L, Lorente G, Duan JX, Cai X, Mowday AM, Guise CP, et al:
Molecular and cellular pharmacology of the hypoxia-activated
prodrug TH-302. Mol Cancer Ther. 11:740–751. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Cavazos DA and Brenner AJ: Hypoxia in
astrocytic tumors and implications for therapy. Neurobiol Dis.
85:227–233. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: European Organisation for Research and Treatment of
Cancer Brain Tumor and Radiotherapy Groups; National Cancer
Institute of Canada Clinical Trials Group: Radiotherapy plus
concomitant and adjuvant temozolomide for glioblastoma. N Engl J
Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Hegi ME, Diserens AC, Gorlia T, Hamou MF,
de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani
L, et al: MGMT gene silencing and benefit from temozolomide in
glioblastoma. N Engl J Med. 352:997–1003. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yeo EJ, Ryu JH, Cho YS, Chun YS, Huang LE,
Kim MS and Park JW: Amphotericin B blunts erythropoietin response
to hypoxia by reinforcing FIH-mediated repression of HIF-1. Blood.
107:916–923. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Kirkpatrick J, Desjardins A, Quinn J, Rich
J, Vredenburgh J, Sathornsumetee S, Gururangan S, Sidor C, Friedman
H and Reardon D: Phase ii open-label, safety, pharmacokinetic and
efficacy study of 2-methoxyestradiol nanocrystal colloidal
dispersion administered orally to patients with recurrent
glioblastoma multiforme. J Clin Oncol (ASCO Annual Meeting abs.).
25:20652007.
|