Introduction
Pancreatic cancer (PC) ranks as the fourth leading
cause of cancer-associated mortality in the USA, with the worst
prognosis of all solid tumors. It is estimated that 40,560/48,960
patients diagnosed with PC in the USA will succumb to this disease
in 2015 (1). Despite advancements in
the understanding of the genetics of PC and application of combined
chemotherapy and targeted biological agents, the management of this
lethal malignancy remains one of the greatest oncological
challenges (2). Currently, the only
successful treatment for a local pancreatic tumor is surgery, and
adjuvant chemotherapy after surgery is indicated to delay relapse,
but the effect is limited (3). For
those who have advanced and metastatic PC, chemotherapy is a
primary treatment and gemcitabine has become the most popular
first-line therapy for the treatment of pancreatic ductal
adenocarcinoma (PDAC) (4,5). However, the response ratio of PDAC to
gemcitabine in clinical research is <25%, and those patients
showing initial response generally develop drug resistance during
therapy (6,7). The development of rapid resistance to
gemcitabine may be due to either stem-like subpopulations of tumor
cells, which have innate resistance to chemotherapy, or be caused
by molecular changes in cancer cells, such as alternations of
transport and metabolism of gemcitabine or the upregulation of
phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) or the
DNA repair pathway (8–12). In addition to monotherapy, there are
clinical treatments with gemcitabine in combination with other
biological or chemotherapeutical targeted agents, such as the
epidermal growth factor receptor (EGFR) inhibitors erlotinib,
tipifarnib and gefitinib (13,14), but
the combinations are limited, with unsatisfactory efficacy. Only
patients developing a rapid response upon erlotinib could benefit
from EGFR-targeted therapy (13,15).
Overall, there is an urgent requirement to identify novel
chemotherapeutical agents or an effective combination scheme for
this malignancy.
Arenobufagin is a cardiac glycoside agent and one of
the main active ingredients of toad secretions. Secretions of the
postauricular and skin glands of Bufo gargarizans and
Duttaphrynus melanostictus have been used as a traditional
Chinese medicine (TCM) in numerous diseases, including cancers,
heart failure and sore throat (16).
A previous study suggested cardiac glycosides as potent inhibitors
of cancer cell growth (17). Previous
studies have demonstrated that arenobufagin is a potent
Na+-K+ pump inhibitor that depresses the
delayed rectifier K+ current in myocytes (18,19). It
has been reported that arenobufagin can suppress cell adhesion,
migration and invasion and induce apoptosis and autophagy via
inhibition of the PI3K/Akt/mammalian target of rapamycin pathway in
a human hepatoma cell line (20,21) as
well as block vascular endothelial growth factor (VEGF)-mediated
angiogenesis to prevent carcinogenesis (22). However, to the best of our knowledge,
the effects and the mechanism of arenobufagin in PC cells have not
been studied.
To uncover the effect of arenobufagin on PC cells
and the assistance to first-line medicine, the
gemcitabine-resistant pancreatic carcinoma cell line Panc-1 and the
gemcitabine-sensitive cell line ASPC-1 were used in the present
study.
In the current study, it was found that arenobufagin
effectively suppressed the proliferation of PC cells by blocking
the phosphorylation of both Akt and extracellular signal-regulated
kinases (Erk), as well as inducing G2/M phase cell cycle arrest and
apoptosis in PC cells. In order to find a new strategy for
combination therapy, the present study determined the effect of
arenobufagin in combination with gemcitabine or 5-fluorouracil
(5-FU), revealing a significant impact on cell proliferation. These
results indicate that arenobufagin may be used as a potential
adjuvant to overcome the resistance to chemotherapy in PC.
Materials and methods
Materials
Arenobufagin was isolated from toad secretions, as
previously described (23).
Gemcitabine and 5-FU were purchased from Sigma-Aldrich (Merck KGaA,
Darmstadt, Germany). All 3 agents were dissolved in dimethyl
sulfoxide (DMSO) as a stock solution (10 mM) and stored at −20°C.
The culture media containing different concentrations of these
agents were freshly prepared for each experiment. The final
concentration of DMSO was <0.1%. Propidium iodide (PI) for cell
cycle analysis was purchased from Sigma-Aldrich (Merck KGaA).
Rabbit monoclonal antibodies against Akt (#4691), phosphorylated
AktSer473 (p-Akt) (#4060), Erk1/2 (#4695),
phosphorylated Erk1/2 (p-Erk; #4370), EGFR (#4267), phosphorylated
epidermal growth factor receptorTyr1068 (p-EGFR; #3777),
caspase-3 (#9665), poly (ADP-ribose) polymerase (PARP; #9542),
cleaved PARP (Asp214; #5625), caspase-9 (#9508) and cleaved
caspase-9 (Asp330; #7237), mouse monoclonal GAPDH (#97166) and
β-actin (#3700), and anti-mouse (#7076) and anti-rabbit (#7074)
IgG-horseradish peroxidase (HRP)-linked antibodies were purchased
from Cell Signaling Technology, Inc. (Danvers, MA, USA).
Cell lines and cell culture
The human PC Panc-1 and ASPC-1 cell lines were
obtained from the American Type Culture Collection (Manassas, VA,
USA). These two cell lines were incubated in Dulbecco's modified
Eagle's medium (DMEM; Gibco, Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS)
and 1% (v/v) penicillin-streptomycin at 37°C under 5%
CO2.
MTT assay
The viability of the Panc-1 and ASPC-1 cells was
detected using MTT assay. Cells were planted on 96-well plates with
3 replicates at a density of 5×103 cells per well. After
culturing in DMEM medium containing 10% FBS for 12 h to obtain a
confluent monolayer, the medium was replaced with DMEM containing
5% FBS and arenobufagin at different concentrations (0, 1, 10 and
100 nM) for 24, 48 and 96 h. Subsequently, 10 µl MTT (5 mg
ml−1 in PBS) was added to each well at the indicated
time points. The culture medium was removed and MTT formazan was
dissolved in 150 µl DMSO per well 4 h later. The plates were
agitated for 15 min, and OD490nm was measured using an
absorbance reader.
Clonogenic survival assay
Cell proliferation ability was measured using a
colony formation assay. Approximately 500 cells were seeded in each
well on a 12-well plate as a single cell suspension. After 24 h,
different concentrations of arenobufagin (0, 5, 10 and 50 nM) were
added and the cells continued to be maintained at 37°C with a
humidified atmosphere of 5% CO2 for 15–20 days. Visible
colonies were then stained with crystal violet and manually
counted.
Western blot analysis
Cells were treated with arenobufagin (0, 5 and 10
nM) at the indicated time (0, 0.5 and 1 h). Whole-cell extracts
were prepared using radioimmunoprecipitation assay buffer
supplemented with proteinase inhibitors (Sigma-Aldrich, Merck KGaA)
at 4°C. Following centrifugation at 13,000 × g for 20 min, the
supernatant was collected and quantified using the bicinchoninic
acid protein assay. Total protein (50 µg per well) was separated
using 8–12% SDS-PAGE and transferred to nitrocellulose transfer
membranes. The membranes were blocked in TBST (10 mM Tris-HCl, pH
7.4; 150 mM NaCl; 0.1% Tween-20) with 5% non-fat milk for 2 h. The
membranes were then incubated with specific primary antibodies (all
antibodies were diluted at a ratio of 1:1,000) overnight at 4°C,
followed by treatment with HRP-linked secondary antibodies
(24). The protein bands were
visualized using ECL agent (Thermo Fisher Scientific, Inc.) and
detected using a Gel imaging system (Bio-Rad Laboratories, Inc.,
Hercules, California, USA).
Detection of cell cycle
Cells were seeded on a 6-well plate and treated with
100 nM arenobufagin for 24 h. The cells were then harvested by
trypsin (up to 5×106 cells), washed twice with PBS, and
fixed with cold 70% ethanol at 4°C overnight. The cells were
stained with 50 µg/ml PI [PBS 480 µl; 5 µl PI (5 mg/ml); 5 µl RNase
(10 mg/ml); 10 µl Triton-100 (10%)] at 37°C for 30 min and were
kept in the dark. The cells were then suspended in PBS and cell
cycle distribution was detected. Samples were analyzed using a
FACSC flow cytometer. Additional analyses were processed by Flow Jo
software (Tree Star, Inc., Ashland, OR, USA) (24).
Statistical analysis
The data were analyzed by Student's t-test or
one-way analysis of variance (Dunnett's test and Least Significant
Difference test). P<0.05 was considered to indicate a
statistically significant difference. All analysis was performed
using SPSS13.0 (SPSS, Inc., Chicago, IL, USA).
Results
Arenobufagin inhibited survival and
proliferation of PC cells
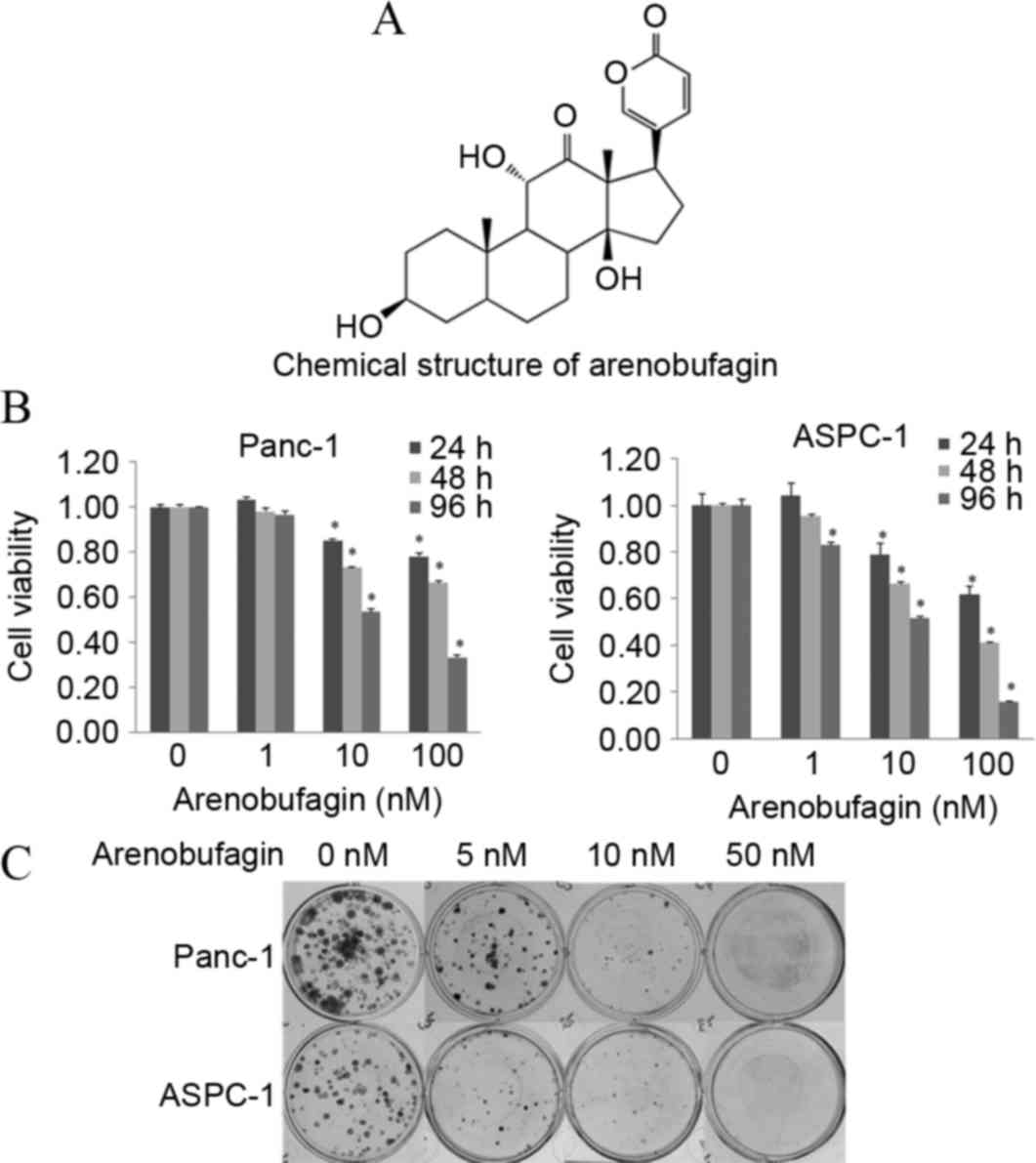
The chemical structure of arenobufagin is shown in
Fig. 1A. The cytotoxic efficacy of
arenobufagin was first tested on the PC Panc-1 and ASPC-1 cell
lines by MTT analysis. Subsequent to treatment with different
concentrations (0–100 nM) of arenobufagin for 24, 48 and 96 h, cell
proliferation was significantly suppressed in a dose- and
time-dependent manner, as shown in Fig.
1B. To confirm the inhibition of cellular proliferation, the
present study processed the colony formation assay (Fig. 1C). The MTT assay and the colony
formation assay showed that cellular viability and proliferation
could be inhibited by arenobufagin even at a concentration as low
as 1 nM.
Arenobufagin suppressed the
phosphorylation of Akt, Erk and EGFR
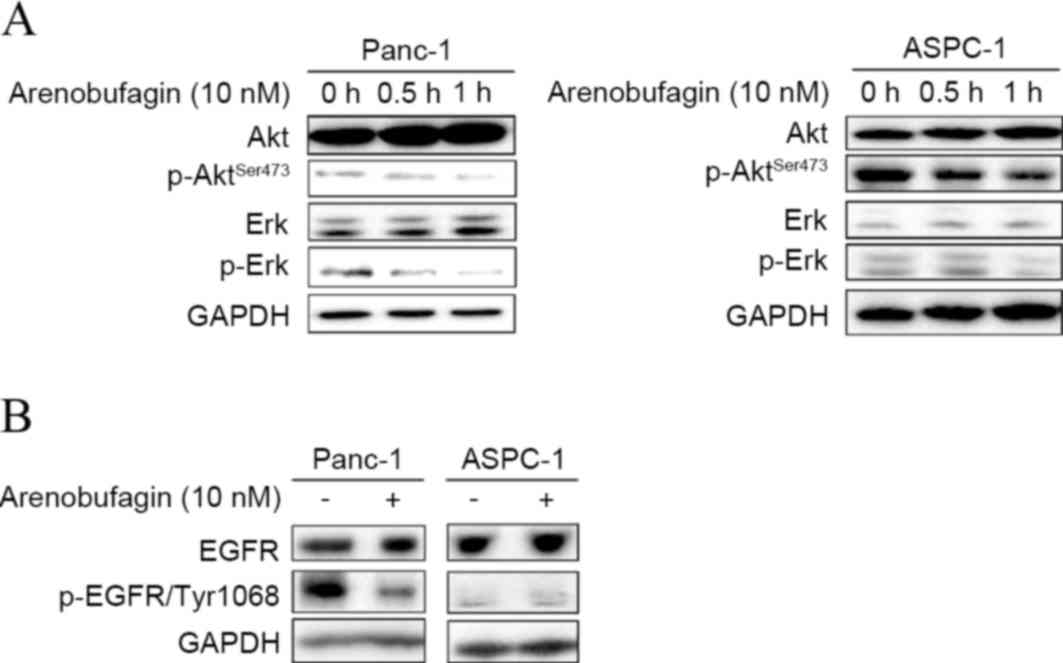
The present study investigated whether arenobufagin
could inhibit key downstream pathways of cell survival and
proliferation, and whether they play a role in
arenobufagin-mediated cell death. As shown in Fig. 2A, western blot analysis showed that
the phosphorylation of Akt and Erk decreased in ASPC-1 and Panc-1
cell lines within 1 h when treated with arenobufagin at 10 nM. To
uncover the mechanism of arenobufagin on PC cells, the present
study examined the state of EGFR. Subsequent to treatment with
arenobufagin at 10 nM for 0 and 0.5 h, the levels of p-EGFR
decreased in the Panc-1 cell line. There was no evident change in
the gemcitabine-sensitive ASPC-1 cells, but it should be noted that
the initial level of p-EGFR was low in the ASPC-1 cell line, as
shown in Fig. 2B.
Arenobufagin induced cell cycle G2/M
phase arrest and G0/S phase decline
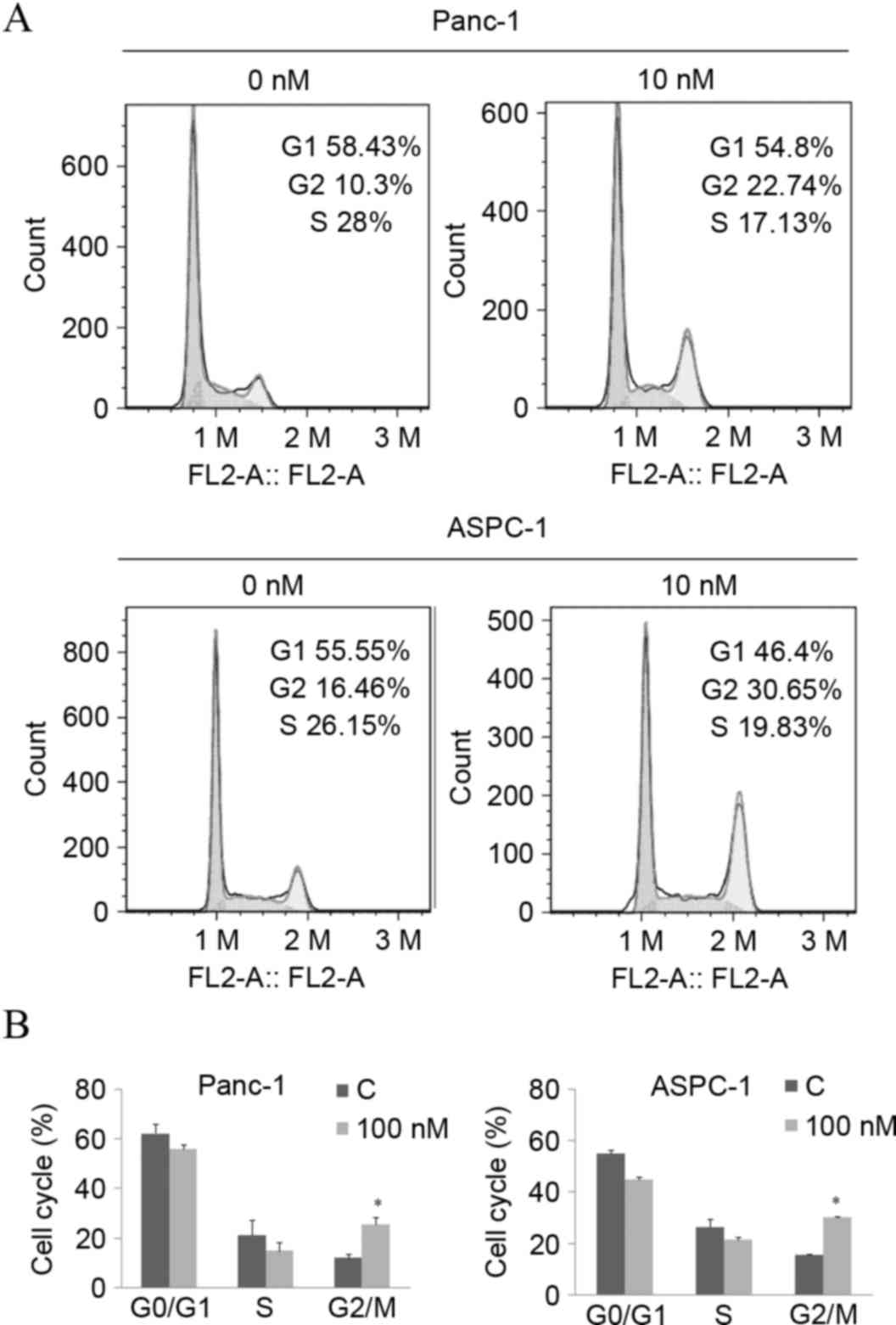
The inhibition of cellular proliferation is usually
caused by cell cycle arrest and cell death. Given the significant
decrease in cell viability, a dose of 10 nM of arenobufagin was
used to explore the underlying mechanism of arenobufagin-induced
inhibition on cellular proliferation. Subsequent to treatment with
the indicated concentration for 24 h, the percentage of cells in
the G2/M phase increased from 10.3–22.74% in Panc-1 cells and
16.46–30.65% in ASPC-1 cells, along with a decrease in the S phase,
as shown in Fig. 3A and B. The
statistical analysis also showed that the increase in the G2/M
phase percentage was significant (P<0.05).
Arenobufagin affected the
apoptosis-related pathway
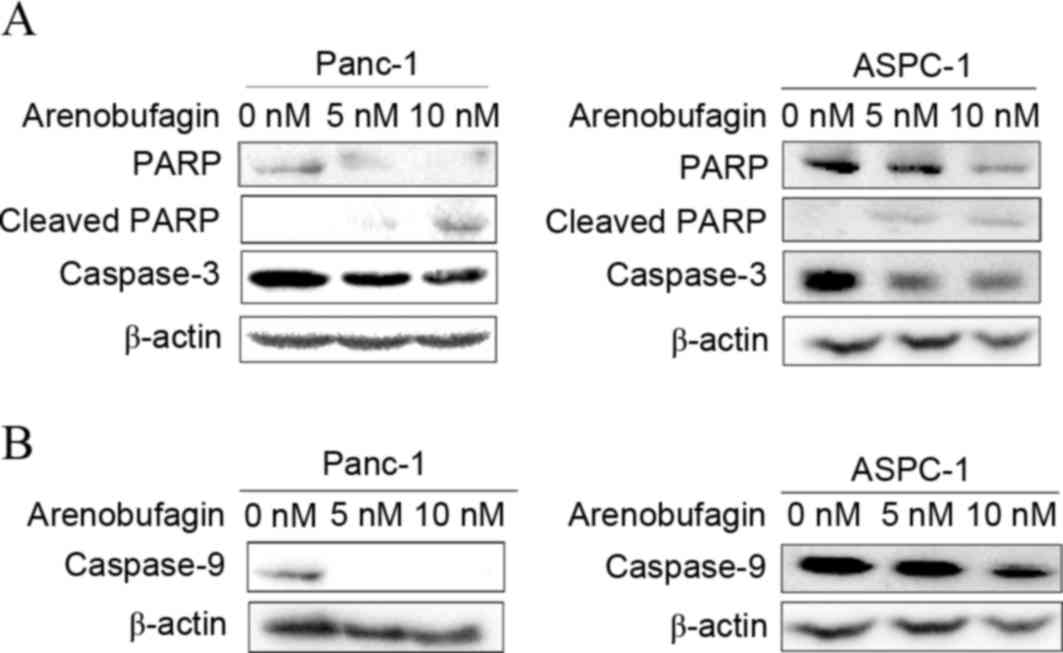
In addition to cell cycle arrest, the effect of
arenobufagin on the apoptosis pathway in PC cells was examined.
Western blot analysis revealed not only specific cleavage of PARP
but also a decrease in PARP, pro-caspase-3 and pro-caspase-9 was
induced by arenobufagin treatment (Fig.
4A). The decrease in caspase-9 (Fig.
4B) indicated that the mechanism of apoptosis may be associated
with a mitochondrial-dependent pathway.
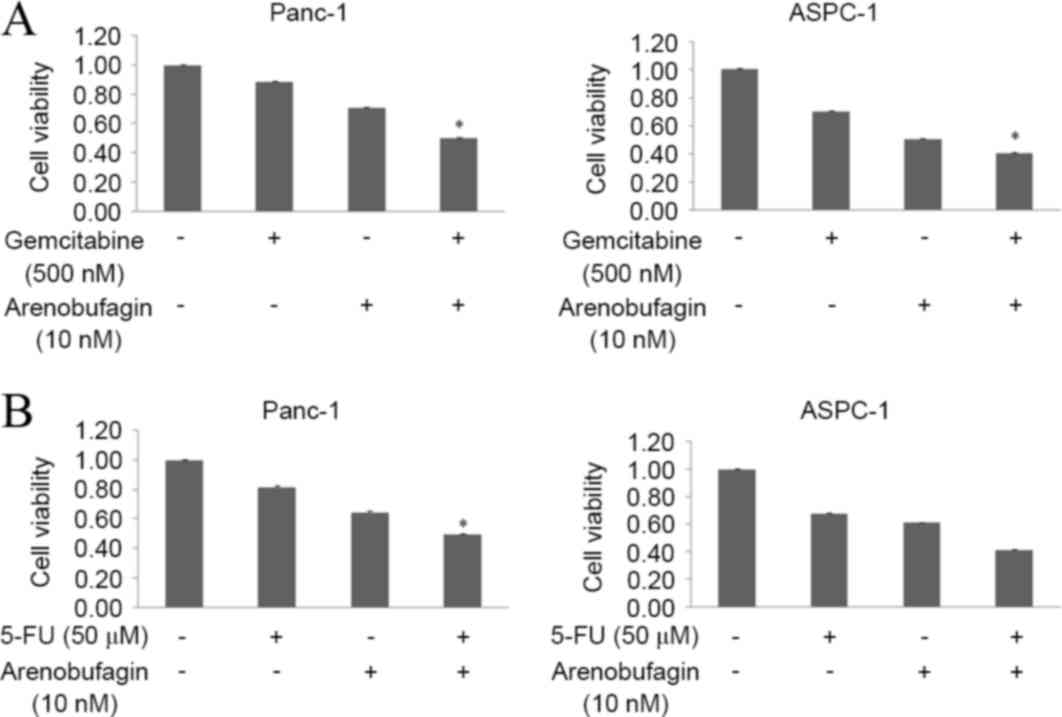
Arenobufagin significantly enhanced
the effects of both gemcitabine and 5-FU
The present study assessed the effect of
arenobufagin on the inhibition of proliferation induced by
gemcitabine in the gemcitabine-resistantPanc-1 and
gemcitabine-sensitive ASPC-1cell lines. The two cell lines were
treated with arenobufagin and gemcitabine at the indicated
concentrations, either alone or in combination for 48 h. The
combination of arenobufagin and gemcitabine inhibited the growth of
Panc-1 and ASPC-1 cells more than either of the agentsalone, with
the cell viability of Panc-1 cells declining from 89% in the
gemcitabine group and 71% in the arenobufagin group to 50% in the
combination treatment group. In the ASPC-1 cell line, the viability
of cells declined from 71% in the gemcitabine group and 51% in the
arenobufagin group to 41% in the combination treatment group. The
decline between the single treatment groups and the combination
groups was statistically significant (P<0.05; Fig. 5A). In addition, the present study
identified that the combination treatment of arenobufagin at 10 nM
and 5-FU at 50 µM for 48 h significantly enhanced (P<0.05) the
effect of 5-FU, with the cell viability of Panc-1 cells decreasing
between 82% in the 5-FU group and 50% in the combination group. The
viability of ASPC-1 cells decreased between 68% in the 5-FU group
to 42% in the combination treatment group (Fig. 5B).
Discussion
Arenobufagin, an effective cardiac glycoside, is one
of the most active compounds found in toad secretions, and is
listed in the Chinese Pharmacopoeia (20). Arenobufagin has been used to treat
hepatic carcinoma in TCM. Certain cardiac glycosides have been
reported as potent inhibitors of cancer cell growth (17). To the best of our knowledge, the
present study demonstrated for the first time that arenobufagin is
an effective agent in PC cell lines, including the drug-resistant
Panc-1cell line.
K-Ras has previously been identified as a small
GTPase that is mutated in 90% of human pancreatic carcinomas
(25). Certain genetic studies have
shown that K-Ras activation and mutation is necessary for the
initiation of PC (26–28), and an inducible pancreas-specific
expression system was used to show that K-Ras expression is also
required for tumor maintenance (29).
These mutations lock K-Ras and its downstream proteins, such as Akt
and Erk, in a constitutively activated form. In turn, this leads to
enhanced cell proliferation and a growth advantage to the cancer
cell, as well as playing a key role in tumorigenesis and resistance
to standard therapies, such as chemotherapy and radiation (30,31). Thus,
the associated signaling pathways are critical targets for which
specific inhibitors are expected to exert antitumor efficacy. It is
notable that arenobufagin could evidently downregulate the
phosphorylation of Erk and Akt in the K-Ras mutant Panc-1 cell
line, suggesting that arenobufagin may be an efficient inhibitor in
Ras mutation cancer cell lines. In addition, the present study
showed that this downregulation was more evident on the level of
p-Erk compared with p-Akt when treated with arenobufagin, which is
consistent with previous studies (32).
A new study recently showed that EGFR signaling is
essential for K-Ras oncogene-driven PDAC (33). EGFR belongs to the tyrosine kinase
receptor erbB family, and is important in tumor growth, metastasis
and disease recurrence (34,35). Overexpression of EGFR commonly occurs
in PC, and this overexpression is associated with a poor outcome
(36–38). Although EGFR inhibitors have exerted
significant clinical benefits, clinical research has shown that
only patients who developed a rapid response upon erlotinib
treatment benefit from EGFR inhibitors (13), which suggests a limited efficacy of
EGFR inhibition and the requirement for agents targeting multiple
signaling pathways. Considering the inhibitory effects of
arenobufagin on the phosphorylation of Erk and Akt in K-Ras mutant
Panc-1 cell lines and the cross-talk between EGFR and K-Ras, it was
hypothesized that a possible target by which arenobufagin may
affect cell growth and death was EGFR. The present study detected
significant decreases of phosphorylated EGFR subsequent to
treatment with arenobufagin in PC cells, thus confirming that
arenobufagin is an efficient agent for the treatment of Ras mutated
PC.
Previously, it has been reported that arenobufagin
induces apoptosis in hepatocellular carcinoma cells. Similar to the
present findings for arenobufagin, certain studies have reported
that bufalin, another representative cardiac glycoside compound
from secreted toad toxins, inhibited cell proliferation in various
cancer cells and induced apoptosis and cell cycle arrest in PC
cells (21,39–41). The
exact mechanism by which arenobufagin induces apoptosis in
pancreatic cells is unclear. The present finding on the reduction
of caspase-9 indicated that arenobufagin may induce apoptosis via a
mitochondrial pathway in PC cells. In addition, to the best of our
knowledge, the present data showed for the first time that
arenobufagin induced cell cycle G2/M phase arrest and a decrease in
the population of cells in the S phase, inhibiting the
proliferation of PC cells.
Several studies have indicated that chemotherapeutic
and biological targeted agents could be used in combination with
clinical chemotherapeutic drugs, such as gemcitabine and 5-FU to
overcome drug resistance and improve the efficiency of treatments
(42,43). The classic model for PDAC treatment is
treatment with gemcitabine as a single agent or in combination with
EGFR inhibitors. Since it has been reported that the levels of EGFR
and Erk phosphorylation in PDAC were increased in Panc-1 and BXPC-1
cell lines when treated with gemcitabine, this may be a mechanism
of drug resistance (15). The
phosphorylation of Erk is associated with cell proliferation,
differentiation, migration and transcription. Notably, the present
western blot analysis showed that arenobufagin could significantly
downregulate the phosphorylation of EGFR, Erk and Akt. Cell growth
analysis testified to the effect of arenobufagin in combination
with gemcitabine or 5-FU, and the results demonstrated that the
involvement of arenobufagin could significantly enhance the
inhibiting effect of either gemcitabine or 5-FU against PC cells.
Thus, the present results indicated that arenobufagin could be used
as a new drug to enhance the effect of gemcitabine through
inhibiting EGFR, Erk, Akt and NF-κB associated pathways.
In summary, the present study determined that
arenobufagin could cause cell cycle G2/M phase arrest and apoptosis
in PC cells, and downregulate the levels of p-Erk, p-Akt and p-EGFR
even in drug-resistant Panc-1 cell line, which could in turn offset
the adverse effects of gemcitabine. Thus, the present study
identified arenobufagin as a potential effective agent with a
marked effect on pancreatic cancer cells, and therefore causing us
to consider arenobufagin as a promising candidate for combination
therapy in PC.
Acknowledgements
This study was supported by funds from the Liaoning
Province Natural Science Foundation of China (grant no. 2013023043)
and National Natural Science Foundation of China (grant no.
510575).
Glossary
Abbreviations
Abbreviations:
|
5-FU
|
5-fluorouracil
|
|
EGFR
|
epidermal growth factor receptor
|
|
PC
|
pancreatic carcinoma
|
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
PI
|
propidium iodide
|
|
PI3K
|
phosphatidylinositol 3-kinase
|
|
TCM
|
traditional Chinese medicine
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hidalgo M: Pancreatic cancer. N Engl J
Med. 362:1605–1617. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vincent A, Herman J, Schulick R, Hruban RH
and Goggins M: Pancreatic cancer. Lancet. 378:607–620. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Burris HA III, Moore MJ, Andersen J, Green
MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo
AM, Tarassoff P, et al: Improvements in survival and clinical
benefit with gemcitabine as first-line therapy for patients with
advanced pancreas cancer: A randomized trial. J Clin Oncol.
15:2403–2413. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Berlin J and Benson AB III: Chemotherapy:
Gemcitabine remains the standard of care for pancreatic cancer. Nat
Rev Clin Oncol. 7:135–137. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ducreux M, Boige V and Malka D: Treatment
of advanced pancreatic cancer. Semin Oncol. 34 (2 Suppl 1):S25–S30.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ying JE, Zhu LM and Liu BX: Developments
in metastatic pancreatic cancer: Is gemcitabine still the standard?
World J Gastroenterol. 18:736–745. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davidson JD, Ma L, Flagella M, Geeganage
S, Gelbert LM and Slapak CA: An increase in the expression of
ribonucleotide reductase large subunit 1 is associated with
gemcitabine resistance in non-small cell lung cancer cell lines.
Cancer Res. 64:3761–3766. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Itoi T, Sofuni A, Fukushima N, Itokawa F,
Tsuchiya T, Kurihara T, Moriyasu F, Tsuchida A and Kasuya K:
Ribonucleotide reductase subunit M2 mRNA expression in pretreatment
biopsies obtained from unresectable pancreatic carcinomas. J
Gastroenterol. 42:389–394. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakano Y, Tanno S, Koizumi K, Nishikawa T,
Nakamura K, Minoguchi M, Izawa T, Mizukami Y, Okumura T and Kohgo
Y: Gemcitabine chemoresistance and molecular markers associated
with gemcitabine transport and metabolism in human pancreatic
cancer cells. Br J Cancer. 96:457–463. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hagmann W, Jesnowski R and Löhr JM:
Interdependence of gemcitabine treatment, transporter expression,
and resistance in human pancreatic carcinoma cells. Neoplasia.
12:740–747. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ng SS, Tsao MS, Nicklee T and Hedley DW:
Wortmannin inhibits pkb/akt phosphorylation and promotes
gemcitabine antitumor activity in orthotopic human pancreatic
cancer xenografts in immunodeficient mice. Clin Cancer Res.
7:3269–3275. 2001.PubMed/NCBI
|
|
13
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fountzilas G, Bobos M, Kalogera-Fountzila
A, Xiros N, Murray S, Linardou H, Karayannopoulou G, Koutras AK,
Bafaloukos D, Samantas E, et al: Gemcitabine combined with
gefitinib in patients with inoperable or metastatic pancreatic
cancer: A phase II Study of the Hellenic Cooperative Oncology Group
with biomarker evaluation. Cancer Invest. 26:784–793. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Venkatasubbarao K, Peterson L, Zhao S,
Hill P, Cao L, Zhou Q, Nawrocki ST and Freeman JW: Inhibiting
signal transducer and activator of transcription-3 increases
response to gemcitabine and delays progression of pancreatic
cancer. Mol Cancer. 12:1042013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liang Y, Liu AH, Qin S, Sun JH, Yang M, Li
P and Guo DA: Simultaneous determination and pharmacokinetics of
five bufadienolides in rat plasma after oral administration of
Chansu extract by SPE-HPLC method. J Pharm Biomed Anal. 46:442–448.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang Y, Lonard DM, Yu Y, Chow DC, Palzkill
TG, Wang J, Qi R, Matzuk AJ, Song X, Madoux F, et al: Bufalin is a
potent small-molecule inhibitor of the steroid receptor
coactivators SRC-3 and SRC-1. Cancer Res. 74:1506–1517. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cruz Jdos S and Matsuda H: Arenobufagin, a
compound in toad venom, blocks Na(+)-K+ pump current in cardiac
myocytes. Eur J Pharmacol. 239:223–226. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Cruz Jdos S and Matsuda H: Depressive
effects of arenobufagin on the delayed rectifier K+ current of
guinea-pig cardiac myocytes. Eur J Pharmacol. 266:317–325. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cao HH, Zhang DM, Liu JS, Hou CY, Kurihara
H and Ye WC: Inhibitory effect of arenobufagin on the adhesion,
invasion and migration of human hepatoma carcinoma cells. Chinese
Pharmacol Bull. 27:19–23. 2011.
|
|
21
|
Zhang DM, Liu JS, Deng LJ, Chen MF, Yiu A,
Cao HH, Tian HY, Fung KP, Kurihara H, Pan JX and Ye WC:
Arenobufagin, a natural bufadienolide from toad venom, induces
apoptosis and autophagy in human hepatocellular carcinoma cells
through inhibition of PI3K/Akt/mTOR pathway. Carcinogenesis.
34:1331–1342. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li M, Wu S, Liu Z, Zhang W, Xu J, Wang Y,
Liu J, Zhang D, Tian H, Li Y and Ye W: Arenobufagin, a
bufadienolide compound from toad venom, inhibits VEGF-mediated
angiogenesis through suppression of VEGFR-2 signaling pathway.
Biochem Pharmacol. 83:1251–1260. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li J, Ma X, Li F, Wang J, Chen H, Wang G,
Lv X, Sun C and Jia J: Preparative separation and purification of
bufadienolides from Chinese traditional medicine of ChanSu using
high-speed counter-current chromatography. J Sep Sci. 33:1325–1330.
2010.PubMed/NCBI
|
|
24
|
Yuan Y, Qin L, Liu D, Wu RC, Mussi P, Zhou
S, Songyang Z and Xu J: Genetic Screening Reveals an Essential Role
of p27kip1 in Restriction of Breast Cancer Progression. Cancer Res.
67:8032–8042. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Thomas RK, Baker AC, Debiasi RM, Winckler
W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L,
et al: High-throughput oncogene mutation profiling in human cancer.
Nat Genet. 39:347–351. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aguirre AJ, Bardeesy N, Sinha M, Lopez L,
Tuveson DA, Horner J, Redston MS and DePinho RA: Activated Kras and
Ink4a/Arf deficiency cooperate to produce metastatic pancreatic
ductal adenocarcinoma. Genes Dev. 17:3112–3126. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hingorani SR, Petricoin EF, Maitra A,
Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD,
Hitt BA, et al: Preinvasive and invasive ductal pancreatic cancer
and its early detection in the mouse. Cancer Cell. 4:437–450. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hingorani SR, Wang L, Multani AS, Combs C,
Deramaudt TB, Hruban RH, Rustgi AK, Chang S and Tuveson DA:
Trp53R172H and KrasG12D cooperate to promote chromosomal
instability and widely metastatic pancreatic ductal adenocarcinoma
in mice. Cancer Cell. 7:469–483. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Collins MA, Bednar F, Zhang Y, Brisset JC,
Galbán S, Galbán CJ, Rakshit S, Flannagan KS, Adsay NV and di
Magliano Pasca M: Oncogenic Kras is required for both the
initiation and maintenance of pancreatic cancer in mice. J Clin
Invest. 122:639–653. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shaw RJ and Cantley LC: Ras, PI(3)K and
mTOR signalling controls tumour cell growth. Nature. 441:424–430.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sweet RW, Yokoyama S, Kamata T, Feramisco
JR, Rosenberg M and Gross M: The product of ras is a GTPase and the
T24 oncogenic mutant is deficient in this activity. Nature.
311:273–275. 1984. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hofmann I, Weiss A, Elain G, Schwaederle
M, Sterker D, Romanet V, Schmelzle T, Lai A, Brachmann SM,
Bentires-Alj M, et al: K-RAS mutant pancreatic tumors show higher
sensitivity to MEK than to PI3K inhibition in vivo. PLoS One.
7:e441462012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Navas C, Hernandez-Porras I, Schuhmacher
AJ, Sibilia M, Guerra C and Barbacid M: EGF receptor signaling is
essential for k-ras oncogene-driven pancreatic ductal
adenocarcinoma. Cancer Cell. 22:318–330. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pryczynicz A, Guzińska-Ustymowicz K,
Czyzewska J and Kemona A: Expression of epidermal growth factors
and apoptosis markers in pancreatic ductal adenocarcinoma. Folia
Histochem Cytobiol. 47:667–671. 2009.PubMed/NCBI
|
|
35
|
Weiss GA, Rossi MR, Khushalani NI, Lo K,
Gibbs JF, Bharthuar A, Cowell JK and Iyer R: Evaluation of
phosphatidylinositol-3-kinase catalytic subunit (PIK3CA) and
epidermal growth factor receptor (EGFR) gene mutations in
pancreaticobiliary adenocarcinoma. J Gastrointest Oncol. 4:20–29.
2013.PubMed/NCBI
|
|
36
|
Barr S, Thomson S, Buck E, Russo S, Petti
F, Sujka-Kwok I, Eyzaguirre A, Rosenfeld-Franklin M, Gibson NW,
Miglarese M, et al: Bypassing cellular EGF receptor dependence
through epithelial-to-mesenchymal-like transitions. Clin Exp
Metastasis. 25:685–693. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Miyabayashi K, Ijichi H, Mohri D, Tada M,
Yamamoto K, Asaoka Y, Ikenoue T, Tateishi K, Nakai Y, Isayama H, et
al: Erlotinib prolongs survival in pancreatic cancer by blocking
gemcitabine-induced MAPK signals. Cancer Res. 73:2221–2234. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Walters DM, Lindberg JM, Adair SJ, Newhook
TE, Cowan CR, Stokes JB, Borgman CA, Stelow EB, Lowrey BT,
Chopivsky ME, et al: Inhibition of the growth of patient-derived
pancreatic cancer xenografts with the MEK inhibitor trametinib is
augmented by combined treatment with the epidermal growth factor
receptor/HER2 inhibitor lapatinib. Neoplasia. 15:143–155. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang L, Nakaya K, Yoshida T and Kuroiwa
Y: Induction by bufalin of differentiation of human leukemia cells
HL60, U937, and ML1 toward macrophage/monocyte-like cells and its
potent synergistic effect on the differentiation of human leukemia
cells in combination with other inducers. Cancer Res. 52:4634–4641.
1992.PubMed/NCBI
|
|
40
|
Masuda Y, Kawazoe N, Nakajo S, Yoshida T,
Kuroiwa Y and Nakaya K: Bufalin induces apoptosis and influences
the expression of apoptosis-related genes in human leukemia cells.
Leuk Res. 19:549–556. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yu CH, Kan SF, Pu HF, Chien Jea E and Wang
PS: Apoptotic signaling in bufalin- and cinobufagin-treated
androgen-dependent and -independent human prostate cancer cells.
Cancer Sci. 99:2467–2476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zalatnai A and Molnár J: Review. Molecular
background of chemoresistance in pancreatic cancer. In Vivo.
21:339–347. 2007.PubMed/NCBI
|
|
43
|
Mimeault M, Hauke R and Batra SK: Recent
advances on the molecular mechanisms involved in the drug
resistance of cancer cells and novel targeting therapies. Clin
Pharmacol Ther. 83:673–691. 2008. View Article : Google Scholar : PubMed/NCBI
|