Introduction
Hepatocellular carcinoma (HCC), a highly lethal
malignancy, has an increasing worldwide incidence and is the third
most frequent cause of cancer-associated mortality (1,2). It is
estimated that >700,000 new cases are diagnosed each year
(3). Infection with hepatitis B or C
viruses, alcohol-associated cirrhosis and non-alcoholic
steatohepatitis have been described as risk factors (4,5). In
addition, the mortality rate almost equals the incidence rate in
the majority of countries (4,5). Therefore, more research is required to
identify novel effective treatments for HCC and to elucidate the
underlying molecular mechanisms of HCC progression.
Extensive studies have been previously conducted and
advances have been made in the identification of genes and pathways
associated with HCC. Yong et al (6) demonstrated that spalt-like transcription
factor 4 is associated with poor prognosis and may serve as a
potential target for the therapy of HCC. Downregulation of
microRNA-195 (miR-195) and miR-497 may affect molecular pathways
associated with cell cycle progression, leading to the abnormal
cellular proliferation observed in hepatocarcinogenesis (7). Cillo et al (8) demonstrated that the transcriptional and
post-transcriptional deregulation of homeobox A13 may be involved
in HCC through mRNA nuclear export of eukaryotic translation
initiation factor 4E-dependent transcripts. Revill et al
(9), using integrative genomic
analysis, revealed that sphingomyelin phosphodiesterase 3 and
neurofilament heavy polypeptide are tumor suppressor genes in HCC.
Yoshikawa et al (10) reported
that suppressor of cytokine signaling 1 and Janus kinase 2 may
serve as potential therapeutic targets for the treatment of HCC. In
addition, inhibition of the rapidly accelerated
fibrosarcoma/mitogen-activated protein kinase (MAPK)/extracellular
signal-regulated kinase pathway inhibited tumor angiogenesis and
induced apoptosis in a HCC model (11). Xu et al (12) demonstrated that miR-122 induces
apoptosis and inhibits cellular proliferation in HCC by directly
targeting the Wnt/β-catenin signaling pathway. Furthermore,
ubiquitin D, an oncogene located at 6q21.3, promotes hepatitis B
virus-associated HCC progression via the protein kinase B/glycogen
synthase kinase 3β, signaling pathway (13). The tyrosine-protein kinase Met pathway
is also involved in the pathogenesis of HCC (14). Additional studies indicated that a
number of other pathways, including the hedgehog and the ρ GTPase
signaling pathways, are associated with the progression of HCC
(15,16). Even though a number of genes and
pathways have been associated with HCC, the underlying molecular
mechanisms of HCC progression have not been completely identified
yet. Therefore, further research is required.
In the present study, the expression profile of the
GSE49515 dataset was obtained and analyzed, and the differentially
expressed genes (DEGs) between HCC and healthy samples were
identified. Furthermore, functional enrichment analysis and
protein-protein interaction (PPI) network analysis were performed.
In contrast to the previous studies of Shi et al (17) and Jiang et al (18), which used the same dataset to identify
genes and pathways associated with HCC, the present study also
investigated gene and drug interactions using the comparative
toxicogenomics database (CTD). The aim of the present study was to
identify key genes and pathways associated with HCC progression and
investigate potential compounds leading to HCC carcinogenesis.
Materials and methods
Expression profile data
The GSE49515 dataset originally derived from the
study by Shi et al (17) was
obtained from the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo/). The dataset contains
the expression profile of 26 samples of peripheral blood
mononuclear cells, including 10 HCC samples, 3 pancreatic carcinoma
samples, 3 gastric carcinoma samples and 10 healthy samples. In the
present study, the 10 HCC and 10 healthy samples were used to
analyze the mRNA expression profile of HCC. These data were
analyzed using the GPL570 Affymetrix Human Genome U133 Plus 2.0
Array platform (Affymetrix; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA).
Data preprocessing
The mRNA expression profile data were preprocessed
using the Robust Multi-Array Average algorithm in the Affy software
package (19), and subsequently, the
expression matrix was generated. If several probes mapped to one
gene symbol, then the mean value was set as the final expression
value of this gene. A total of 20,108 genes were obtained.
DEG analysis
The limma software package (20) within Bioconductor was used to identify
the DEGs in HCC samples compared with healthy samples. The P-values
of DEGs were calculated using Student's t-test (21) in the limma software package and
adjusted according to the Benjamini-Hochberg (BH) procedure
(22).
|log2(fold-change)|≥1 and BH_P<0.05 were used as
threshold criteria.
Functional enrichment analysis
The Biological Networks Gene Ontology tool (BiNGO)
(23) is a Cytoscape plugin used to
assess the overrepresentation of Gene Ontology (GO) terms in
biological networks. The Kyoto Encyclopedia of Genes and Genomes
(KEGG) database was used to assign related gene categories into
their associated pathways (24). The
Database for Annotation, Visualization and Integrated Discovery
(DAVID), an integrated data-mining environment, was used for
pathway enrichment analysis (25). GO
annotation and KEGG pathway enrichment analysis were performed
using BINGO and DAVID. P<0.05 was used as the threshold
criterion.
PPI network analysis
The Michigan Molecular Interactions database (MiMI)
plugin (26) within the Cytoscape
software platform (v2.7.0) (27) was
used to perform PPI visual network analysis. The MiMI plugin
integrates data from several databases, including the Biomolecular
Interaction Network Database, the Biological General Repository for
Interaction Datasets, the Human Protein Reference Database, the
Molecular Interaction Database, InterPro and SwissProt. The hub
nodes were identified by calculating the degree value using the
degree-sorted tool in Cytoscape. Highest degree hub nodes indicate
key genes with important physiological regulation roles.
Gene and chemical interaction
analysis
CTD (28,29) is a publicly available resource that
provides manually curated information about chemical-gene
interactions and chemical/gene-disease associations derived from
microarray data and published literature. In addition, CTD provides
chemical-gene interaction information for various diseases in
vertebrates and invertebrates (30).
The chemical-gene interaction data for HCC were obtained from the
CTD database to investigate the therapeutic efficacy of several
drugs.
Results
DEG analysis
A total of 302 DEGs, including 231 downregulated and
71 upregulated, were identified in HCC samples compared with
healthy samples.
Functional enrichment analysis
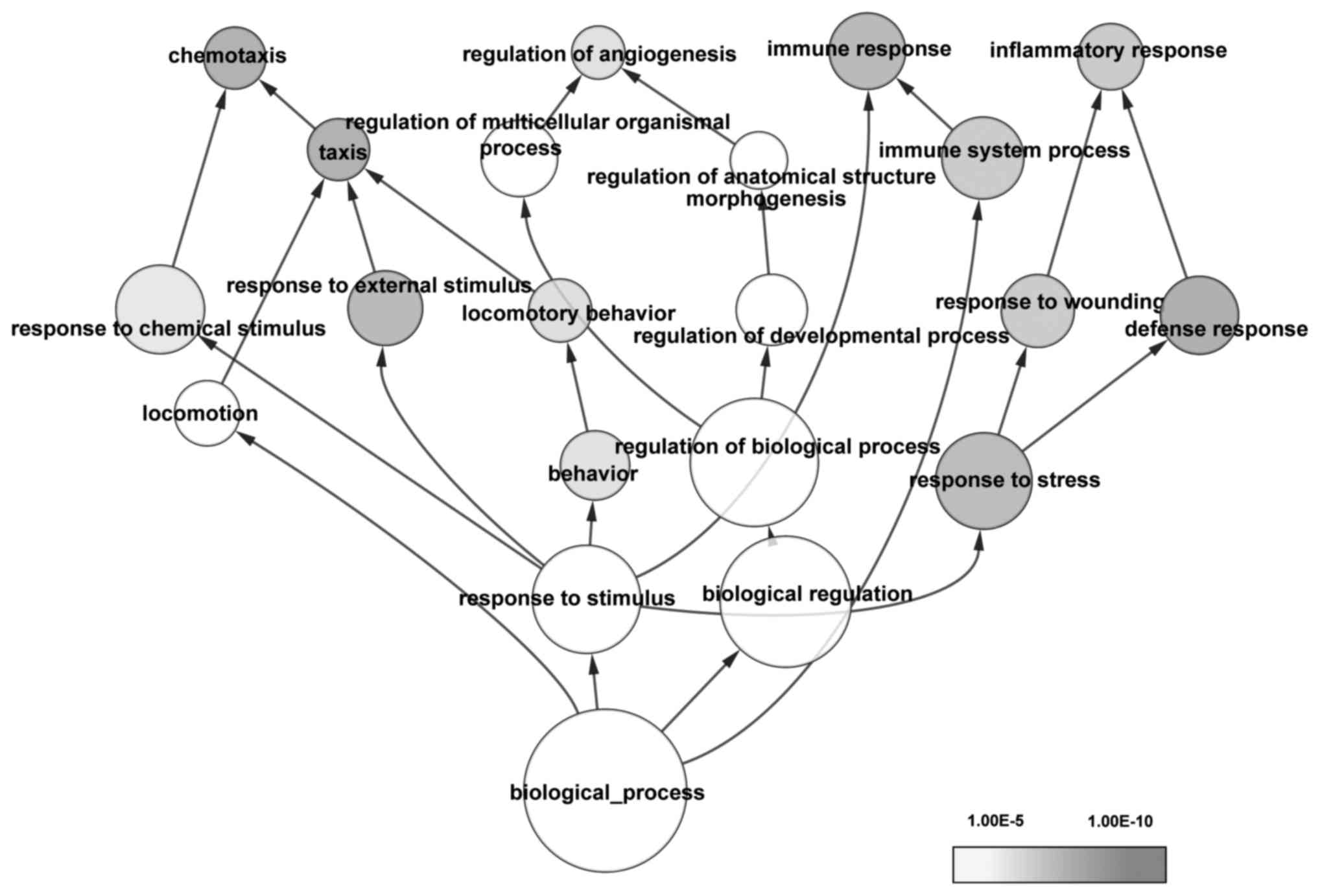
GO and KEGG pathway enrichment analyses were
performed. The overrepresented GO terms (adjusted
P<1.00×10−5) were mainly associated with drug
reactions, immune response and cellular processes associated with
stress (Fig. 1). The most
significantly enriched pathways were cytokine-cytokine receptor
interaction, the chemokine signaling pathway, the Toll-like
receptor signaling pathway and the MAPK signaling pathway (Table I).
 | Table I.Significantly enriched pathways of
differentially expressed genes. |
Table I.
Significantly enriched pathways of
differentially expressed genes.
| KEGG term | Count | P-value | Genes |
|---|
|
hsa04060:Cytokine-cytokine receptor
interaction | 23 |
1.31×10−10 | TNFSF4, CXCL5,
IL8, CCR1, CXCL2, TNFRSF17, FASLG, CXCR2, PF4, PF4V1, CCL4, CXCL10,
OSM, TNFRSF1A, TNFSF10, CCR6, CCR5, VEGFA, CX3CR1, IFNG, CCR2,
IL1B, EGF |
| hsa04062:Chemokine
signaling pathway | 14 |
1.07×10−5 | IL8, CXCL5,
CCR1, CXCL2, PF4, CXCR2, GNG11, PF4V1, CCL4, CXCL10, CCR6, CCR5,
CX3CR1, CCR2 |
| hsa04620:Toll-like
receptor signaling pathway | 10 |
3.96×10−5 | FOS, IL8, JUN,
TLR1, IL1B, TLR4, TLR7, CCL4, TLR8, CXCL10 |
| hsa04621:NOD-like
receptor signaling pathway | 6 |
3.55×10−3 | NLRC4, IL8,
CXCL2, IL1B, RIPK2, CARD6 |
| hsa04010:MAPK
signaling pathway | 11 |
1.29×10−2 | TNFRSF1A, FOS,
PLA2G4A, DUSP2, JUN, IL1B, FASLG, EGF, GADD45B, DDIT3,
DUSP6 |
| hsa05120:Epithelial
cell signaling in Helicobacter pylori infection | 5 |
2.63×10−2 | IL8, JUN, HBEGF,
CXCR2, ATP6V1B2 |
| hsa05219:Bladder
cancer | 4 |
3.24×10−2 | CDKN1A, IL8,
VEGFA, EGF |
PPI network analysis
The PPI network analysis was performed using the
MiMI Cytoscape plugin. A total of 47 nodes, including 20
overexpressed and 27 underexpressed genes, were identified
(Fig. 2). A total of 13 highest
degree proteins, including toll-like receptor 1 (TLR1), TLR4, TLR7,
TLR8, receptor-interacting serine/threonine kinase 2, FBJ murine
osteosarcoma viral oncogene homolog (FOS), FOS-like antigen 2, fas
ligand, chemokine ligand 4, tyrosine 3-monooxygenase/tryptophan
5-monooxygenase activation protein γ, cyclin-dependent kinase
inhibitor 1α, hypoxia inducible factor 1α and DNA-damage-inducible
transcript 3 (DDIT3), were identified as hub nodes and may
potentially be associated with HCC carcinogenesis.
Gene and chemical interaction
analysis
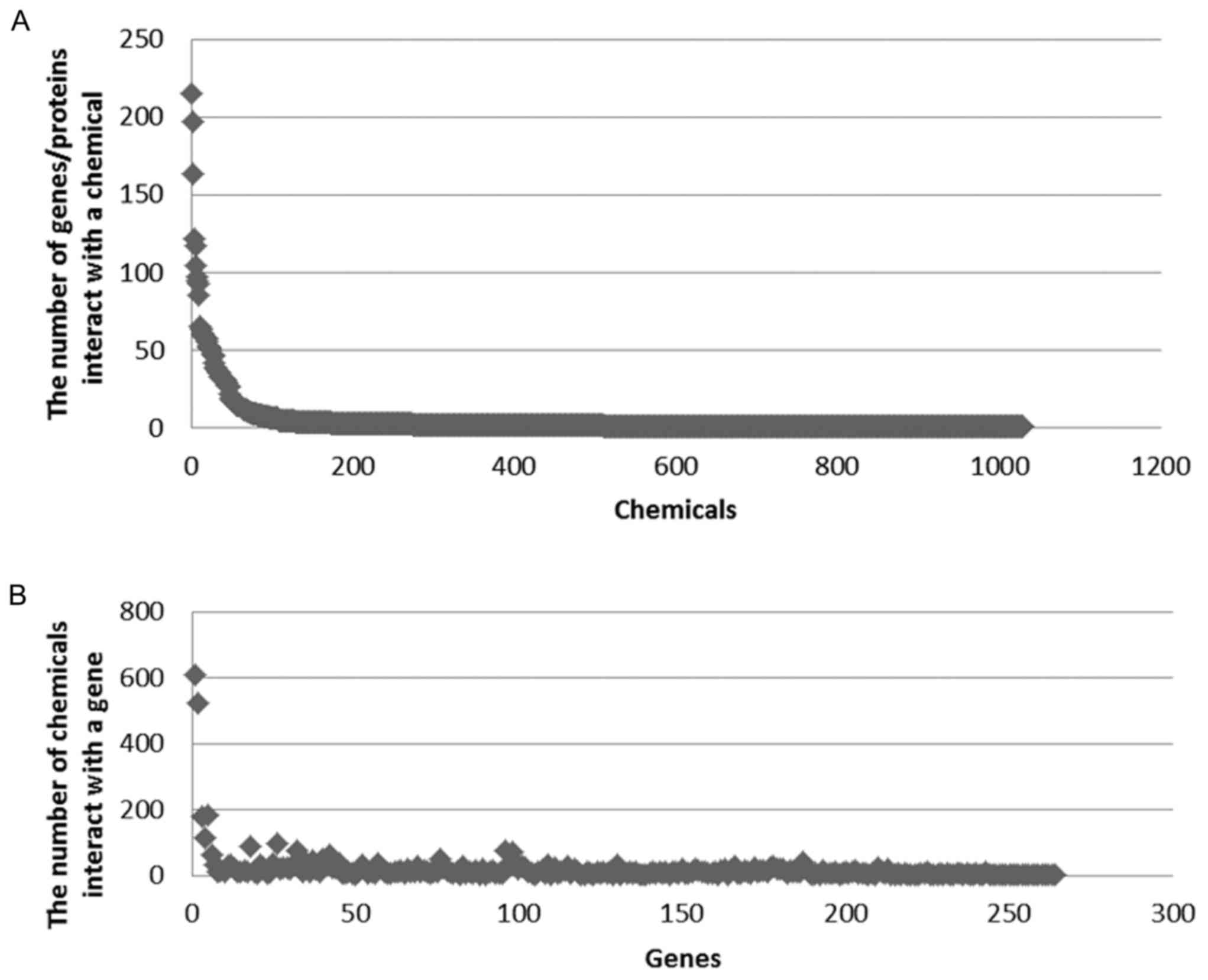
A total of 264 key genes, 1,030 small molecule
compounds and 5,037 small molecule compounds and mRNA interaction
association pairs were identified in the CTD database.
Subsequently, the number of compounds targeting the same gene and
the number of genes targeting the same compound were counted
(Fig. 3A and B). A number of genes
were targeted by several small molecule compounds. It was observed
that >500 compounds were targeted by two genes (FUN, 611;
FOS, 521) and >200 genes were targeted by two compounds
(tetrachlorodibenzodioxin, 215; benzo(a)pyrene, 197).
Discussion
HCC is the most common primary liver malignancy
characterized by a multifaceted molecular pathogenesis (31). In the present study, a total of 302
DEGs, including 231 downregulated and 71 upregulated genes, were
identified in HCC samples compared with healthy samples. Using KEGG
pathway analysis, it was demonstrated that the cytokine-cytokine
receptor interaction and chemokine signaling pathways were the
major enriched pathways. In addition, a total of 13 highest degree
proteins, including FOS and DDIT3, were identified as hub nodes in
PPI network analysis. Furthermore, >500 compounds were targeted
by FUN and FOS, and >200 genes were targeted by
2,3,7,8-tetrachlorodibenzodioxin and benzo(α)pyrene in the analysis
of gene and chemical interactions.
In the present study, FOS was identified as a
hub node in the PPI network analysis, and >500 compounds were
targeted by FOS in the analysis of gene and chemical
interactions. One study using immunohistochemical analysis
demonstrated that c-FOS serves a key role in HCC
pathogenesis (32). The upregulation
of c-FOS and Jun proto-oncogene mediated by protein kinase R
promotes HCC proliferation (33). In
addition, Fan et al (34)
demonstrated that suppression of c-FOS, mediated by miR-139
downregulation, promotes HCC metastasis. miR-101 inhibits the
expression of the FOS oncogene in HCC, suppressing
hepatocyte growth factor-induced cellular migration and invasion
(35). Additionally, Shen et
al (36) reported that
recombinant adeno-associated virus carrying Vastatin inhibited
tumor metastasis and reduced the expression of phosphoenolpyruvate
carboxykinase 1, jagged 2 and c-FOS in HCC, inhibiting the
cellular metabolism, Notch and activator protein-1 signaling
pathways, respectively. This suggests that FOS may serve a
critical role in the progression of HCC.
One additional study demonstrated that
FUS-DDIT3 and DDIT3 may serve an important role in
the induction of a liposarcoma phenotype (37). Kåbjörn Gustafsson et al
(38) reported a dual promoting and
inhibiting role in the formation of liposarcoma morphology mediated
by DDIT3. DDIT3 and lysine acetyltransferase 2A proteins
regulate the expression of tumor necrosis factor receptor
superfamily, member 10α (TNFRSF10A) and TNFRSF10B in endoplasmic
reticulum-associated, stress-induced apoptosis in human lung cancer
cells (39). In addition, long
non-coding RNA HOXA genes cluster antisense RNA 2 promotes
proliferation of gastric cancer via silencing the expression of
p21, polo-like kinase 3 and DDIT3 (40). Furthermore, it has also been
demonstrated that DDIT3 is associated with the development
of several types of cancer, including myxoid liposarcoma and
squamous cell carcinoma (41,42). Therefore, DDIT3 serves an
important role in several cancer types. In the present study,
DDIT3 was overexpressed in HCC samples and indicated to be a
hub node in PPI network analysis, suggesting that DDIT3 may
be associated with HCC.
A number of studies have also demonstrated that the
cytokine-cytokine receptor interaction pathway may be involved in
HCC carcinogenesis (43–46). In the study by Hsu et al
(47), it was reported that the
cytokine-cytokine receptor interaction pathway is associated with
HCC. Furthermore, Ryschich et al (48) demonstrated that the chemokine
signaling pathway is involved in liver carcinogenesis, while
another study indicated its involvement in organ-specific
metastatic growth of cancer cells (49). Accordingly, the present study
demonstrated that the cytokine-cytokine receptor interaction and
chemokine signaling pathways were the major enriched pathways in
KEGG pathway analysis. Therefore, these pathways may be involved in
the development of HCC.
Analysis of gene and chemical interactions in the
present study revealed that >200 genes were targeted by
2,3,7,8-tetrachlorodibenzodioxin and benzo(α)pyrene. Previously, it
has been demonstrated, using in vivo models, that
2,3,7,8-tetrachlorodibenzodioxin may cause cancer (50). 2,3,7,8-Tetrachlorodibenzodioxin
promotes liver tumor growth via an aryl hydrocarbon and
TNF/interleukin-1 receptor-dependent manner (51). One study reported that benzo(α)pyrene
inhibits cell adhesion and promotes cell migration and invasion in
HCC (52). Furthermore, a
case-control study demonstrated that exposure to benzo(α)pyrene
increases the risk of developing HCC (53). Therefore, exposure to
2,3,7,8-tetrachlorodibenzodioxin or benzo(α)pyrene may be
associated with hepatocarcinogenesis.
In conclusion, FOS, DDIT3, the
cytokine-cytokine receptor interaction pathway and the chemokine
signaling pathway may serve critical roles in the development of
HCC. Exposure to 2,3,7,8-tetrachlorodibenzodioxin or benzo(α)pyrene
may cause hepatocarcinogenesis. However, the lack of experimental
verification and the relatively small sample size are major
limitations of the present study. Therefore, further research is
required to verify the present findings.
References
|
1
|
Arzumanyan A, Reis HM and Feitelson MA:
Pathogenic mechanisms in HBV-and HCV-associated hepatocellular
carcinoma. Nat Rev Cancer. 13:123–135. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
El-Serag HB: Epidemiology of viral
hepatitis and hepatocellular carcinoma. Gastroenterology. 142:1264.
e1–1273. e1. 2012. View Article : Google Scholar
|
|
3
|
Bruix J, Gores GJ and Mazzaferro V:
Hepatocellular carcinoma: Clinical frontiers and perspectives. Gut.
63:844–855. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bruix J and Sherman M; American
Association for the Study of Liver Diseases, : Management of
hepatocellular carcinoma: An update. Hepatology. 53:1020–1022.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
European association for the study of the
liver and European organisation for research and treatment of
cancer: EASL-EORTC clinical practice guidelines: Management of
hepatocellular carcinoma. J Hepatol. 56:908–943. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yong KJ, Gao C, Lim JS, Yan B, Yang H,
Dimitrov T, Kawasaki A, Ong CW, Wong KF, Lee S, et al: Oncofetal
gene SALL4 in aggressive hepatocellular carcinoma. N Engl J Med.
368:2266–2276. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Furuta M, Kozaki K, Tanimoto K, Tanaka S,
Arii S, Shimamura T, Niida A, Miyano S and Inazawa J: The
tumor-suppressive miR-497-195 cluster targets multiple cell-cycle
regulators in hepatocellular carcinoma. PLoS One. 8:e601552013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cillo C, Schiavo G, Cantile M, Bihl MP,
Sorrentino P, Carafa V, D' Armiento M, Roncalli M, Sansano S,
Vecchione R, et al: The HOX gene network in hepatocellular
carcinoma. Int J Cancer. 129:2577–2587. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Revill K, Wang T, Lachenmayer A, Kojima K,
Harrington A, Li J, Hoshida Y, Llovet JM and Powers S: Genome-wide
methylation analysis and epigenetic unmasking identify tumor
suppressor genes in hepatocellular carcinoma. Gastroenterology.
145:1424–1435. e1-25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris CC and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xu J, Zhu X, Wu L, Yang R, Yang Z, Wang Q
and Wu F: MicroRNA-122 suppresses cell proliferation and induces
cell apoptosis in hepatocellular carcinoma by directly targeting
Wnt/β-catenin pathway. Liver Int. 32:752–760. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu L, Dong Z, Liang J, Cao C, Sun J, Ding
Y and Wu D: As an independent prognostic factor, FAT10 promotes
hepatitis B virus-related hepatocellular carcinoma progression via
Akt/GSK3β pathway. Oncogene. 33:909–920. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goyal L, Muzumdar MD and Zhu AX: Targeting
the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res.
19:2310–2318. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang Y, Han C, Lu L, Magliato S and Wu T:
Hedgehog signaling pathway regulates autophagy in human
hepatocellular carcinoma cells. Hepatology. 58:995–1010. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ma W, Wong CC, Tung EK, Wong CM and Ng IO:
RhoE is frequently down-regulated in hepatocellular carcinoma (HCC)
and suppresses HCC invasion through antagonizing the
Rho/Rho-Kinase/Myosin phosphatase target pathway. Hepatology.
57:152–161. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Shi M, Chen MS, Sekar K, Tan CK, Ooi LL
and Hui KM: A blood-based three-gene signature for the non-invasive
detection of early human hepatocellular carcinoma. Eur J Cancer.
50:928–936. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang JX, Yu C, Li ZP, Xiao J, Zhang H,
Chen MY and Sun CY: Insights into significant pathways and gene
interaction networks in peripheral blood mononuclear cells for
early diagnosis of hepatocellular carcinoma. J Cancer Res Ther.
12:981–989. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Smyth GK: Limma: Linear models for
microarray dataBioinformatics and computational biology solutions
using R and Bioconductor. Springer; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
21
|
Smyth GK: Linear models and empirical
Bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article32004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ferreira JA: The Benjamini-Hochberg method
in the case of discrete test statistics. Int J Biostat. 3:Article
112007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Altermann E and Klaenhammer TR:
PathwayVoyager: Pathway mapping using the Kyoto encyclopedia of
genes and genomes (KEGG) database. BMC Genomics. 6:602005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Huang da W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao J, Ade AS, Tarcea VG, Weymouth TE,
Mirel BR, Jagadish HV and States DJ: Integrating and annotating the
interactome using the MiMI plugin for cytoscape. Bioinformatics.
25:137–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mattingly CJ, Colby GT, Forrest JN and
Boyer JL: The comparative toxicogenomics database (CTD). Environ
Health Perspect. 111:793–795. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wiegers TC, Davis AP, Cohen KB, Hirschman
L and Mattingly CJ: Text mining and manual curation of
chemical-gene-disease networks for the comparative toxicogenomics
database (CTD). BMC Bioinformatics. 10:3262009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Davis AP, Wiegers TC, Roberts PM, King BL,
Lay JM, Lennon-Hopkins K, Sciaky D, Johnson R, Keating H, Greene N,
et al: A CTD-Pfizer collaboration: Manual curation of 88,000
scientific articles text mined for drug-disease and drug-phenotype
interactions. Database (Oxford). 2013:bat0802013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Whittaker S, Marais R and Zhu AX: The role
of signaling pathways in the development and treatment of
hepatocellular carcinoma. Oncogene. 29:4989–5005. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moghaddam SJ, Haghighi EN, Samiee S,
Shahid N, Keramati AR, Dadgar S and Zali MR: Immunohistochemical
analysis of p53, cyclinD1, RB1, c-fos and N-ras gene expression in
hepatocellular carcinoma in Iran. World J Gastroenterol.
13:588–593. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Watanabe T, Hiasa Y, Tokumoto Y, Hirooka
M, Abe M, Ikeda Y, Matsuura B, Chung RT and Onji M: Protein kinase
R modulates c-Fos and c-Jun signaling to promote proliferation of
hepatocellular carcinoma with hepatitis C virus infection. PLoS
One. 8:e677502013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fan Q, He M, Deng X, Wu WK, Zhao L, Tang
J, Wen G, Sun X and Liu Y: Derepression of c-Fos caused by
MicroRNA-139 down-regulation contributes to the metastasis of human
hepatocellular carcinoma. Cell Biochem Funct. 31:319–324. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li S, Fu H, Wang Y, Tie Y, Xing R, Zhu J,
Sun Z, Wei L and Zheng X: MicroRNA-101 regulates expression of the
v-fos FBJ murine osteosarcoma viral oncogene homolog (FOS) oncogene
in human hepatocellular carcinoma. Hepatology. 49:1194–1202. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Shen Z, Yao C, Wang Z, Yue L, Fang Z, Yao
H, Lin F, Zhao H, Sun YJ, Bian XW, et al: Vastatin, an Endogenous
Antiangiogenesis Polypeptide That Is Lost in Hepatocellular
Carcinoma, Effectively Inhibits Tumor Metastasis. Mol Ther.
24:1358–1368. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Engström K, Willén H, Kåbjörn-Gustafsson
C, Andersson C, Olsson M, Göransson M, Järnum S, Olofsson A,
Warnhammar E and Aman P: The myxoid/round cell liposarcoma fusion
oncogene FUS-DDIT3 and the normal DDIT3 induce a liposarcoma
phenotype in transfected human fibrosarcoma cells. Am J Pathol.
168:1642–1653. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kåbjörn Gustafsson C, Engström K and Åman
P: DDIT3 expression in liposarcoma development. Sarcoma.
2014:9546712014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li T, Su L, Lei Y and Liu X, Zhang Y and
Liu X: DDIT3 and KAT2A proteins regulate TNFRSF10A and TNFRSF10B
expression in endoplasmic reticulum stress-mediated apoptosis in
human lung cancer cells. J Biol Chem. 290:11108–11118. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Xie M, Sun M, Zhu YN, Xia R, Liu YW, Ding
J, Ma HW, He XZ, Zhang ZH, Liu ZJ, et al: Long noncoding RNA
HOXA-AS2 promotes gastric cancer proliferation by epigenetically
silencing P21/PLK3/DDIT3 expression. Oncotarget. 6:33587–33601.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Narendra S, Valente A, Tull J and Zhang S:
DDIT3 gene break-apart as a molecular marker for diagnosis of
myxoid liposarcoma-assay validation and clinical experience. Diagn
Mol Pathol. 20:218–224. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Huang Y, Chuang AY, Romano RA, Liégeois
NJ, Sinha S, Trink B, Ratovitski E and Sidransky D: Phospho-∆
Np63α/NF-Y protein complex transcriptionally regulates DDIt3
expression in squamous cell carcinoma cells upon cisplatin
exposure. Cell Cycle. 9:328–338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Riehle KJ, Campbell JS, McMahan RS,
Johnson MM, Beyer RP, Bammler TK and Fausto N: Regulation of liver
regeneration and hepatocarcinogenesis by suppressor of cytokine
signaling 3. J Exp Med. 205:91–103. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Taub R: Liver regeneration: From myth to
mechanism. Nat Rev Mol Cell Biol. 5:836–847. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Calvisi DF, Ladu S, Gorden A, Farina M,
Conner EA, Lee JS, Factor VM and Thorgeirsson SS: Ubiquitous
activation of Ras and Jak/Stat pathways in human HCC.
Gastroenterology. 130:1117–1128. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Niwa Y, Kanda H, Shikauchi Y, Saiura A,
Matsubara K, Kitagawa T, Yamamoto J, Kubo T and Yoshikawa H:
Methylation silencing of SOCS-3 promotes cell growth and migration
by enhancing JAK/STAT and FAK signalings in human hepatocellular
carcinoma. Oncogene. 24:6406–6417. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Hsu CN, Lai JM, Liu CH, Tseng HH, Lin CY,
Lin KT, Yeh HH, Sung TY, Hsu WL, Su LJ, et al: Detection of the
inferred interaction network in hepatocellular carcinoma from EHCO
(Encyclopedia of hepatocellular carcinoma genes online). BMC
Bioinformatics. 8:662007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Ryschich E, Lizdenis P, Ittrich C, Benner
A, Stahl S, Hamann A, Schmidt J, Knolle P, Arnold B, Hämmerling GJ,
et al: Molecular fingerprinting and autocrine growth regulation of
endothelial cells in a murine model of hepatocellular carcinoma.
Cancer Res. 66:198–211. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chambers AF, Groom AC and MacDonald IC:
Dissemination and growth of cancer cells in metastatic sites. Nat
Rev Cancer. 2:563–572. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Clemens MW: Reply: Association between
agent orange exposure and nonmelanotic invasive skin cancer: A
pilot study. Plast Reconstr Surg. 135:234e–235e. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kennedy GD, Nukaya M, Moran SM, Glover E,
Weinberg S, Balbo S, Hecht SS, Pitot HC, Drinkwater NR and
Bradfield CA: Liver tumor promotion by
2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl
hydrocarbon receptor and TNF/IL-1 receptors. Toxicol Sci.
140:135–143. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ba Q, Li J, Huang C, Qiu H, Li J, Chu R,
Zhang W, Xie D, Wu Y and Wang H: Effects of benzo [a] pyrene
exposure on human hepatocellular carcinoma cell angiogenesis,
metastasis, and NF-κB signaling. Environ Health Perspect.
123:246–254. 2015.PubMed/NCBI
|
|
53
|
Su Y, Zhao B, Guo F, Bin Z, Yang Y, Liu S,
Han Y, Niu J, Ke X, Wang N, et al: Interaction of benzo [a] pyrene
with other risk factors in hepatocellular carcinoma: A case-control
study in Xiamen, China. Ann Epidemiol. 24:98–103. 2014. View Article : Google Scholar : PubMed/NCBI
|