Introduction
Chronic myelogenous leukemia (CML) is a
hematopoietic stem cell disorder characterized by the Philadelphia
chromosome, resulting from a t(9;22) reciprocal chromosomal
translocation (1,2). t(9;22) occurs in the juxtaposition of
the Abelson (ABL1) oncogene, a tyrosine kinase (TK) located on
chromosome 9, to the breakpoint cluster region (BCR) gene located
on chromosome 22, leading to aberrantly increased TK activity
(3,4).
Mechanisms that have been attributed to BCR-ABL-positive cells and
been implicated in the pathogenesis of CML include increased
proliferation, increased resistance to apoptosis, and an alteration
of cell adhesion properties (1). The
disease progresses through three phases: Chronic, accelerated and
blast crisis, with disease progression likely due to an
accumulation of additional genetic aberrations (5).
In May of 2001, the Food and Drug Administration
approved imatinib mesylate (STI-571, Gleevec, Glivec), a selective
inhibitor of the BCR-ABL TK, for the first-line treatment of
patients with CML (6). Imatinib, a
2-phenyl amino pyrimidine, is a TK inhibitor (TKI) with activity
against BCR-ABL. All active sites of TKs have a binding site for
ATP. Imatinib mesylate works by binding close to the ATP site,
locking it in a closed or self-inhibited conformation and,
therefore, inhibiting the enzyme activity of the protein (7). Inhibition of BCR-ABL kinase activity by
imatinib results in the transcriptional modulation of genes
involved in the control of the cell cycle, cell adhesion and
cytoskeleton organization (1).
Imatinib functions by revoking the effects of the BCR-ABL
oncoprotein through inhibiting BCR-ABL autophosphorylation and
substrate phosphorylation, thus blocking proliferation and inducing
apoptosis (8). Nonetheless, in
certain patients, resistance to imatinib may occur. In addition to
imatinib mesylate, various TKIs have recently emerged on the
market, including bosutinib, dasatinib, nilotinib and ponatinib
(9). These drugs are used following
the failure of imatinib treatment due to resistance, disease
progression or intolerance (9). Point
mutations within the kinase domain (KD) of BCR-ABL, clonal
chromosomal evolution, BCR-ABL amplification, pharmacogenomic
variations, or activation of signaling shortcuts have all been
implicated in imatinib mesylate resistance (10). Previous studies have reported that
~20% of patients fail therapy with imatinib mesylate due to point
mutations within the BCR/ABL KD (11). The emergence of point mutations in the
BCR-ABL KD is a primary cause of imatinib resistance in patients
with CML, and >90 types of KD mutations have been reported,
particularly in the accelerated and blast crisis phases (4,8,12). These mutations may alter the BCR-ABL
KD structure and impair the imatinib-binding affinity (4,12). Point
mutations in the KD occur in 30–90% of patients who develop
resistance to the drug (13). The
sites of the KD mutations are predominantly clustered within nine
amino acid positions, including T315I, Y253H/F, M351T, G250E,
E255K/V, F359V and H396R, with varying sensitivities to imatinib
(14). Less sensitive or resistant to
BCR-ABL point mutations for dasatinib include Q252H, E255K/V,
V299L, F317L, T315A and T315I; for nilotinib, they include E255K/V
or F359C/V, Y253H and T315 (15). The
T315I mutation was detected in 15% of 112 patients with CML who
failed to respond to imatinib mesylate treatment (13). The T315I mutation is only sensitive to
ponatinib (16).
To detect genetic anomalies in CML, the following
methods may be used: Direct sequencing, denaturing high performance
liquid chromatography (dHPLC), pyro-sequencing, ultra-deep
sequencing (UDS), allele-specific oligonucleotide (ASO)-PCR
(17) and denaturing gradient gel
electrophoresis (DGGE) (18).
The present study was designed to determine if and
what types of KD mutations were present in the 45 Turkish patients
with CML enrolled, all of whom were resistant to imatinib. The aim
was to detect the frequency of BCR-ABL KD mutations using a
sequence analysis method to evaluate the clinical significance of
the identified mutation. Furthermore, the current study intended to
emphasize the importance of mutation analysis and support using the
ASO-PCR method in drug selection and disease follow-up.
Materials and methods
Patients
The present study was performed retrospectively on
peripheral blood (PB) samples obtained from 45 patients enrolled
between January 2001 and December 2015 in the Department of
Hematology at Marmara University School of Medicine (Istanbul,
Turkey). In total, 45 BCR-ABL-positive patients with CML who were
resistant or intolerant to imatinib were included in the present
study. Patients were treated with imatinib or nilotinib or
dasatinib. In total, 13 patients exhibited hematological and
cytogenetic responses to imatinib, whereas 32 patients demonstrated
no major molecular response and were subsequently switched to a 2nd
generation TKI (nilotinib or dasatinib). Recently diagnosed chronic
phase CML patients were administered imatinib (400 mg/day orally).
Patients who demonstrated resistance to imatinib were administered
nilotinib (100 mg/day orally) or dasatinib (100 mg/day orally).
Patients had a median age of 46.6 (range, 25–87). In total, 45
patients, including 43 in the chronic phase, 1 in the accelerated
phase, and 1 in blast crisis (transformed to acute myeloid
leukemia), were analyzed for the presence of mutations using direct
sequencing. The study protocol was approved by the Local Ethics
Committee of Marmara University (Istanbul, Turkey). Once written
informed consent was obtained, 10 ml PB samples were taken from all
patients. Patients' clinical characteristics are provided in
Table I.
 | Table I.Clinical characteristics of patients
with CML carrying MT and WT ABL KDs. |
Table I.
Clinical characteristics of patients
with CML carrying MT and WT ABL KDs.
| Characteristics | Mutant type | Wild type | P-value |
|---|
| Sex |
|
|
|
| Male | 6 | 15 | NS |
|
Female | 5 | 10 |
|
| Median age,
years | 48 | 50 | 0.94 |
| Median WBC counts
(×109/l)a |
6.000 |
5.650 | 0.41 |
| Median PLT counts
(×109/l)a | 245.000 | 224.000 | 0.42 |
| Median Hb
(g/dl)a | 12 | 12.25 | 0.52 |
| IS at mutation
detection | 4.43 | 0.47 | 0.85 |
Amplification of BCR-ABL KD by nested
PCR
RNA was isolated from peripheral blood leukocytes
using a 5 PRIME-Perfect Pure RNA Purification kit (Thermo Fisher
Scientific Inc., Waltham, MA, USA), according to the manufacturer's
protocol. cDNA was synthesized using the Bio-Speedy First Strand
cDNA Synthesis kit (Bioeksen R&D Technologies Ltd., Istanbul,
Turkey) from 1 ng total RNA in a 20 µl reaction mixture, according
to the manufacturer's protocol.
BCR-ABL1 transcripts were detected using nested
polymerase chain reaction (PCR) using specific primers for p210
transcripts. PCR products were prepared using a pair of primers
designed to cover BCR exon 13 and ABL exon 10 with the expected
products of 1,643 bp. The first-round of amplification was
performed using a forward primer (5′-ACAGCATTCCGCTGACCATCAATAAG-3′)
and a reverse primer (5′-ATGGTCCAGAGGATCGCTCTCT-3′) as previously
described (19). PCR reactions were
performed in a final volume of 20 µl, containing 2 µl cDNA sample,
2X reaction buffer, 4 mM MgSO4, 20 mM KCI, 0.4 mM dNTPs, 0.5 µM
each of the forward and reverse primers and 5 U/µl polymerase
(Bio-Speedy Proof-Reading DNA Polymerase Pre-Mix kit; (Bioeksen
R&D Technologies Ltd.). The first step of nested PCR was
conducted under touchdown PCR conditions: 2 min of initial
denaturation at 95°C, followed by a touchdown protocol of 14 cycles
of 30 sec at 95°C, 30 sec at 67°C and 3 min at 72°C, and then 24
cycles of 30 sec at 95°C, 30 sec at 60°C and 3 min at 72°C, with a
final extension for 5 min at 72°C (19).
The second round of amplification was performed
using two primer pairs (Bioeksen R&D Technologies Ltd.). Abl
fragment 1 (Abl-1) amplification was performed using the following
primers: Abl-1F 5′-TGGTTCATCATCATTCAACGGTGG-3′ and Abl-1R
5′-TCTGAGTGGCCATGTACAGCAGC-3′ (product 447 bp, spanning codons
206–346). Abl fragment 2 (Abl-2) amplification was performed using
the following primers: Abl-2F 5′-TCATGACCTACGGGAACCTC-3′ and Abl-2R
5′-ATACTCCAAATGCCCAGACG-′3 (product 333 bp, spanning codons
293–428) (20). PCR reactions were
performed in a total volume of 20 µl, containing 2 µl first-round
PCR product, 5 U/µl Taq DNA polymerase (Bioeksen R&D
Technologies Ltd.), 2X Phusion buffer, 4 mM MgSO4, 20 mM KCI, 0.4
mM dNTPs and 0.5 µM of each primer.
Conditions for the second step were as follows: 30
sec of initial denaturation at 98°C, amplification for 40 cycles of
10 sec at 98°C, 30 sec at 60°C and 40 sec at 72°C, with a final
extension step of 5 min at 72°C (4).
The nested PCR products (5 µl loaded per well) were then resolved
via 2% agarose gel electrophoresis and visualized using ethidium
bromide. Quantification of PCR bands densitometry was performed
using ImageJ 1.41o software (National Institutes of Health,
Bethesda, MD, USA).
BCR-ABL KD mutations screening by
Sanger sequencing
PCR products from nested PCR reactions were analyzed
by Sanger sequencing. The PCR products were sent to Macrogen Europe
(Amsterdam, The Netherlands) for Sanger sequencing. ABL fragment 1
(spanning codons 206–346) and ABL fragment 2 (spanning codons
293–428) of BCR-ABL KD were sequenced. The results were compared
with the ABL-1 sequence (GenBank: EU216071.1).
ASO-PCR assay for E255K mutations
Genomic DNA (gDNA) was extracted from peripheral
blood samples using a MagNA Pure LC DNA Isolation kit (Roche
Molecular Diagnostics, Pleasanton, CA, USA). The quantity and ratio
of absorbance at 260 and 280 nm (A260/280) of the purified gDNA was
determined with a Qubit® Fluorometer (Thermo Fisher
Scientific, Inc.).
BCR-ABL transcripts were detected in three patients
by ASO-PCR using allele-specific primers for p210, according to the
E255K mutation. To amplify the E255K mutation, the forward
5′-GCGGGGGCCAGTACGGGA-3′ and reverse 5′-GCCAATGAAGCCCTCGGAC-3′
primers were used, as previously described (21). The thermocycling conditions used were
as follows: 5 min at 94°C followed by 35 cycles of denaturation at
94°C for 30 sec, annealing at 60°C for 30 sec, extension at 72°C
for 45 sec, and a final extension for 5 min at 72°C. The ASO-PCR
products (5 µl loaded per well) then resolved via 2% agarose gel
electrophoresis and visualized with ethidium bromide. One
peripheral blood sample was obtained from a patient with CML
exhibiting wild type for ABL mutations and used as a negative
control. Quantification of PCR bands densitometry was performed
using ImageJ 1.41o software (National Institutes of Health).
Results were confirmed using sequencing analysis.
Statistical analysis
Statistical data were analyzed using GraphPad Prism
6.0 (GraphPad Software, Inc., La Jolla, CA, USA). The associations
between BCR-ABL KD mutations and the clinical parameters were
determined using Fisher's exact test, and an unpaired t-test when
required. All tests were two-tailed, and P<0.05 was considered
to indicate a statistically significant difference.
Results
Frequency of BCR-ABL KD mutations
The six types of ABL KD mutations detected by Sanger
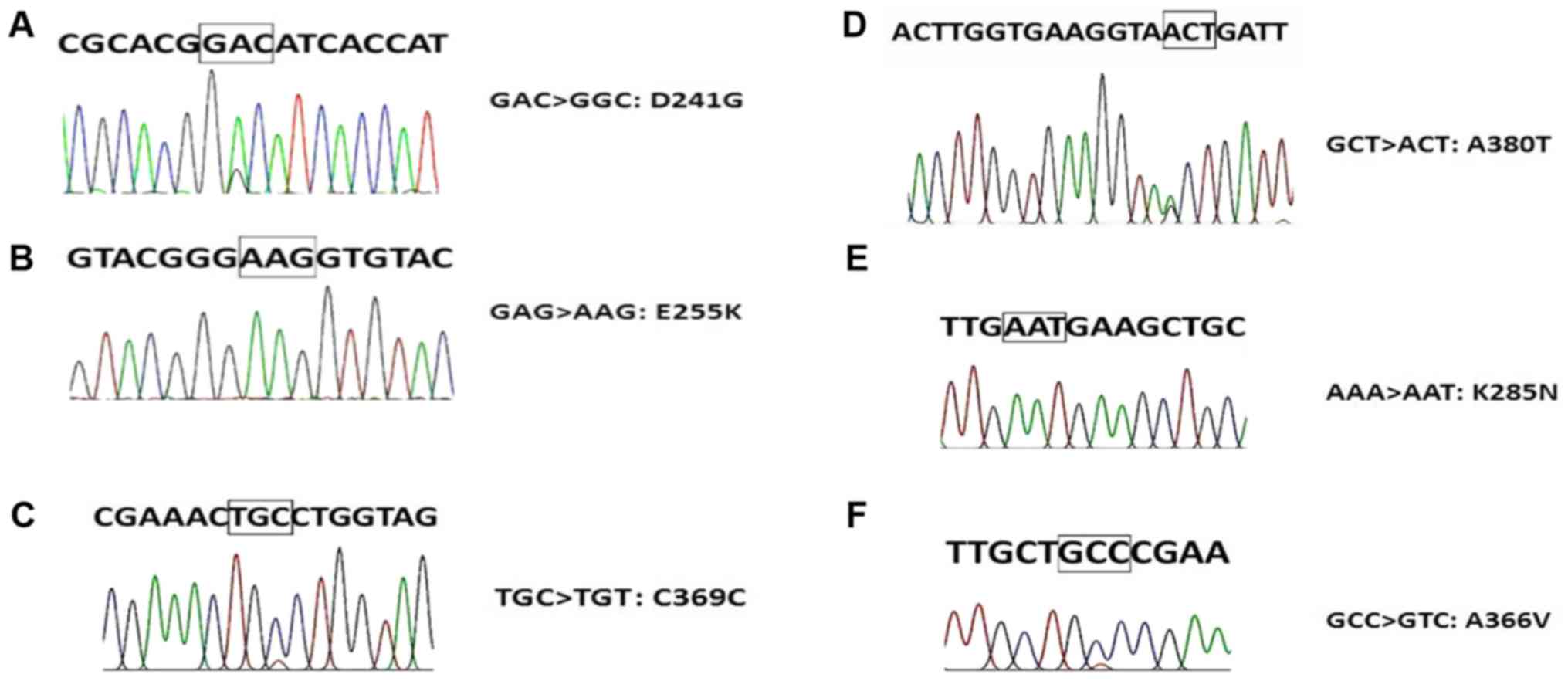
sequencing in the present study are demonstrated in Fig. 1. In total, 11/45 (24.44%) patients had
mutations that were detected by Sanger sequencing. An E255K
mutation was detected in 4 patients (8.8%), D241G mutation was
detected in 2 patients (4.4%), C369C mutation was detected in 2
patients (4.4%), K285N mutation was detected in 1 patient (2.2%),
A380T mutation was detected in 1 patient (2.2%) and an A366V
mutation was detected in 1 patient (2.2%) (Table II). There was no significant
association between age, sex, median white blood cell count,
platelet count and haemoglobin count, International Scale value
(16) and mutation (Table I).
| Table II.Sanger-sequencing detected mutations
and their properties. |
Table II.
Sanger-sequencing detected mutations
and their properties.
| Patient code | Mutation | Amino acid
changes | BCR/ABL % (IS) | BCR/ABL type | Exon |
Treatmenta |
|---|
| 3 | D241G
(heterozygous) | GACàGGC
(A>G) | 42.9 | p210 | Exon 4
(p-loop) | Imatinib |
| 4 | E255K
(homozygous) | GAGàAAG
(G>A) | 0.151 | p210 | Exon 4
(p-loop) | Glivec |
| 5 | E255K
(homozygous) | GAGàAAG
(G>A) | 4.43 | p210 | Exon 4
(p-loop) | Nilotinib |
| 7 | E255K
(homozygous) | GAGàAAG
(G>A) | 0.81 | p210 | Exon 4
(p-loop) | Imatinib |
| 9 | D241G
(heterozygous) | GACàGGC
(A>G) | 8.35 | p210 | Exon 4
(p-loop) | Dasatinib |
| 10 | E255K
(homozygous) | GAGàAAG
(G>A) | 38.4 | p210 | Exon 4
(p-loop) | Nilotinib |
| 28 | K285N
(homozygous) | AAAàAAT
(A>T) | 0.362 | p210 | Exon 5 (IM
B.S.) | Imavec |
| 14 | C369C
(heterozygous) | TGCà TGT
(C>T) | 0.0013 | p210 | Exon 7
(C-loop) | Dasatinib |
| 24 | A380T
(heterozygous) | GCTàACT
(G>A) | 0.15 | p210 | Exon 7
(A-loop) | Dasatinib |
| 15 | C369C
(heterozygous) | TGCà TGT
(C>T) | 0.02 | p210 | Exon 7
(C-loop) | Dasatinib |
| 40 | A366V
(heterozygous) | GCCàGTC
(C>T) | 0.003 | p210 | Exon 7
(C-loop) | Nilotinib |
Verification of the E255K mutation
using ASO-PCR
Sequencing data confirmed the presence of an E255K
mutation in three previously Sanger sequencing-detected patients,
using ASO-PCR. Of note, one patient with the E255K mutation
succumbed to CML due to disease progression during the study, thus
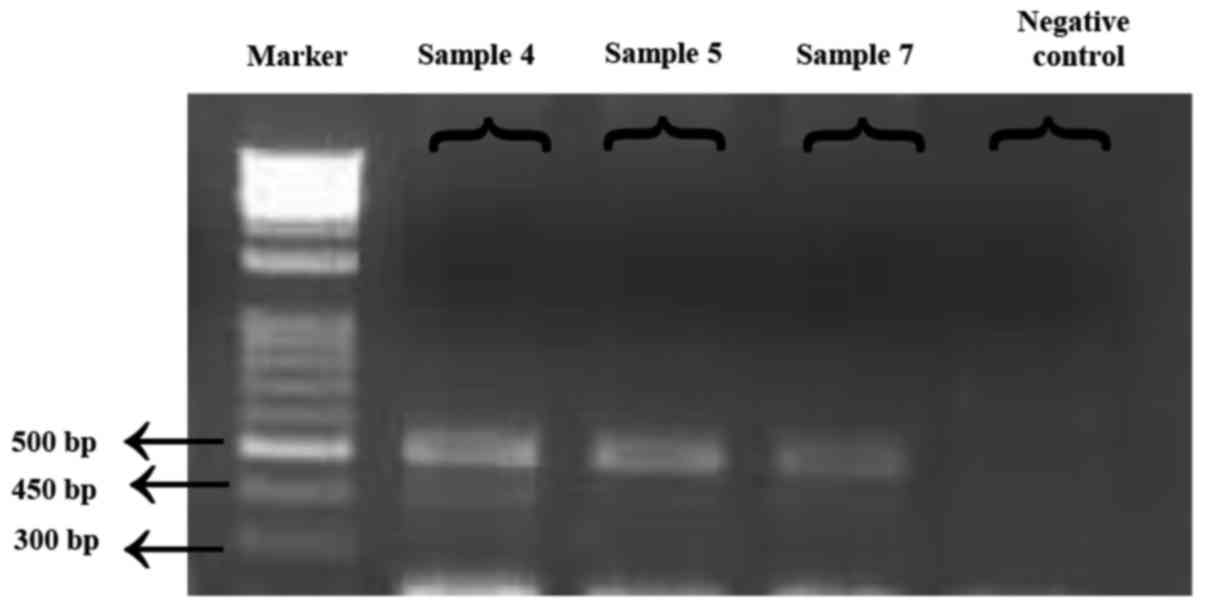
ASO-PCR could not be performed for this patient. Fig. 2 presents the monitoring data (obtained
from electrophoresis results) for the detected E255K mutation. DNA
molecules were visualized using ultraviolet light following
staining with ethidium bromide.
Discussion
Imatinib mesylate is a first line therapy for
patients with CML, a selective inhibitor of the BCR/ABL TK
(6). Point mutations within the KD of
BCR-ABL have been previously associated with promoting drug
resistance. Furthermore, additional resistance mechanisms
including; clonal chromosomal evolution, BCR-ABL amplification,
pharmacogenomic variations are present in patients with and without
kinase domain mutations (10).
However, the exact contribution(s) to resistance are yet to be
determined. Additionally, the inconsistent use of prescription
drugs by patients results in resistance development (10).
Unidentified mutations in a given sequence may be
detected using methods such as direct sequencing, DGGE and dHPLC;
previously recognized mutations can also be identified with methods
including ASO-PCR and RFLP. Direct sequencing is the most
widespread method applied in the routine monitoring of patients,
but it has limited sensitivity (10–25%). DGGE is a powerful
technique, which may detect single base mutations up to 100%
sensitivity; however, its sensitivity decreases for fragments
larger than 500 bp, requiring the use of specialized equipment.
dHPLC is a simple method for detecting low-level mutations which is
more sensitive when compared with direct sequencing, and it may
recognize sequence variations (17).
However, dHPLC is not as common as direct sequencing and it may
generate false negative results in cases with a vast number of
mutant subclones. ASO-PCR has been designed to detect specific
mutations; therefore, it is a troublesome method to use in
screening for more than one mutation, and is therefore not used
frequently (17). In a previous study
that used the dHPLC method in Turkey (22), 8 types of mutations were detected, all
of which are different from the 6 types of mutations detected in
the present study. As ASO-PCR primers should be specifically
designed for each individual mutation, ASO-PCR may be considered a
troublesome method in this respect. The use of the ASO-PCR method
may be advantageous for detecting the most common mutations in the
disease, as ASO-PCR assays have the best sensitivity for detecting
mutations even following dilutions in the 10,000-fold range
(23). ASO-PCR is also a more
sensitive method when compared with dHPLC and direct sequencing.
ASO-PCR assays have various advantages, including quicker analysis
results, lower costs and an easy protocol (23). For this reason, an ASO-PCR assay was
used in the present study for detecting the E255K mutation; the
results of ASO-PCR mutation analysis were compared with those from
direct sequencing. The current study focused on the frequency and
types of KD mutations in imatinib-resistant Turkish patients with
CML, and described the occurrence of mutations in the BCR/ABL KD in
11/45 patients with CML, detected by Sanger sequencing. Using two
different methodological approaches, namely Sanger sequencing and
ASO-PCR, the E255K mutation was identified in 4/45 patients with
CML.
The BCR/ABL KD mutation frequency was 24.44% (11/45)
in the present study. Compared with the literature, it was observed
that the frequency of BCR/ABL KD mutations (24.44%) present in the
current study was slightly lower compared with that stated in
previous studies, which reported the overall incidence of KD
mutations as 33% in Thailand (4), 44%
in Korea (23), 41.53% in India
(24) and 30% in Germany (25); however, it was slightly increased
compared with, though similar to, the previously recorded 23% in a
Turkish population (22).
In the present study, 6 diverse types of mutations
were detected, including E255K, D241G, C369C, K285N, A380T and
A366V. Erbilgin et al (22)
detected 8 types of mutations, including M244V, G250E, Y253H,
V304I, F359C, K357R, V304I and K357R. The mutations detected in
these two studies were all distinct from each other, demonstrating
that there is a wide spectrum of ABL KD region mutations in the
Turkish population of patients with CML.
The most resistant mutation T315I could not be
detected in any of the 45 patients with CML in the present study;
therefore, the results are not concordant with the literature with
respect to the T315I mutation. The E255K mutation is located in
exon 4 (P-loop region) on ABL, as confirmed by the My Cancer Genome
database (https://www.mycancergenome.org). The E255K mutation
results in an amino acid substitution at position 255 in BCR-ABL,
from a glutamate to a lysine. The E255K mutation frequency was
determined to be 8.8%. In the OncQuest and GIMEMA studies, the
E255K mutation frequency was reported to be 6.9 and 16.5%,
respectively. With respect to the E255K mutation, the results of
the present study are consistent with previous literature. The
E255K mutation has been associated with imatinib and nilotinib
resistance in patients with CML (8).
Dasatinib therapy is recommended in the presence of an E255K
mutation, according to the European LeukemiaNet recommendations
(8).
D241G point mutations, targeting amino acids located
near the P-loop, have been previously described (26). A366V mutations (substitution) at the
C-loop have also been described in a prior study (27) and confirmed by the Catalogue of
Somatic Mutations in Cancer database (http://cancer.sanger.ac.uk/cosmic). According to
localization, these mutations may be associated with drug
resistance and could affect the binding mechanisms, but further
analysis is required for confirmation. The remaining four mutations
defined in the current study have been previously described in
association with imatinib mesylate resistance (8). The K285N mutation was determined to
confer a lower imatinib binding affinity. In a previous in
vitro trial, it was demonstrated that K285N mutants had a lower
binding affinity for imatinib when compared with the native type
(28), and exhibited resistance to
imatinib (29). The A380T mutation
was detected at the A-loop in one patient, who received dasatinib
treatment due to imatinib-resistance. The A380T mutation was
previously detected as an ABL kinase mutation in imatinib-resistant
patients with CML (30,31). Additionally, C369C is available in the
Catalogue of Somatic Mutations in Cancer database (http://cancer.sanger.ac.uk/cosmic). It is a
silent mutation; therefore, it has not been discussed in the
literature to the best of our knowledge.
ASO-PCR experiments were performed using forward and
reverse primers, and reliable results were obtained via these
means. The ASO-PCR assay exhibits numerous advantages including an
increase in sensitivity, short analysis time, lower costs, and a
simple procedure. Kang et al (23) previously identified 11 types of
mutations with a 97% sensitivity using the ASO-PCR method. Iqbal
et al (21) identified 4
different types of mutations using the ASO-PCR method. Thus, it was
concluded that the ASO-PCR method is a reliable tool for routine
research.
By determining the mutational profile of patients,
the optimal treatment (2nd generation TKIs, alloSCT, etc.) for each
patient may be selected. The ASO-PCR assay has a high sensitivity
for the identified mutations. The results suggest that using
ASO-PCR assays will be beneficial in the routine monitoring of
mutations, particularly for commonly identified mutations in
patients with CML.
Acknowledgements
The present study was supported by the Scientific
Research Projects Committee (grant no. SAG-C-YLP-110315-0057) at
Marmara University.
Glossary
Abbreviations
Abbreviations:
|
CML
|
chronic myeloid leukemia
|
|
TKI
|
tyrosine kinase inhibitor
|
|
KD
|
kinase domain
|
|
Ph
|
Philadelphia chromosome
|
|
BCR
|
breakpoint cluster region
|
|
ABL
|
Abelson
|
|
ASO-PCR
|
allele-specific oligonucleotide
polymerase chain reaction
|
References
|
1
|
Deininger MW, Goldman JM and Melo JV: The
molecular biology of chronic myeloid leukemia. Blood. 96:3343–3356.
2000.PubMed/NCBI
|
|
2
|
Corbin AS, La Rosée P, Stoffregen EP,
Druker BJ and Deininger MW: Several Bcr-Abl kinase domain mutants
associated with imatinib mesylate resistance remain sensitive to
imatinib. Blood. 101:4611–4614. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Litzow MR: Imatinib resistance, obstacles
and opportunities. Arch Pathol Lab Med. 130:669–679.
2006.PubMed/NCBI
|
|
4
|
Wongboonma W, Thongnoppakhun W and
Auewarakul CU: BCR-ABL kinase domain mutations in tyrosine kinase
inhibitors-naïve and -exposed Southeast Asian chronic myeloid
leukemia patients. Exp Mol Pathol. 92:259–265. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sawyers CL: Chronic myeloid leukemia. N
Engl J Med. 340:1330–1340. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
O'Brien S, Berman E, Borghaei H, DeAngelo
DJ, Devetten MP, Devine S, Erba HP, Gotlib J, Jagasia M, Moore JO,
et al: NCCN clinical practice guidelines in oncology: Chronic
myelogenous leukemia. J Natl Compr Canc Netw. 7:984–1023. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iqbal N and Iqbal N: Imatinib: A
breakthrough of targeted therapy in cancer. Chemother Res Pract.
2014:3570272014.PubMed/NCBI
|
|
8
|
Soverini S, Hochhaus A, Nicolini FE,
Gruber F, Lange T, Saglio G, Pane F, Müller MC, Ernst T, Rosti G,
et al: BCR-ABL kinase domain mutation analysis in chronic myeloid
leukemia patients treated with tyrosine kinase inhibitors:
Recommendations from an expert panel on behalf of European Leukemia
net. Blood. 118:1208–1215. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ursan ID, Jiang R, Pickard EM, Lee TA, Ng
D and Pickard AS: Emergence of BCR-ABL kinase domain mutations
associated with newly diagnosed chronic myeloid leukemia: A
meta-analysis of clinical trials of tyrosine kinase inhibitors. J
Manag Care Spec Pharm. 21:114–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
La Rosée P and Deininger MW: Resistance to
imatinib: Mutations and beyond. Semin Hematol. 47:335–343. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vaidya S, Vundinti BR, Shanmukhaiah C,
Chakrabarti P and Ghosh K: Evolution of BCR/ABL gene mutation in
CML is time dependent and dependent on the pressure exerted by
tyrosine kinase inhibitor. PLoS One. 10:e01148282015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang Z, Liu Z, Wu X, Chu S, Wang J, Yuan
H, Roth M, Yuan YC, Bhatia R and Chen WY: Correction: ATRA-induced
cellular differentiation and CD38 expression inhibits acquisition
of BCR-ABL mutations for CML acquired resistance. PLoS Genet.
10:e10044142014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cortes J, Jabbour E, Kantarjian H, Yin CC,
Shan J, O'Brien S, Garcia-Manero G, Giles F, Breeden M, Reeves N,
et al: Dynamics of BCR-ABL kinase domain mutations in chronic
myeloid leukemia after sequential treatment with multiple tyrosine
kinase inhibitors. Blood. 110:4005–4011. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Apperley JF: Part I: Mechanisms of
resistance to imatinib in chronic myeloid leukaemia. Lancet Oncol.
8:1018–1029. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jabbour E, Hochhaus A, Cortes J, La Rosée
P and Kantarjian HM: Choosing the best treatment strategy for
chronic myeloid leukemia patients resistant to imatinib: Weighing
the efficacy and safety of individual drugs with BCR-ABL mutations
and patient history. Leukemia. 24:6–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baccarani M, Castagnetti F, Gugliotta G
and Rosti G: A review of the european leukemianet recommendations
for the management of CML. Ann Hematol. 94 Suppl 2:S141–S147. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ai J and Tiu RV: Practical management of
patients with chronic myeloid leukemia who develop tyrosine kinase
inhibitor-resistant BCR-ABL1 mutations. Ther Adv Hematol.
5:107–120. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sorel N, Roy L, Martineau G, Guilhot F,
Turhan AG and Chomel JC: Sequential emergence of ABL-kinase
mutations with loss of unmutated BCR-ABL allele during targeted
therapies of CML. Blood. 108:1782–1783. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ernst T, Erben P, Muller MC, Paschka P,
Schenk T, Hoffmann J, Kreil S, La Rosée P, Hehlmann R and Hochhaus
A: Dynamics of BCR-ABL mutated clones prior to hematologic or
cytogenetic resistance to imatinib. Haematologica. 93:186–192.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Deininger MW, McGreevey L, Willis S,
Bainbridge TM, Druker BJ and Heinrich MC: Detection of ABL kinase
domain mutations with denaturing high performance liquid
chromatography. Leukemia. 18:864–871. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Iqbal Z, Aleem A, Iqbal M, Naqvi MI, Gill
A, Taj AS, Qayyum A, ur-Rehman N, Khalid AM, Shah IH, et al:
Sensitive detection of pre-existing BCR-ABL kinase domain mutations
in CD34+ cells of newly diagnosed chronic-phase chronic myeloid
leukemia patients is associated with imatinib resistance:
Implications in the post-imatinib era. PLoS One. 8:e557172013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Erbilgin Y, Çatal S, Eşkazan AE, Hatirnaz
Ö, Soysal T and Özbek U: ABL gene kinase domain mutation scanning
by denaturing high performance liquid chromatography sequencing
method. Turk J Hematol. 28:97–102. 2011. View Article : Google Scholar
|
|
23
|
Kang HY, Hwang JY, Kim SH, Goh HG, Kim M
and Kim DW: Comparison of allele specific
oligonucleotide-polymerase chain reaction and direct sequencing for
high throughput screening of ABL kinase domain mutations in chronic
myeloid leukemia resistant to imatinib. Haematologica. 91:659–662.
2006.PubMed/NCBI
|
|
24
|
Srivastava S and Dutt S: Imatinib mesylate
resistance and mutations: An Indian experience. Indian J Med
Paediatr Oncol. 34:213–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sherbenou DW, Hantschel O, Kaupe I, Willis
S, Bumm T, Turaga LP, Lange T, Dao K, Press RD, Druker BJ, et al:
BCR-ABL SH3-SH2 domain mutations in chronic myeloid leukemia
patients on imatinib. Blood. 116:3278–3285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gruber F, Hansen H, Olsen M, Eggen L,
Skogen B, Gedde-Dahl T, Lundan T, Porkka K, Simonsson B, Markevärn
B, et al: Quantitative analyses of BCR-ABL mutations associated
with imatinib treatment in CML. Blood. 106:20002005.
|
|
27
|
Khorashad JS, Kelley TW, Szankasi P, Mason
CC, Soverini S, AdriaL T, Eide CA, Zabriskie MS, Lange T, Estrada
JC, et al: BCR-ABL1 compound mutations in tyrosine kinase
inhibitor-resistant CML: Frequency and clonal relationships. Blood.
121:489–498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rajasekaran R, Doss PDC, Prasad GA and
Sethumadhavan R: In silico identification and analysis of drug
resistant mutants of ABL tyrosine kinase based on detrimental
missense mutations. Curr Signal Trans Ther. 6:396–404. 2011.
View Article : Google Scholar
|
|
29
|
von Bubnoff N, Veach DR, van der Kuip H,
Aulitzky WE, Sänger J, Seipel P, Bornmann WG, Peschel C, Clarkson B
and Duyster J: A cell-based screen for resistance of
Bcr-Abl-positive leukemia identifies the mutation pattern for
PD166326, an alternative Abl kinase inhibitor. Blood.
105:1652–1659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Soverini S, Baccarani M, Iacobucci I and
Martinelli G: Resistance to tyrosine kinase inhibitors in
Philadelphia chromosome-positive leukemias: Which mutations matter?
Clinical Leukemia. 1:223–228. 2007. View Article : Google Scholar
|
|
31
|
Chomel JC, Sorel N and Turhan AG:
ABL-Kinase mutations in progenitors and stem cells from chronic
myeloid leukemia patients. Stem Cells Cancer Stem Cells. 8:305–315.
2012. View Article : Google Scholar
|