Introduction
Ovarian cancer is the most aggressive gynecologic
cancer and is thus the main cause of cancer-associated mortality in
women worldwide (1,2). According to a report by the American
Cancer Society, the standard treatment approach is a platinum
compound, for example cisplatin or carboplatin, in combination with
a taxane, for example paclitaxel or docetaxel; however, intrinsic
or acquired tumor chemoresistance remains a serious clinical
problem and a major obstacle to successful therapy (3). Although rates of response of the primary
tumor to taxane- and platinum-based therapy are high, ~20% of cases
remain non-responsive and the majority of patients will relapse and
ultimately succumb to drug-resistant disease (4). The anticancer effects of platinum-based
antineoplastics are mediated by their ability to generate
irreparable intrastrand DNA crosslinks/adducts, which result in the
induction of apoptosis and initiation of oxidative and endoplasmic
reticulum stress (5). A number of
factors lead to drug resistance, including enhanced drug efflux,
drug inactivation, modifications of drug targets, processing of
drug-induced damage and avoidance of apoptosis (6,7).
Furthermore, platinum resistance in cancer arises from numerous
adaptive mechanisms, including decreased cellular uptake, enhanced
DNA repair and tolerance and inactivation by glutathione; however,
the overall molecular mechanisms require further elucidation
(4).
Shikonin is a natural compound obtained from the
roots of Lithospermum erythrorhizon, Arnebia euchroma and
Onosma paniculata (8–10). Shikonin has anti-inflammatory,
anti-oxidant, anticancer, wound-healing and anti-microbial effects
(11). Shikonin induces cancer cell
death via various mechanisms, including suppression of the activity
of protein tyrosine kinases and DNA topoisomerases, which have an
important function in gene regulation and the inhibition of
expression of tumor necrosis factor receptor-associated protein 1.
The anticancer effect of shikonin is associated with enhanced
expression of tumor protein p53 and suppression of cancer cell
glycolysis through targeting of pyruvate kinase M2 (11,12).
Previous studies have demonstrated that shikonin induces apoptosis
in keratinocyte HaCaT cells, human stomach carcinoma AGS cells,
colon cancer cells, lung cancer cells, human medullary thyroid
carcinoma cells, human promyelocytic leukemia HL-60 cells, and
human cervical cancer HeLa cells (13–20). The
present study investigated the apoptotic effects of shikonin on
cisplatin-resistant human ovarian cancer A2780 (A2780-CR)
cells.
The epithelial-mesenchymal transition (EMT), a
feature of aggressive tumors, is characterized by decreased
epithelial (E-)cadherin expression and increased neural
(N-)cadherin expression, which contributes to a stroma-oriented
cellular adhesion profile with enhanced cancer cell motility and
invasive features. These events have already been demonstrated in
several cancer cell lines including breast, prostate and colorectal
adenocarcinoma (21). Shikonin was
reported to stimulate EMT in skin wound-healing (22). On the other hand, shikonin inhibits
the migration and invasion of breast cancer and glioblastoma cells
(23,24). Therefore, the present study sought to
clarify the effects of shikonin on the migratory capacity of
ovarian cancer cells. On the basis of its cytotoxic and
anti-migratory effects, shikonin may be an alternative drug to be
used during chemotherapy to treat this type of tumor.
Materials and methods
Reagents
Shikonin
[5,8-dihydroxy-2-(1-hydroxy-4-methylpent-3-enyl)naphthalene-1,4-dione]
was obtained from Cayman Chemical Company (Ann Arbor, MI, USA).
MTT, Hoechst 33342, trypan blue solution, Triton X-100 solution and
the anti-actin antibody (catalog no. A2066) were obtained from
Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). 5,5′,
6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine
iodide (JC-1) was obtained from Invitrogen (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The anti-phosphorylated c-Jun
N-terminal kinase (phospho-JNK) (catalog no. 9255), anti-JNK
(catalog no. 9252), anti-phospho-p38 (catalog no. 9211), and
anti-caspase-3 (catalog no. 9662) antibodies, SB203580 (p38
inhibitor), and U0126 (extracellular signal-regulated kinase (ERK)
inhibitor) were provided by Cell Signaling Technology, Inc.
(Danvers, MA, USA). The anti-B-cell lymphoma-2 (Bcl-2)-associated X
protein (Bax) (catalog no. sc-7480), anti-Bcl-2 (catalog no.
sc-492), anti-caspase-9 (catalog no. sc-7885),
anti-phospho-extracellular-related kinase (ERK) (catalog no.
sc-7383), anti-ERK (catalog no. sc-154), anti-p38 (catalog no.
sc-535), anti-E-cadherin (catalog no. sc-7870), and anti-N-cadherin
(catalog no. sc-7939) antibodies were obtained from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The JNK inhibitor SP600125
was purchased from Selleck Chemicals (Houston, TX, USA). All other
chemicals and reagents used were of analytical grade.
Cell culture
Cisplatin-sensitive and -resistant human ovarian
carcinoma A2780 cells were purchased from Sigma-Aldrich; Merck
KGaA). Paclitaxel-sensitive and -resistant (PR) human ovarian
carcinoma SKOV3 cells were obtained from the MD Anderson Cancer
Center (Houston, TX, USA). Cells were incubated at 37°C in a
humidified atmosphere of 5% CO2 and cultured in
Dulbecco's modified Eagle's medium containing 10% heat-inactivated
fetal bovine serum (both, Gibco; Thermo Fisher Scientific, Inc.),
streptomycin (100 mg/ml), and penicillin (100 U/ml).
Cell viability
To determine the effect of shikonin on cell
viability, A2780, A2780-CR, SKOV3 and SKOV3-PR cells were seeded in
24-well plates at a density of 1×105 cells/ml and
treated with 0.0, 2.5, 5.0, 10.0, 20.0, 30.0 or 40.0 µM shikonin
for 48 h. Cells were treated with shikonin under room-temperature
conditions and subsequently incubated at 37°C. To investigate the
protective effect of mitogen-activated protein kinase (MAPK)
inhibitors against shikonin-induced cell death, A2780-CR cells were
pretreated with 10.0 µM SB203580, U0126, or SP600125 at room
temperature, incubated for 1 h at 37°C, and subsequently treated
with 9.0 µM shikonin and incubated for another 24 h. MTT stock
solution (125 µl; 2 mg/ml) was then added into each well to attain
a total reaction volume of 500 µl. Following incubation for 4 h at
37°C, plates were centrifuged at 200 × g for 10 min at room
temperature and the supernatants were aspirated. The formazan
crystals in each well were dissolved in 300 µl dimethyl sulfoxide,
and absorbance at 540 nm was read on a scanning multi-well
spectrophotometer (25). From the
cell viability at various concentrations of shikonin, the half
maximal inhibitory concentration (IC50) value was
determined.
Colony formation
To examine the inhibitory effect of shikonin on
colony-forming ability, A2780-CR cells were seeded in 60-mm dishes
at a density of 400 cells/dish and treated with 9 µM shikonin on
day 7 of incubation. The medium was changed every 3 days. After 11
days, cells were fixed using 70% ethanol for 10 min at room
temperature and stained with trypan blue solution (0.4%) at room
temperature until blue colonies became visible. Colony-forming
units (CFUs) were considered formed if they were visible to the
naked eye. CFUs were imaged and counted using Image J software
(version 1.47; National Institutes of Health, Bethesda, MD,
USA).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) assay
To visualize apoptotic bodies, A2780-CR cells were
seeded in chamber slides at a density of 1×105 cells/ml,
incubated for 24 h, and then treated with 9 µM shikonin for 24 h.
Slides were then prepared according to the protocol of the DeadEnd
Colorimetric TUNEL system (Promega Corporation, Madison, WI, USA).
Cells were fixed in 10% buffered formalin for 25 min at room
temperature, washed twice with PBS for 5 min at room temperature
and permeabilized by immersing the slides in 0.2% Triton X-100
solution in PBS for 5 min at room temperature. Subsequently, cells
were washed twice with PBS for 5 min at room temperature, covered
with 100 µl equilibration buffer provided in a DeadEnd colorimetric
TUNEL system (Promega Corporation) and equilibrated at room
temperature for 10 min. The rTdT reaction mix (containing 98%
equilibration buffer, 1% biotinylated nucleotide mix and 1% rTdT
enzyme, all provided in a DeadEnd colorimetric TUNEL system) was
added to the sections on a slide and the slide was covered with a
plastic coverslip and incubated at 37°C for 60 min inside a
humidified chamber. The plastic coverslips were removed and the
reactions were terminated by immersing the slides in 2X
saline-sodium citrate (SSC; made by dilution of 20X SSC available
in a DeadEnd colorimetric TUNEL system with deionized water) for 15
min at room temperature. Following washing the slides three times
in PBS for 5 min at room temperature, the slides were immersed in
0.3% H2O2 in PBS for between 3 and 5 min at
room temperature, and washed three times in PBS. Subsequently,
horseradish peroxidase-labeled streptavidin in a DeadEnd
colorimetric TUNEL system (dilution, 1:500 in PBS) was added to
each slide and the slides were incubated for 30 min at room
temperature. Slides were washed three times in PBS and the
chromogen solution (50 µl DAB Substrate 20X buffer, 950 µl
deionized water, 50 µl DAB 20X chromogen and 50 µl
H2O2 20X, all except the deionized water
provided in a DeadEnd colorimetric TUNEL system) were added to each
slide and developed until there was a light brown background. Cells
were washed four times in deionized water and the stained cells
were mounted onto microscope slides in faramount aqueous mounting
medium (Dako; Agilent Technologies, Inc., Santa Clara, CA, USA).
Microscopic images of 5 fields of view per group (magnification,
×20) were captured using the laser scanning microscope 5 PASCAL
program (Carl Zeiss, AG, Oberkochen, Germany) on a confocal
fluorescent microscope.
Nuclear staining with Hoechst
33342
A2780-CR cells were treated with 9 µM shikonin and
incubated for 24 h. Next, 1.5 µl Hoechst 33342 (stock solution, 10
mg/ml), a DNA-specific fluorescent dye, was added to each well and
incubated for 10 min at 37°C. The stained cells were then observed
under a fluorescence microscope, which was equipped with a Cool
SNAP-Pro color digital camera and microscopic images were collected
using a laser scanning microscope 5 PASCAL program. The index of
apoptotic bodies was identified by dividing the number of observed
apoptotic bodies (calculated manually for each microscopic image)
by the number of unmodified nuclei observed on the same image. This
method was also used to visualize the protective effect of MAPK
inhibitors against shikonin-induced apoptosis. In this case, cells
were pretreated with 10 µM SB203580, U0126, or SP600125 and then
treated with 9 µM shikonin 1 h following this, as aforementioned.
They were then stained with Hoechst 33342, as described.
Mitochondrial membrane potential
(Δψm)
For image analysis, following incubation and
treatment with 9 µM shikonin, A2780-CR cells were stained with JC-1
(10 µg/ml) for 15 min at 37°C and affixed to microscope slides in
mounting medium. The intensity of fluorescence emission in the red
and green range was assessed using a confocal microscope and the
Laser Scanning Microscope 5 PASCAL program. This experiment was
validated using FAC-SCalibur flow cytometer (BD Biosciences,
Franklin Lakes, NJ, USA). Shikonin (9 µM)-treated or untreated
A2780-CR cells were harvested, washed, suspended in PBS containing
JC-1 (10 µg/ml), incubated for 15 min at 37°C and analyzed using
flow cytometry. The intensity of emission in the green range was
assessed based on histograms generated by CellQuest Pro 5.1
software (Becton-Dickinson, Bedford, MA, USA) and ModFit LT 3.2
software (Verity Software House, Topsham, ME, USA).
Western blotting
A2780-CR cells were harvested, washed with PBS,
lysed on ice for 30 min using PRO-PREP™ protein extraction solution
(Intron Biotechnology, Inc., Seongnam, South Korea), and then
centrifuged at 13,000 × g for 15 min at 4°C. The supernatants were
collected, and the protein concentrations were measured using the
Bio-Rad protein assay reagent kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). and equalized across samples by addition of
suitable volumes of 5X SDS loading dye and PBS. Aliquots of the
lysates (40 µg of protein) were boiled for 5 min and
electrophoresed in 10%, 12, or 15% SDS-PAGE. The proteins in the
gels were transferred onto nitrocellulose membranes (Bio-Rad
Laboratories, Inc.), which were blocked with 2% fetal bovine serum
for 1 h at room temperature and then incubated with the appropriate
primary antibodies (dilution, 1:1,000) for 1 h at room temperature
and subsequently overnight at 4°C to achieve optimal binding. Actin
or COX4 were used as the loading control for total or mitochondrial
protein fractions, respectively. The membranes were further
incubated with secondary horseradish peroxidase-conjugated goat
anti-rabbit immunoglobulin G (IgG; catalog no. G-21234) or goat
anti-mouse IgG (catalog no. G-21040) antibodies (Pierce; Thermo
Fisher Scientific, Inc.; dilution, 1:5,000) for 1 h at room
temperature. Protein bands were visualized using an enhanced
chemiluminescence western blotting detection kit (GE Healthcare
Life Sciences, Little Chalfont, UK) and then exposed to X-ray
film.
Cell migration assay
A2780-CR cells were seeded in 100-mm dishes at a
density of 1×104 cells/ml and incubated for 24 h. Next,
following treatment with 9 µM shikonin, a line (diameter, 1.5 mm)
was scratched across the bottom of each dish. After 48 h
incubation, this area was observed under a fluorescence
microscope.
Statistical analysis
All measurements were performed in triplicate, and
all values represent the mean ± standard error of the mean. The
results were assessed using a one-way analysis of variance with
Tukey's post-hoc test to analyze the differences. P<0.05 was
considered to indicate a statistically significant difference.
Results
Cytotoxic effect of shikonin on
ovarian cancer cells
Shikonin exerted a potent, dose-dependent cytotoxic
effect on cisplatin-sensitive and -resistant human ovarian
carcinoma A2780 cells (Fig. 1A). The
IC50 of shikonin was 11 µM for A2780 cells and 9 µM for
A2780-CR cells. Thus, cisplatin-sensitive and cisplatin-resistant
cells were highly sensitive to this compound. In addition, the
effect of shikonin on the viability of paclitaxel-sensitive and
-resistant human ovarian carcinoma SKOV3 cells was assessed
(Fig. 1A). SKOV3-PR cells
demonstrated lower sensitivity to shikonin than SKOV3 cells
(IC50 value 16 and 11 µM, respectively). Since
cisplatin-resistant cells exhibited the greatest sensitivity to
shikonin, A2780-CR cells were used in future experiments. To
confirm the cytotoxic effect of shikonin on A2780-CR cells, the
colony-forming ability was determined (Fig. 1B). Treatment with 9 µM shikonin
significantly reduced the colony-forming ability of A2780-CR
compared with control cells. Thus, 9 µM shikonin was used for
subsequent studies of A2780-CR cells.
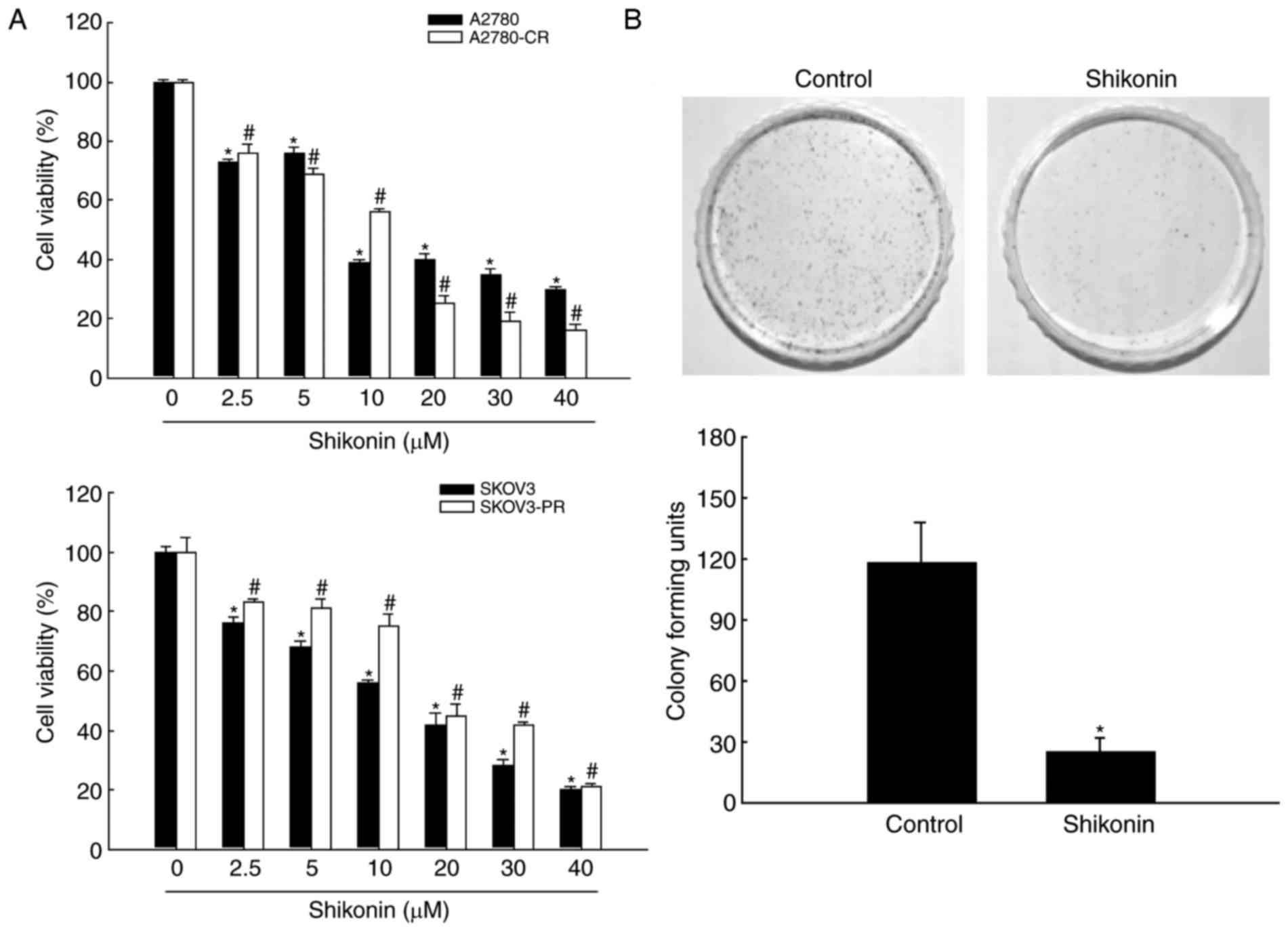 | Figure 1.Shikonin induces death of ovarian
cancer cells. (A) A2780, A2780-CR, SKOV3 and SKOV3-PR cells were
treated with 0.0, 2.5, 5.0, 10.0, 20.0, 30.0 or 40.0 µM shikonin
for 48 h. Cell viability was measured using the MTT assay. (B) For
the colony formation assay, A2780-CR cells were cultured for 11
days and treated with 9 µM shikonin on the seventh day. Colonies
containing >400 cells were counted. *P<0.05 and
#P<0.05, compared with the respective control groups.
CR, cisplatin-resistant; PR, paclitaxel-resistant. |
Induction of apoptosis by shikonin in
cisplatin-resistant ovarian cancer cells
To verify whether the cytotoxic properties of
shikonin were caused by apoptosis, the degree of apoptotic body
formation and the extent of DNA condensation were visualized using
the TUNEL assay and Hoechst 33342 nuclear staining, respectively.
Shikonin-treated cells exhibited a greater number of apoptotic
bodies than control cells (Fig. 2A).
Furthermore, the degree of DNA condensation and nuclear
fragmentation were also significantly higher in the
shikonin-treated group (Fig. 2B).
Involvement of the mitochondrial
pathway in shikonin-induced apoptosis
As indicated by confocal microscopy, the
mitochondria in control cells demonstrated bright red JC-1
fluorescence, signifying Δψm polarization. By contrast,
shikonin-treated cells had an attenuated red mitochondrial
fluorescence and an enhanced green fluorescence, indicating
Δψm depolarization (Fig.
2C). Similar data were obtained by flow cytometry (Fig. 2D), confirming that shikonin decreased
Δψm. The expression of proteins associated with
mitochondria-induced apoptosis were also investigated. Treatment
with shikonin decreased the level of the
pro-survival/anti-apoptotic protein Bcl-2 and increased the level
of pro-apoptotic protein Bax. Shikonin increased the levels of
active (cleaved) caspase-9 and −3 (Fig.
2E). Thus, these data indicate that shikonin induces apoptosis
via the mitochondrial pathway.
The mitochondrial apoptosis pathway is associated
with releasing of cytochrome c from mitochondria into the
cytosol (26); therefore, the levels
of cytochrome c were detected using western blot analysis in
the mitochondrial and cytosolic protein fractions separately
(Fig. 2F). These results confirmed
that this release indeed occurred following shikonin treatment.
Induction of apoptosis by shikonin via
MAPK activation
Shikonin induced the expression of
mitochondria-associated pro-apoptotic proteins; therefore, our
group sought to investigate whether shikonin treatment activated
the MAPK signaling cascade. Previous studies have demonstrated that
shikonin activates the signaling network of ERK and protein kinase
B (11,15). In the present study, western blot
analysis demonstrated that shikonin induced phosphorylation, and
thus activation of, JNK, p38 and ERK in a time-dependent manner
(Fig. 3A), indicating the involvement
of this pathway in shikonin-induced cell death. Next, whether the
specific inhibitors of JNK, p38, and ERK attenuated
shikonin-induced apoptosis was assessed. The MTT assay and Hoechst
33342 nuclear staining indicated that the JNK inhibitor SP600125,
the p38 inhibitor SB203580, and the ERK inhibitor U0126 reduced the
cytotoxic effects of shikonin (Fig. 3B
and C).
Effect of shikonin on the EMT in
cisplatin-resistant ovarian cancer cells
To investigate the effect of shikonin treatment on
the migratory capability of A2780-CR cells, lines (diameter, 1.5
mm) were scratched across the bottom of culture dishes following
treatment with 9 µM shikonin, and the degree of cell migration was
observed after 48 h incubation (Fig.
4A). Whereas the gap width diminished with time in untreated
cells, it increased in shikonin-treated cells. The difference
between the treated and control groups was significant at 48 h.
Thus, shikonin treatment attenuated the migration capability of
A2780-CR cells. To further confirm this result, the expression of
E- and N-cadherin, which are glycoproteins that are required to
initiate and stabilize cell-cell contact, was determined. The
expression of E-cadherin and N-cadherin protein was increased and
decreased, respectively, in the shikonin-treated group compared
with the control group (Fig. 4B).
Discussion
A large proportion of therapeutic failures in the
ovarian cancer treatment are associated with chemoresistance to
cisplatin- or taxol-based therapy (3,4).
Overcoming cisplatin resistance remains difficult (4). The present data study demonstrated that
shikonin reportedly inhibits the proliferation of
cisplatin-resistant human ovarian cancer cells in a dose-dependent
manner. Additionally, we attempted to clarify the molecular
mechanisms by which shikonin induces apoptosis in A2780-CR
cells.
In the present study, shikonin exhibited a greater
cytotoxic effect in cisplatin-resistant cells compared with
parental ovarian cancer A2780 cells, as evidenced by the respective
IC50 values. Considering the antioxidant effect of
shikonin (11), the difference in
sensitivity to shikonin between A2780 and A2780-CR cells may be due
to its reactive oxygen species (ROS)-scavenging properties.
Parental and cisplatin-resistant ovarian cancer cells have
different redox statuses, with basic level of ROS in A2780-CR cells
was higher than that in parental A2780 cells, with shikonin
reducing the enhanced ROS levels in A2780-CR cells (data not
shown). This disorder in redox balance of A2780-CR cells may
provide further cellular stress and lead to cytotoxicity. This
assumption is consistent with the results of other studies, which
demonstrate an association between chemotherapy resistance and
enhanced ROS levels in different cancer cells (27–29).
Apoptosis is a type of physiological cell death that is associated
with specific morphological and metabolic changes, massive nuclear
fragmentation, chromatin condensation and the activation of
specific proteins (30). Therefore,
the stimulation of apoptosis remains important in cancer
eradication. Data generated in the present study indicated that the
numbers of apoptotic bodies and TUNEL-positive cells increased
following shikonin treatment, representing the cytotoxic effect of
this compound that is mediated through the apoptotic pathway
(Fig. 2).
Mitochondria and cytochrome c, a key electron
carrier in the electron transport chain, have a crucial function in
apoptosis induced by various stimuli (26). During the apoptotic process the Δψm is
destroyed, leading to the leakage of cytochrome c from mitochondria
into the cytosol. This process is regulated by the
pro-survival/anti-apoptotic protein Bcl-2 and pro-apoptotic protein
Bax, and results in the activation and heptamerization of the
adaptor molecule apoptotic protease-activating factor 1 (Apaf-1),
forming the apoptosome complex. Apaf-1 then cleaves the preform of
caspase-9 to initiate the caspase cascade, resulting in apoptosis
(31). Therefore,
mitochondria-mediated apoptosis involves upregulation of Bax,
downregulation of Bcl-2, disruption of mitochondrial membrane
permeability, leakage of cytochrome c into the cytosol, the
formation of apoptotic bodies, and initiation of the caspase
cascade. Upon shikonin treatment, mitochondrial membrane
depolarization was observed. The level of Bcl-2 was decreased and
that of Bax was increased. These changes in protein expression
altered the mitochondria membrane permeability and caused release
of cytochrome c into the cytosol, inducing apoptosis via
caspase-9 and −3 cleavage (Fig. 2).
This indicates that shikonin initiates mitochondria-mediated
apoptosis.
Multiple studies have demonstrated that the
cytotoxic proprieties of the majority of anticancer drugs are
associated with the regulation of MAPK-family protein activity
(32–34). The present study also investigated the
involvement of MAPKs in shikonin-induced apoptosis. Levels of
phosphorylated JNK, p38, and ERK were increased following shikonin
treatment in a time-dependent manner, and specific inhibitors of
these kinases attenuated the decrease in A2780-CR cell viability
caused by shikonin (Fig. 3). These
results suggested that MAPK-mediated pathways are activated in
shikonin-induced apoptosis of A2780-CR cells.
EMT is associated with tumor metastasis (35,36). EMT
promotes cancer cell migration and invasion and has a crucial
function in the invasion and metastasis of epithelial cancer
specifically (35,36). EMT is associated with the
downregulation of epithelial markers (including E-cadherin and
β-catenin) and upregulation of interstitial markers (including
N-cadherin and fibronectin). Alterations in N-cadherin levels, as
well as the associated EMT E-cadherin/N-cadherin switching, are
considered to be risk markers for tumor progression and invasion
(37). These results have been
previously assessed for gastric, prostate and oral carcinoma cell
lines (38,39). E-cadherin/N-cadherin switching is
essential for increased cell motility, but not for the
morphological alterations that occur in the EMT process (37). There are limited reports regarding EMT
in ovarian cancer. Ovarian cancer cells easily invade and diffuse
in the pelvic cavity, and this results in the high mortality rate
of ovarian cancer (2,3,36). EMT is
an early event in the invasion and metastasis of cancer cells,
including ovarian cancer cells (36,40). The
present study revealed that the migration of A2780-CR cells was
significantly decreased upon shikonin treatment, indicating that
EMT decreases cell polarity and normal cell-cell adhesion, induces
the loss of epithelial characteristics and improves the invasion of
the cells. The level of E-cadherin (an epithelial marker) was
enhanced, while the level of N-cadherin (an interstitial marker)
was decreased following shikonin treatment, indicating that
shikonin attenuates EMT.
Taken together, these results suggested that
A2780-CR ovarian cancer cells are highly sensitive to shikonin,
which activates apoptosis and attenuates EMT. Therefore, shikonin
should be considered a promising chemotherapeutic candidate to
overcome cisplatin resistance in ovarian cancer.
Acknowledgements
The present study was supported by grants from
National Research Foundation of Korea funded by the Korean
Government (grant nos. NRF-2016R1A2B4007934 and
NRF-2015M2A2A7061657).
References
|
1
|
Liu X, Gao Y, Lu Y, Zhang J, Li L and Yin
F: Oncogenes associated with drug resistance in ovarian cancer. J
Cancer Res Clin Oncol. 141:381–395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Suh DH, Kim M, Kim HJ, Lee KH and Kim JW:
Major clinical research advances in gynecologic cancer in 2015. J
Gynecol Oncol. 27:e532016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
American Cancer Society, . Chemotherapy
for ovarian cancer. http://www.cancer.org/cancer/ovariancancer/detailedguide/ovarian-cancer-treating-chemotherapy2014
February 4–2016
|
|
4
|
Huang H, Tong TT, Yau LF, Chen CY, Mi JN,
Wang JR and Jiang ZH: LC-MS based sphingolipidomic study on A2780
human ovarian cancer cell line and its taxol-resistant strain. Sci
Rep. 6:346842016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galluzzi L, Senovilla L, Vitale I, Michels
J, Martins I, Kepp O, Castedo M and Kroemer G: Molecular mechanisms
of cisplatin resistance. Oncogene. 31:1869–1883. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hiss D: Optimizing molecular-targeted
therapies in ovarian cancer: The renewed surge of interest in
ovarian cancer biomarkers and cell signaling pathways. J Oncol.
2012:7379812012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kim H, Park GS, Lee JE and Kim JH: A
leukotriene B4 receptor-2 is associated with paclitaxel resistance
in MCF-7/DOX breast cancer cells. Br J Cancer. 109:351–359. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim DK, Seo EJ, Choi EJ, Lee SI, Kwon YW,
Jang IH, Kim SC, Kim KH, Suh DS, Seong-Jang K, et al: Crucial role
of HMGA1 in the self-renewal and drug resistance of ovarian cancer
stem cells. Exp Mol Med. 48:e2552016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang WR, Zhang Y and Tang X: Shikonin
inhibits the proliferation of human lens epithelial cells by
inducing apoptosis through ROS and caspase-dependent pathway.
Molecules. 19:7785–7797. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu T, Ma C, Yang L, Wang W, Sui X, Zhao C
and Zu Y: Optimization of shikonin homogenate extraction from
Arnebia euchroma using response surface methodology. Molecules.
18:466–481. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Andújar I, Ríos JL, Giner RM and Recio MC:
Pharmacological properties of shikonin - A review of literature
since 2002. Planta Med. 79:1685–1697. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Han CT, Kim MJ, Moon SH, Jeon YR, Hwang
JS, Nam C, Park CW, Lee SH, Na JB, Park CS, et al: Acute and 28-day
subacute toxicity studies of hexane extracts of the roots of
Lithospermum erythrorhizon in Sprague-Dawley rats. Toxicol Res.
31:403–414. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
He G, He G, Zhou R, Pi Z, Zhu T, Jiang L
and Xie Y: Enhancement of cisplatin-induced colon cancer cells
apoptosis by shikonin, a natural inducer of ROS in vitro and in
vivo. Biochem Biophys Res Commun. 469:1075–1082. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jeung YJ, Kim HG, Ahn J, Lee HJ, Lee SB,
Won M, Jung CR, Im JY, Kim BK, Park SK, et al: Shikonin induces
apoptosis of lung cancer cells via activation of FOXO3a/EGR1/SIRT1
signaling antagonized by p300. Biochim Biophys Acta.
1863:2584–2593. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jing H, Sun W, Fan J, Zhang Y, Yang J, Jia
J, Li J, Guo J, Luo S and Zheng Y: Shikonin induces apoptosis of
HaCaT cells via the mitochondrial, Erk and Akt pathways. Mol Med
Rep. 13:3009–3016. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ko H, Kim SJ, Shim SH, Chang H and Ha CH:
Shikonin induces apoptotic cell death via regulation of p53 and
Nrf2 in AGS human stomach carcinoma cells. Biomol Ther (Seoul).
24:501–509. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lu D, Qian J, Li W, Feng Q, Pan S and
Zhang S: β-hydroxyisovaleryl-shikonin induces human cervical cancer
cell apoptosis via PI3K/AKT/mTOR signaling. Oncol Lett.
10:3434–3442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tang X, Zhang C, Wei J, Fang Y, Zhao R and
Yu J: Apoptosis is induced by shikonin through the mitochondrial
signaling pathway. Mol Med Rep. 13:3668–3674. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Trivedi R, Müller GA, Rathore MS, Mishra
DP and Dihazi H: Anti-leukemic activity of shikonin: Role of ERP57
in shikonin induced apoptosis in acute myeloid leukemia. Cell
Physiol Biochem. 39:604–616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei Y, Li M, Cui S, Wang D, Zhang CY, Zen
K and Li L: Shikonin inhibits the proliferation of human breast
cancer cells by reducing tumor-derived exosomes. Molecules.
21(pii): E7772016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Christiansen JJ and Rajasekaran AK:
Reassessing epithelial to mesenchymal transition as a prerequisite
for carcinoma invasion and metastasis. Cancer Res. 66:8319–8326.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yin SY, Peng AP, Huang LT, Wang YT, Lan CW
and Yang NS: The phytochemical shikonin stimulates
epithelial-mesenchymal transition (EMT) in skin wound healing. Evid
Based Complement Alternat Med. 2013:2627962013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Thakur R, Trivedi R, Rastogi N, Singh M
and Mishra DP: Inhibition of STAT3, FAK and Src mediated signaling
reduces cancer stem cell load, tumorigenic potential and metastasis
in breast cancer. Sci Rep. 5:101942015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang FY, Hu Y, Que ZY, Wang P, Liu YH,
Wang ZH and Xue YX: Shikonin inhibits the migration and invasion of
human glioblastoma cells by targeting phosphorylated β-catenin and
phosphorylated PI3K/Akt: A potential mechanism for the anti-glioma
efficacy of a traditional Chinese herbal medicine. Int J Mol Sci.
16:23823–23848. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Carmichael J, DeGraff WG, Gazdar AF, Minna
JD and Mitchell JB: Evaluation of a tetrazolium-based semiautomated
colorimetric assay: Assessment of chemosensitivity testing. Cancer
Res. 47:936–942. 1987.PubMed/NCBI
|
|
26
|
Garrido C, Galluzzi L, Brunet M, Puig PE,
Didelot C and Kroemer G: Mechanisms of cytochrome c release from
mitochondria. Cell Death Differ. 13:1423–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bi W, Wang Y, Sun G, Zhang X, Wei Y, Li L
and Wang X: Paclitaxel-resistant HeLa cells have up-regulated
levels of reactive oxygen species and increased expression of taxol
resistance gene 1. Pak J Pharm Sci. 27:871–878. 2014.PubMed/NCBI
|
|
28
|
Okon IS and Zou MH: Mitochondrial ROS and
cancer drug resistance: Implications for therapy. Pharmacol Res.
100:170–174. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wangpaichitr M, Wu C, Li YY, Nguyen DJM,
Kandemir H, Shah S, Chen S, Feun LG, Prince JS, Kuo MT and Savaraj
N: Exploiting ROS and metabolic differences to kill cisplatin
resistant lung cancer. Oncotarget. 8:49275–49292. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Peitsch MC, Polzar B, Stephan H, Crompton
T, MacDonald HR, Mannherz HG and Tschopp J: Characterization of the
endogenous deoxyribonuclease involved in nuclear DNA degradation
during apoptosis (programmed cell death). EMBO J. 12:371–377.
1993.PubMed/NCBI
|
|
31
|
Desagher S and Martinou JC: Mitochondria
as the central control point of apoptosis. Trends Cell Biol.
10:369–377. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun Y, Liu WZ, Liu T, Feng X, Yang N and
Zhou HF: Signaling pathway of MAPK/ERK in cell proliferation,
differentiation, migration, senescence and apoptosis. J Recept
Signal Transduct Res. 35:600–604. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Habli Z, Toumieh G, Fatfat M, Rahal ON and
Gali-Muhtasib H: Emerging cytotoxic alkaloids in the battle against
cancer: Overview of molecular mechanisms. Molecules. 22(pii):
E2502017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xing K and Lisong S: Molecular targeted
therapy of cancer: The progress and future prospect. Front Lab Med.
1:69–75. 2017. View Article : Google Scholar
|
|
35
|
Chen ZS, Ling DJ, Zhang YD, Feng JX, Zhang
XY and Shi TS: Octamer-binding protein 4 affects the cell biology
and phenotypic transition of lung cancer cells involving
β-catenin/E-cadherin complex degradation. Mol Med Rep.
11:1851–1858. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stewart CJ and McCluggage WG:
Epithelial-mesenchymal transition in carcinomas of the female
genital tract. Histopathology. 62:31–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maeda M, Johnson KR and Wheelock MJ:
Cadherin switching: Essential for behavioral but not morphological
changes during an epithelium-to-mesenchyme transition. J Cell Sci.
118:873–887. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
DI Domenico M, Pierantoni GM, Feola A,
Esposito F, Laino L, DE Rosa A, Rullo R, Mazzotta M, Martano M,
Sanguedolce F, et al: Prognostic significance of N-Cadherin
expression in oral squamous cell carcinoma. Anticancer Res.
31:4211–4218. 2011.PubMed/NCBI
|
|
39
|
Liu LK, Jiang XY, Zhou XX, Wang DM, Song
XL and Jiang HB: Up-regulation of vimentin and aberrant expression
of E-cadherin/beta-catenin complex in oral squamous cell
carcinomas: Correlation with the clinicopathological features and
patient outcome. Mod Pathol. 23:213–224. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Powell CD, Paullin TR, Aoisa C, Menzie CJ,
Ubaldini A and Westerheide SD: The heat shock transcription factor
HSF1 induces ovarian cancer epithelial-mesenchymal transition in a
3D spheroid growth model. PLoS One. 11:e01683892016. View Article : Google Scholar : PubMed/NCBI
|

















