Introduction
Among the most common types of cancer in males,
prostate cancer (PC) is the second leading cause of
cancer-associated mortality in the USA in 2011 (1). Currently, the first line of treatment
for prostate cancer is surgery, however, it is not so ideal
following the operation. Gene-targeted therapy has provided a new
perspective on cancer research, and the potential of tumor protein
p53 (p53) in cancer treatment has become an increasingly prominent
research theme (2–4). It has been well established that p53 is
a tumor suppressor gene and that p53 mutations occur in 50% tumor
cells (5). Upon DNA-damage by
radiation or other factors, p53 activates p21, which functions in
DNA repair (6). At the S-stage of the
cell cycle, damaged DNA cannot be repaired and p53 regulates cell
apoptosis (7,8). Previous studies have demonstrated that
the focal adhesion kinase (FAK)/Src proto-oncogene, non-receptor
tyrosine kinase (Src) pathway serves an important role in cell
proliferation, differentiation, migration and survival, and is
closely associated with the development, metastasis and prognosis
of various types of cancer (9,10).
However, it remains unknown how p53 functions in the proliferation
of PC cells, and whether the FAK/Src pathway is activated in the
process. Therefore, the expression of p53 in PC cells its mechanism
in the occurrence and development of PC was analyzed in the present
study.
Materials and methods
Cell culture
RWPE-1 normal prostate cell line and the PC cell
line, DU145, were purchased from Shanghai Institutes for Biological
Sciences, Chinese Academy of Sciences (Shanghai, China). The cells
were cultured in Dulbecco's modified Eagle's medium (DMEM; Hyclone;
GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal calf serum (FBS; Zhejiang Tianhang Biotechnology Co., Ltd.,
Zhejiang, China), 100 U/ml penicillin (Beyotime Institute of
Biotechnology, Shanghai, China) and 100 µg/ml streptomycin
(Beyotime Institute of Biotechnology), at 37°C in 5%
CO2. The cells were passaged every 3–5 days.
Cell transfection
P53 small interfering RNA was designed and
synthesized by GeneChem Inc. (Daejeon, Korea; Gene ID: GCD950481;
sequence, 5′-GCAUGAACCGGAGGCCCAU-3′) and the control siRNA (cat no.
D6145) was purchased from Takara Biotechnology Co., Ltd. (Dalian,
China). The DU145 cells (1×106 cells) were seeded into a
6-well plate 24 h prior to transfection in the logarithmic growth
period, and the DMEM culture medium (Hyclone; GE Healthcare Life
Sciences) was discarded and replaced 2 h prior to transfection. The
cells were then transfected with 5 µl siRNA and 5 µl
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). In the control group, DU145 cells were
transfected with control siRNA under the same experimental
conditions. A total of 4 h later, the medium was replaced and
subsequent experiments were performed. PD184352 (25 µmol/l) and
SP600125 (25 µmol/l) were purchased from Meiyan Biological
Technology Co., Ltd. (Shanghai, China) and applied 24, 48 and 72 h
post-transfection.
Western blotting
The cells were washed twice with PBS and 100 µl 2×
Laemmli sample buffer (Kemin Biological Technology Co., Ltd.,
Shanghai, China) was added to lyse the cells. The samples were
centrifuged for 30 min at 1,800 × g and 4°C, and the supernatants
were collected. Protein concentration was determined using a BCA
bicinchoninic acid assay (Beyotime Institute of Biotechnology),
according to the manufacturer's protocol. The absorbance was
measured at 450 nm using a Benchmark microplate reader (Bio-Rad,
Hercules, CA, USA) to determine protein concentration. The protein
(40 µg) was mixed with SDS loading buffer (Beyotime Institute of
Biotechnology) and heated at 97°C for 3 min. Following protein
separation by 12% SDS-PAGE, the proteins (20 µg per lane) were
transferred into polyvinylidene fluoride membranes. The membranes
were blocked in skimmed milk at room temperature for 2 h, prior to
being washed thrice with TBS containing 0.3% Tween (TBST). The
membranes were incubated with mouse anti-human p53 primary antibody
(cat no. MS-186-B; dilution, 1:500; Gibco; Thermo Fisher
Scientific, Inc.), FAK antibody (cat no. ab40794; dilution, 1:500;
Abcam, Cambridge, UK), Phospho (P)-FAK (Tyr925) polyclonal antibody
(cat no. PA5-17733; dilution, 1:400; Thermo Fisher Scientific,
Inc.), P-FAK (Tyr577) polyclonal antibody (cat no. PA5-37706;
dilution, 1:500; Thermo Fisher Scientific, Inc.), P-FAK (Tyr397)
polyclonal antibody (cat no. 44-624G; dilution, 1:500; Thermo
Fisher Scientific, Inc.), extracellular signal-regulated kinase
(ERK; cat no. ab17942; dilution, 1:400; Abcam), phosphorylated
(p)-ERK (cat no. sc-7383; dilution, 1:400; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and β-actin (cat no. BM0627;
dilution, 1:2,000; Wuhan Boster Biological Technology, Ltd., Wuhan,
China) overnight at 4°C. Subsequent to another 3 washes in TBST, a
horseradish peroxidase-conjugated goat anti-mouse secondary
antibody (cat no. BA1050; dilution, 1:10,000; Wuhan Boster
Biological Technology, Ltd.) was incubated with the membranes at
room temperature for 2 h. Following 3 more washes in TBST, the
protein bands were visualized using an enhanced chemiluminescence
kit (Weipu Jishu, Shanghai, China; http://www.weipujishu.com/), according to the
manufacturer's protocol. Results were analyzed using Image J
software (version 1.38; National Institutes of Health, Bethesda,
MD, USA) and the relative expression of target protein was
calculated.
Cell counting kit-8 (CCK-8) assay
The cell concentration was adjusted to
1×105 cells/ml. A total of 100 µl cell suspension was
added per well in a 96-well plate and incubated at 37°C and in 5%
CO2 for 24 h. Next, after 24 h, 100 µl 10% CCK-8 reagent
(Qianjian Green Sea Treasure Biological Technology Co., Ltd.,
Shanghai, China) was added to each well. After 2 h, 10 µl 0.1 M HCl
was added to each well and the absorbance values were read
immediately at 450 nm.
Invasion assay
A total of 48 h after transfection, the DU145 cells
(1×106 cells) cultured in DMEM (Hyclone; GE Healthcare
Life Sciences) were plated into the upper chamber (1×105
cells) of Transwell plates (Mingyangkehua Biological Technology,
Co., Ltd, Beijing, China) coated with Matrigel, and 200 µl DMEM
medium containing 10% fetal bovine serum was added into the lower
chambers and cultured for 24 h. Hoechst 33258 (5 µg/ml; Shanghai
Yanhui Biotechnology Co., Ltd., Shanghai, China) was incubated at
room temperature with the cells for 2 min. Using an inverted light
microscope (magnification, ×40; TS100; Nikon, Tokyo, Japan), the
cells penetrating the membrane in 10 randomly selected fields of
view were counted. The following formula was used to calculate the
rate of migration inhibition: rate of migration inhibition=(number
migrated cells in the control group-number migrated cells in the
experimental group)/number migrated cells in the control group
×100.
Wound-healing assay
A total of 72 h after transfection, the DU145 cells
cultured in DMEM (Hyclone; GE Healthcare Life Sciences) were plated
in a 6-well plate at 1×105 cells/ml. When confluence
reached 100%, a 10-µl pipette tip was used to wound the cell layer.
The plate was washed 3 times with PBS to remove the cell debris
prior to adding fresh medium for 48 h. Using an inverted light
microscope (magnification, ×40; TS100; Nikon, Tokyo, Japan), the
cells were photographed at 0, 24 and 48 h.
Cell adhesion assay
Transfected cells were placed in a 96-well plate and
incubated with blocking buffer (0.5% bovine serum albumin, PBS pH
7.4 and 0.05% Tween 20; Biogot Technology Co., Ltd., Nanjing,
China) at 37°C in 5% CO2. The cells were washed twice in
PBS prior to fixation in 4% paraformaldehyde at 37°C for 30 min and
a 10 min incubation at 37°C with 0.1% crystal violet. The cells
were then treated with 0.05% Tween-20 for 30 min at room
temperature, and the absorbance values were read at 595 nm.
Statistical analysis
All data are expressed as the mean ± standard
deviation and were analyzed using SPSS 19.0 software (IBM Corp.,
Armonk, NY, USA). Multiple-group comparisons were performed using
one-way analysis of variance and the Least Significant Difference
test. Comparisons between 2 groups were performed using the
unpaired Student's t-test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Protein expression level of p53 in
prostate cancer cells
The expression of p53 was analyzed in normal RWPE-1
cells and PC DU14 cells by western blotting. It was demonstrated
that the p53 protein expression level was significantly increased
in DU145 cells compared with normal control cells (P<0.01;
Fig. 1).
p53 expression is suppressed following
interference using si-p53 in DU145 cells
To assess the efficiency of si-p53 transfection, the
cell viability and protein expression level of p53 were analyzed
24, 48 and 72 h after transfection with 0, 1, 2, 3, 4, 5 and 6 µl
si-p53. It was revealed that 24 h after treatment with 5 µl si-p53,
the viability of DU145 cells declined remarkably compared with
untransfected cells (P<0.01). The result was quite similar
following treatment with 6 µl si-p53 (P>0.05; Fig. 2A). Western blotting revealed that 24 h
after a 5-µl si-p5 treatment, the expression level of p53 was
significantly reduced compared with untransfected cells (P<0.01;
Fig. 2B).
Effect of si-p53 interference on the
proliferation, invasion and adhesion abilities of DU145 cells
The proliferation, migration and adhesion abilities
of cells transfected with si-p53 were significantly reduced
compared with untransfected cells (P<0.01; Fig. 3).
Effect of si-p53 interference on the
FAK-Src signaling pathway
To study the mechanism behind the effect of p53 on
the proliferation, invasion and adhesion abilities of PC cells, the
effect of si-p53 treatment on FAK/Src/mitogen-activated protein
kinase (MAPK) pathway, which is closely associated with cell
adhesion and motility, was investigated. The results revealed that
si-p53 treatment resulted in a significant increase in the
expression levels of FAK, p-FAK, Src, p-ERK and p-janus kinase
(JNK) compared with untreated cells (P<0.05; Fig. 4).
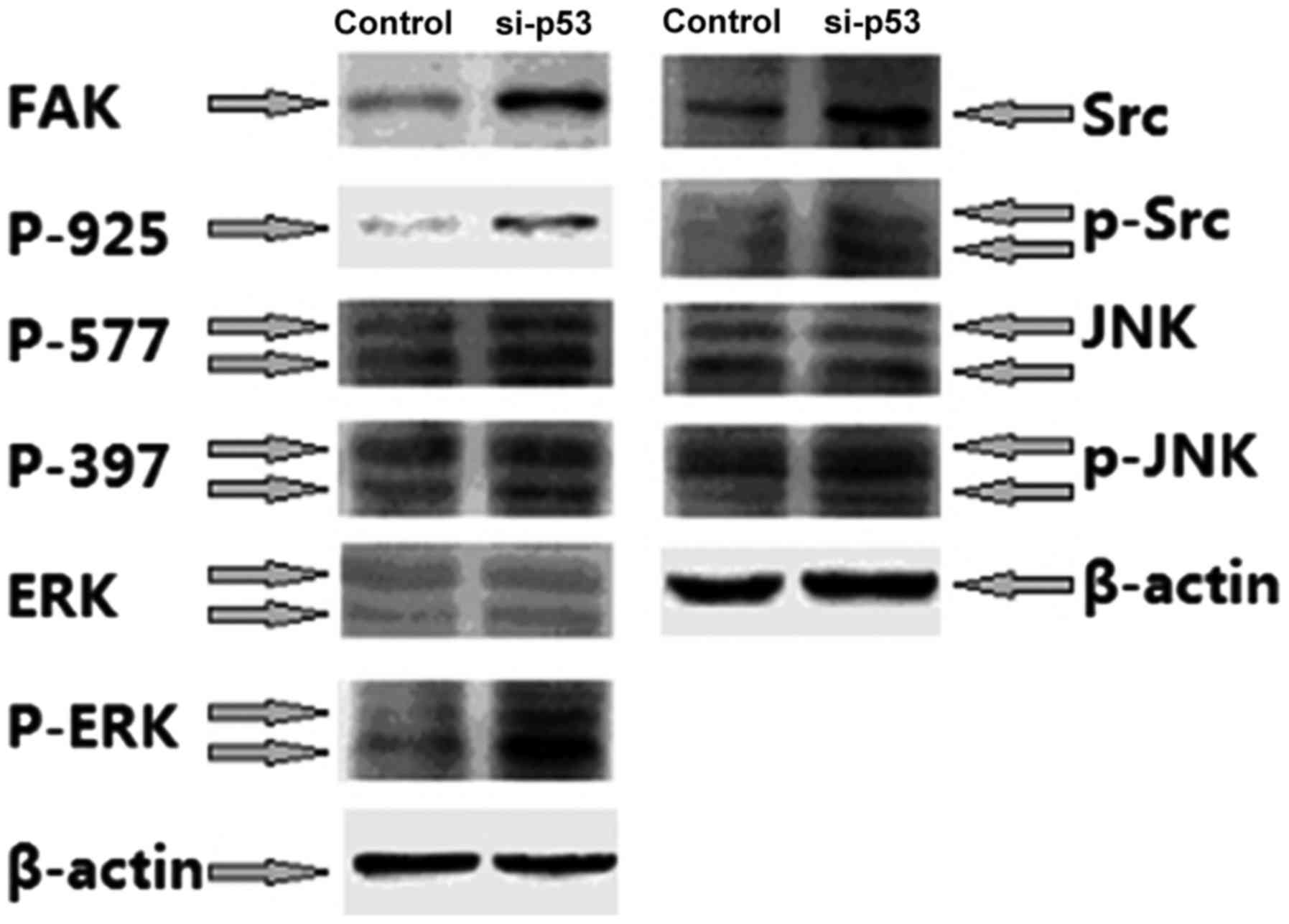 | Figure 4.Western blotting was used to
demonstrate that FAK-Src signaling is activated by si-p53. FAK,
focal adhesion kinase; Src, Src proto-oncogene, non-receptor
tyrosine kinase; si-p53, small interfering RNA targeting p53; p53,
tumor protein p53; ERK, extracellular-regulated kinase; p-,
phosphorylated; JNK, janus kinase; P-925, Phospho-FAK Tyr925;
P-577, Phospho-FAK Tyr577; P-397, Phospho-FAK Tyr397. |
PD184352 and SP600125 treatment alters
the proliferation, migration and adhesion abilities of DU145
cells
Preliminary experiments indicated that 10 ìM
PD184352 and SP600125 inhibitors reduced the proliferation,
invasion and adhesion abilities of PC cells compared with untreated
cells (Fig. 5).
Discussion
In the past ten years, prostate cancer has been
revealed to be the most common type of tumor among males globally
(11). The main cause of prostate
cancer is unknown, however, it has been associated with various
factors, including heredity, environment and sex hormone levels
(10). Although hormonotherapy has
progressed, it effectiveness is limited due to hormone
desensitization (12). Consequently,
the discovery of novel therapeutic targets is urgently
required.
As a negative regulator of cell growth, mutations in
p53 result in dysregulation of the cell cycle, causing abnormal
proliferation and malignant transformation (13). Research has demonstrated that abnormal
expression of p53 in tumor tissue is closely associated with tumor
lymph node metastasis, clinical stage and clinicopathology
(14–16). However, it remains unclear how
abnormal expression of p53 affects the malignant proliferation,
metastasis and differentiation of prostate cancer cells.
The present study demonstrated that p53 interference
may inhibit the proliferation, migration, adhesion and migratory
abilities of DU145 cells. The specific mechanism of these effects
of p53 was also investigated in the present study. FAK is a major
focal ohesion that serves an important role in cell survival and
migration (17). Following external
activation of the FAK pathway, auto-phosphorylation of Tyr397
occurs, followed by the formation of FAK/Src composite, causing the
phosphorylation of Tyr925 and the activation of Ras and MAPK
proteins (18). The results of the
present study indicate that interference with p53 expression causes
FAK/Src pathway activation and increased JNK and ERK
phosphorylation levels. Inhibition of ERK and JNK activity by
PD184352 and SP600125 decreased the proliferation, migration and
adhesion abilities of DU145 cells, implying that p53 controls these
PC-cell functions through the phosphorylation JNK/ERK.
To conclude, high protein expression levels of p53
in PC cells was closely associated with cell proliferation,
migration and adhesion abilities, in which the FAK-Src-MAPK pathway
serves a crucial role. p53 may be an effective anti-cancer target
for suppression of the malignant proliferation of PC cells, and for
prostate cancer gene therapy.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
All data generated or analysed during this study are
included in this published article.
Authors' contributions
JKW and JQZ conceived the study design and drafted
the manuscript. JZ participated in the study design and
coordination. All authors revised and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Zhengzhou Central Hospital Affiliated to Zhengzhou
University (Zhengzhou, China). Written informed consent was gained
from all participants.
Consent for publication
All subjects participating in the present study have
provided consent for the publication of this data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Jiang T, Zhou C, Gu J, Liu Y, Zhao L, Li
W, Wang G, Li Y and Cai L: Enhanced therapeutic effect of cisplatin
on the prostate cancer in tumor-bearing mice by transfecting the
attenuated Salmonella carrying a plasmid co-expressing p53 gene and
mdm2 siRNA. Cancer Lett. 337:133–142. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nande R, Greco A, Gossman MS, Lopez JP,
Claudio L, Salvatore M, Brunetti A, Denvir J, Howard CM and Claudio
PP: Microbubble-assisted p53, RB, and p130 gene transfer in
combination with radiation therapy in prostate cancer. Curr Gene
Ther. 13:163–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang Y, Zhang YX, Kong CZ, Zhang Z and Zhu
YY: Loss of P53 facilitates invasion and metastasis of prostate
cancer cells. Mol Cell Biochem. 384:121–127. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sivoňová MK, Vilèková M, Kliment J,
Mahmood S, Jureèeková J, Dušenková S, Waczulíková I, Slezák P and
Dobrota D: Association of p53 and p21 polymorphisms with prostate
cancer. Biomed Rep. 3:707–714. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gu J, Wang B, Liu Y, Zhong L, Tang Y, Guo
H, Jiang T, Wang L, Li Y and Cai L: Murine double minute 2 siRNA
and wild-type p53 gene therapy interact positively with zinc on
prostate tumours in vitro and in vivo. Eur J Cancer. 50:1184–1194.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xie Q, Lu YY and Yi HF: Expression and
clinical significance of Caspase-3 and P53 in gastric cancer. Chin
J Gastroenterol Hepatol. 23:1287–1289. 2014.
|
|
8
|
Ha US, Bae WJ, Kim SJ, Yoon BI, Hong SH,
Lee JY, Hwang TK, Hwang SY, Wang Z and Kim SW: Anthocyanin induces
apoptosis of DU-145 cells in vitro and inhibits xenograft growth of
prostate cancer. Yonsei Med J. 56:16–23. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Teh BS and Ishiyama H: Hypofractionated
radiotherapy for prostate cancer. Lancet Oncol. 13:5–6. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Henríquez-Hernández LA, Valenciano A,
Foro-Arnalot P, Álvarez-Cubero MJ, Cozar JM, Suárez-Novo JF,
Castells-Esteve M, Fernández-Gonzalo P, De-Paula-Carranza B, Ferrer
M, et al: Genetic variations in genes involved in testosterone
metabolism are associated with prostate cancer progression: A
Spanish multicenter study. Urol Oncol. 33:331.e1–e7. 2015.
View Article : Google Scholar
|
|
11
|
Thomsen FB, Folkvaljon Y, Garmo H,
Robinson D, Loeb S, Ingvar C, Lambe M and Stattin P: Risk of
malignant melanoma in men with prostate cancer: Nationwide,
population-based cohort study. Int J Cancer. 138:2154–2160. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Alva A and Hussain M: Optimal
pharmacotherapeutic management of hormone-sensitive metastatic
prostate cancer. Drugs. 73:1517–1524. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Meek DW: Regulation of the p53 response
and its relationship to cancer. Biochem J. 469:325–346. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh RD, Patel KR and Patel PS: p53
mutation spectrum and its role in prognosis of oral cancer
patients: A study from Gujarat, West India. Mutat Res. 783:15–26.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang G, Li Z, Lin XM, Zhang JH, Cui Y and
Zhao X: Expression and significance of PTEN, S100A4 and p53 protein
in breast invasive ductal carcinoma. Guangdong Med J. 35:3510–3512.
2014.
|
|
16
|
Cai S and Han K: Research on expression
and importance of p53, p16 and VEGF-C in cervical cancer. J Gynecol
Obstet Biol Reprod (Paris). 44:639–645. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Thakur R, Trivedi R, Rastogi N, Singh M
and Mishra DP: Inhibition of STAT3, FAK and Src mediated signaling
reduces cancer stem cell load, tumorigenic potential and metastasis
in breast cancer. Sci Rep. 5:101942015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang H, Zhang SH, He HW, Zhang CX, Yu DK
and Shao RG: Downregulation of G3BPs inhibits the growth, migration
and invasion of human lung carcinoma H1299 cells by suppressing the
Src/FAK-associated signaling pathway. Cancer Gene Ther. 20:622–629.
2013. View Article : Google Scholar : PubMed/NCBI
|


















