Introduction
Erythropoietin-producing hepatocellular (Eph)
receptors and their ligand ephrins are membrane-bound molecules,
which play crucial roles in cell-cell communication (1). Eph receptors constitute the largest
family of receptor tyrosine kinases, and the Eph receptors and
ephrins are divided into subclasses A and B based on their
structure and binding affinities for each other (2). In humans, 16 Eph receptors, including
10 EphA and 6 EphB, and 9 ephrins, including 6 ephrin-A and 3
ephrin-B, have been identified (3).
EphA receptors bind to ephrin-As, and EphB receptors bind to
ephrin-Bs, with a few exceptions (4). As both Eph receptors and ephrins are
expressed on the cell surface, it is necessary for cells to contact
each other to generate Eph/ephrin signals. When they bind each
other, both Eph receptors and ephrins emit signals for regulation
of a wide range of cell phenotypes, including cell morphology,
motility, adhesion, and invasion (4,5).
The roles of the Eph/ephrin system in the
development and progression of cancer vary among the molecules and
cancer types. Upregulated expression of Eph receptors and ephrin
has been observed in many types of cancers, and their
downregulation in cancers has also been reported (5). For example, higher expression of EPHB2
has been reported to be associated with higher stage of cervical
cancer (6) and colorectal cancer
(7), and higher expression of EPHB4
with poor prognosis of ovarian cancer (8) and colon cancer (7). On the other hand, downregulation of
EPHA1 in skin cancer (9) and
colorectal cancer (10), and EphB
receptors in colorectal cancer (11)
and Ephrin-A5 (EFNA5) in glioblastoma (12) have been reported.
Recently we have found that the expression levels of
EPHB2 are significantly higher in mouse cutaneous squamous cell
carcinoma (cSCC) obtained by treating murine dorsal skin using a
two-stage carcinogenesis protocol with
7.12-dimethylbenz(a)anthracene (DMBA) and
12-O-tetradecanoylphorbol-13-acetate (TPA) than in normal skin
(13). In addition, genomic DNA near
the EPHB2 gene has been shown to be hypomethylated in skin cancer
tissues (13). Overexpression of
EPHB2 has also been observed in human cSCC (14), and knockdown of EPHB2 in cSCC cells
derived from cSCC surgical specimens resulted in suppression of
cSCC cell growth in vitro and in vivo (14). It has also been demonstrated that
knockdown of EPHB2 suppressed the expression of genes involved in
cell viability, migration, and invasion (14). These findings indicate that EPHB2
possesses oncogenic properties in epithelial tumors.
Many recent studies have suggested that Eph/ephrin
systems have pivotal roles in epithelial-mesenchymal transition
(EMT) processes (15). Cells
undergoing EMT show reduction in cell-cell adhesion caused by
reduced expression of E-cadherin on the cell surface, and gain
mesenchymal phenotypes with spindle-shaped morphology and increased
potential for migration and invasion (16). Therefore, EMT has been implicated in
the acquisition of an invasive phenotype by cancer cells.
Overexpression of EPHA2 and reduced expression of E-cadherin are
associated with higher stage of gastric cancer (17) and colorectal cancer (18). On the other hand, higher expression
levels of EPHB3 and E-cadherin were reported to be associated with
lower stage of esophageal adenocarcinoma (19). In cervical cancer, forced expression
of EPHB2 induced EMT signature, and silencing of EPHB2 resulted in
the opposite phenotype (6).
Nevertheless, the role of EPHB2 in EMT in SCC is
still unclear. In the present study, we conducted functional
analysis to elucidate whether EPHB2 is involved in the regulation
of EMT in SCC.
Materials and methods
Cell lines and culture conditions
The human skin squamous carcinoma-derived cell line
A431 was obtained from American Type Culture Collection (ATCC,
Manassas, VA, USA). Cells were maintained in DMEM (Nacalai Tesque,
Kyoto, Japan) supplemented with heat-inactivated fetal bovine serum
(FBS; Nichirei Bioscience, Tokyo, Japan) at a final concentration
of 10%, 100 IU/ml of penicillin (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA), and 100 µl/ml of streptomycin (Thermo Fisher
Scientific, Inc.). The cells were maintained at 37°C in a
CO2 incubator with a controlled humidified atmosphere
composed of 95% air and 5% CO2.
Analysis of cell viability
Cells were seeded in 96-well plates at a density of
1×103 cells per well, and cultured for 24 h. The cells
were then transfected with control siRNA or EPHB2 siRNA using
Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. Cell viability was measured by
WST8 assay using Cell Count Reagent CF (Nacalai Tesque) at 24, 48,
and 72 h after transfection.
Wound-healing migration assay
Cells were seeded in 24-well plates at a density of
2×105 cells per well, and transfected with control siRNA
or EPHB2 siRNA at 24 h after seeding. Forty-eight h after
transfection, cell layers were wounded using a Cell Scratcher
Scratch stick (AGC Technoglass, Shizuoka, Japan) and medium was
replaced with 500 µl of fresh medium. After 24 h, cells were
photographed by phase-contrast microscopy, and the width of the
wounded area was measured. The percentage gap size was calculated
by dividing the width at 24 h by the width at 0 h.
Matrigel invasion assay
Cells were seeded into the upper chambers of BD
Matrigel invasion chambers (BD Biosciences, San Jose, CA, USA) at a
density of 2×105 cells per well, and immediately
transfected with control siRNA or EPHB2 siRNA. The lower wells were
filled with culture medium. After 24 h, non-invading cells on the
upper surface of the membrane were removed, and invading cells were
fixed and stained with Diff-Quik solutions (Sysmex, Kobe, Japan).
The number of invading cells in 10 microscopic fields was counted
for each membrane under a light microscope at ×200
magnification.
Analysis of sphere formation
efficiency
Cells were seeded in ultra-low attachment surface
24-well plates (Corning Incorporated, Corning, NY, USA) at a
density of 4×103 cells per well, with MEGM Bullet kit
serum-free medium supplemented with 0.4% BPE, 0.1% hEGF, 0.1%
hydrocortisone, 0.1% GA1000, 0.1% insulin, 1% l-glutamine (Lonza,
Walkersville, MD, USA). To examine the effect of EPHB2 on sphere
formation efficiency, cells were immediately transfected with
control siRNA or EPHB2 siRNA. Five days after seeding, the number
of spheres >100 µm in diameter in eight microscopic fields at
×50 magnification was counted. Representative images were
photographed at ×200 magnification.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cells using RNeasy mini
kits (Qiagen, Inc., Valencia, CA, USA) according to the
manufacturer's instructions. For cDNA synthesis, 500 ng of total
RNA was reverse transcribed using iScript cDNA synthesis system
(Bio-Rad Laboratories, Inc., Hercules, CA, USA). qPCR was performed
using a SYBR Premix Ex Taq™ system according to the manufacturer's
recommendations (Takara Bio, Inc., Otsu, Japan). A mixture of cDNA
derived from total RNA of cells was used as a reference. Then, a
dilution series of the cDNA mixture was prepared and used in
RT-qPCR as the template to obtain a standard curve for each gene.
Three independent measurements were performed and the relative
amounts of the PCR products were quantified via extrapolation from
a standard curve. The sequences of primers used for qPCR-based
amplification were as follows: EPHB2, 5′-TGAGTGCCCTCAGATGGTCAA-3′
(sense) and 5′-AGGGCAGGGTATCACAGTGAATG-3′ (antisense); CDH1,
5′-AATTCCTGCCATTCTGGGGA-3′ (sense) and 5′-TCTTCTCCGCCTCCTTCTTC-3′
(antisense); FN, 5′-CAGTGGGAGACCTCGAGAAG-3′ (sense) and
5′-TCCCTCGGAACATCAGAAAC-3′ (antisense); SNAI1,
5′-CCATGCTCCTCTTTGCTCTC-3′ (sense) and 5′-TACAAAAACCCACGCAGACA-3′
(antisense); ZEB1, 5′-GCCAACAGACCAGACAGTGTT-3′ (sense) and
5′-TCTTGCCCTTCCTTTCCTG-3′ (antisense); ZEB2,
5′-CAAGAGGCGCAAACAAGC-3′ (sense) and 5′-AACCTGTGTCCACTACATTGTCA-3′
(antisense); TWIST, 5′-ACGAGCTGGACTCCAAGATGGCAAG-3′ (sense) and
5′-CATCCTCCAGACCGAGAAGGCGTAG-3′ (antisense); CD44,
5′-GCAGTCAACAGTCGAAGAAGG-3′ (sense) and 5′-TGTCCTCCACAGCTCCATT-3′
(antisense); CD133, 5′-TTGCAATCTCCCTGTT-3′ (sense) and
5′-TTGCTATCTGCCAGTT-3′ (antisense); GAPDH,
5′-GCACCGTCAAGGCTGAGAAC-3′ (sense) and 5′-TGGTGAAGACGCCAGTGGA-3′
(antisense). GAPDH was used as a reference.
Immunoblotting
Cells were lysed in RIPA buffer containing protease
inhibitor cocktail (Nacalai Tesque). The protein concentration of
lysates was measured using Bio-Rad DC kits (Bio-Rad Laboratories,
Inc.). Cell lysates (20 µg of protein) were separated by 4–12%
SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and then
electroblotted onto Immobilon-P membranes (EMD Millipore,
Billerica, MA, USA). Membranes were blocked with Blocking-one
(Nacalai Tesque) overnight at 4°C, and incubated with goat
polyclonal anti-EPHB2 antibody (Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA) or with mouse anti-β-actin antibody (A5441;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) at 4°C. After 24 h
of incubation, membranes were washed with PBS containing 0.1%
Tween-20 (PBS-T), followed by incubation with horseradish
peroxidase-conjugated secondary antibody for rabbit or mouse (GE
Healthcare Life Science, Little Chalfont, UK), for 1 h at room
temperature. The membranes were washed extensively with PBS-T, and
treated with Chemi-Lumi-One Super (Nacalai Tesque) to visualize
immunoreactive signals using LAS4000 (Fujifilm, Tokyo, Japan).
Statistical analysis
Statistical analyses were performed using Student's
t-test with JMP software version 11 (SAS Institute, Inc., Cary, NC,
USA). Data are presented as the mean ± standard deviation from at
least three independent experiments. P<0.05 was considered to
indicate a statistically significant difference.
Results
Knockdown of EPHB2 resulted in
epithelial-mesenchymal transition (EMT)-like morphological changes
of A431 cells
To determine whether EPHB2 could be involved in the
regulation of certain cellular process, we performed siRNA-mediated
knockdown of EPHB2 in SCC-derived A431 cells. As EPHB2 mRNA and
protein expression were sufficiently suppressed by transfection
with EPHB2 siRNA (Fig. 1A and B), we
performed phenotype analysis of cells with EPHB2 knockdown. As
shown in the representative photographs, cells with EPHB2 knockdown
were elongated and isolated compared to control cells showing a
cobblestone-like morphology (Fig.
1C). These phenotypic changes suggested that silencing of EPHB2
induced epithelial-mesenchymal transition (EMT). Meanwhile,
knockdown of EPHB2 in A431 cells had a negligible effect on cell
viability (Fig. 1D), migration
(Fig. 1E) and invasion (Fig. 1F).
 | Figure 1.Knockdown of EPHB2 resulted in the
epithelial-mesenchymal transition-like phenotype in A431 cells. (A
and B) siRNA-mediated silencing of EPHB2. A431 cells were
transfected with EPHB2 siRNA or with control siRNA. At 48 h
post-transfection, total RNA and cell lysates were prepared and
analyzed by (A) reverse transcription-quantitative polymerase chain
reaction and (B) immunoblotting. Data are presented as the mean ±
standard deviation of triplicate experiments. **P<0.01, as
indicated. (C) Representative images of the cells at 48 h
post-transfection (scale bars, 100 µm). EPHB2,
erythropoietin-producing hepatocellular B2; siRNA, small
interfering RNA. Knockdown of EPHB2 resulted in the
epithelial-mesenchymal transition-like phenotype in A431 cells. (D)
Cell viability was measured by WST8 assay at the indicated times
following transfection with control siRNA (dashed line) or EPHB2
siRNA (solid line). (E) Wound-healing assay. At 48 h
post-transfection with control siRNA or EPHB2 siRNA, the cell layer
was scratched, and the medium was replaced with fresh medium (scale
bars, 200 µm). At 24 h post-scratching, the gap size was measured.
Data are presented as the mean ± standard deviation of triplicate
experiments. Representative images of the wounded area are shown.
(F) Matrigel invasion assay. Cells were seeded on invasion
chambers, and immediately transfected with control siRNA or EPHB2
siRNA (scale bars, 100 µm). Following 24 h, the invading cells were
fixed and stained, and the number of stained cells was counted. The
mean ± standard deviation of 10 measurements and representative
images of the invading cells are presented. EPHB2,
erythropoietin-producing hepatocellular B2; siRNA, small
interfering RNA. |
EPHB2 knockdown cells showed
EMT-specific gene expression patterns
As silencing of EPHB2 induced EMT-like morphological
changes in A431 cells, the expression patterns of EMT-specific
markers were examined. As shown in Fig.
2, the expression level of the epithelial marker E-cadherin
(CDH1) was significantly decreased and the mesenchymal marker
fibronectin (FN) was significantly increased in EPHB2 knockdown
cells at 48 h after siRNA transfection. In addition, expression of
transcription factors ZEB1 and ZEB2, that are EMT inducer, were
significantly increased in EPHB2 knockdown cells. Expression level
of another EMT inducer TWIST also tended to increase in EPHB2
silenced cells, but the difference was not significant. On the
other hand, the expression of SNAI1, which is also one of the EMT
inducer, was significantly suppressed in EPHB2 knockdown cells. The
expression profiles of the EMT markers, except for SNAI1, indicated
that knockdown of EPHB2 induces EMT in A431 cells.
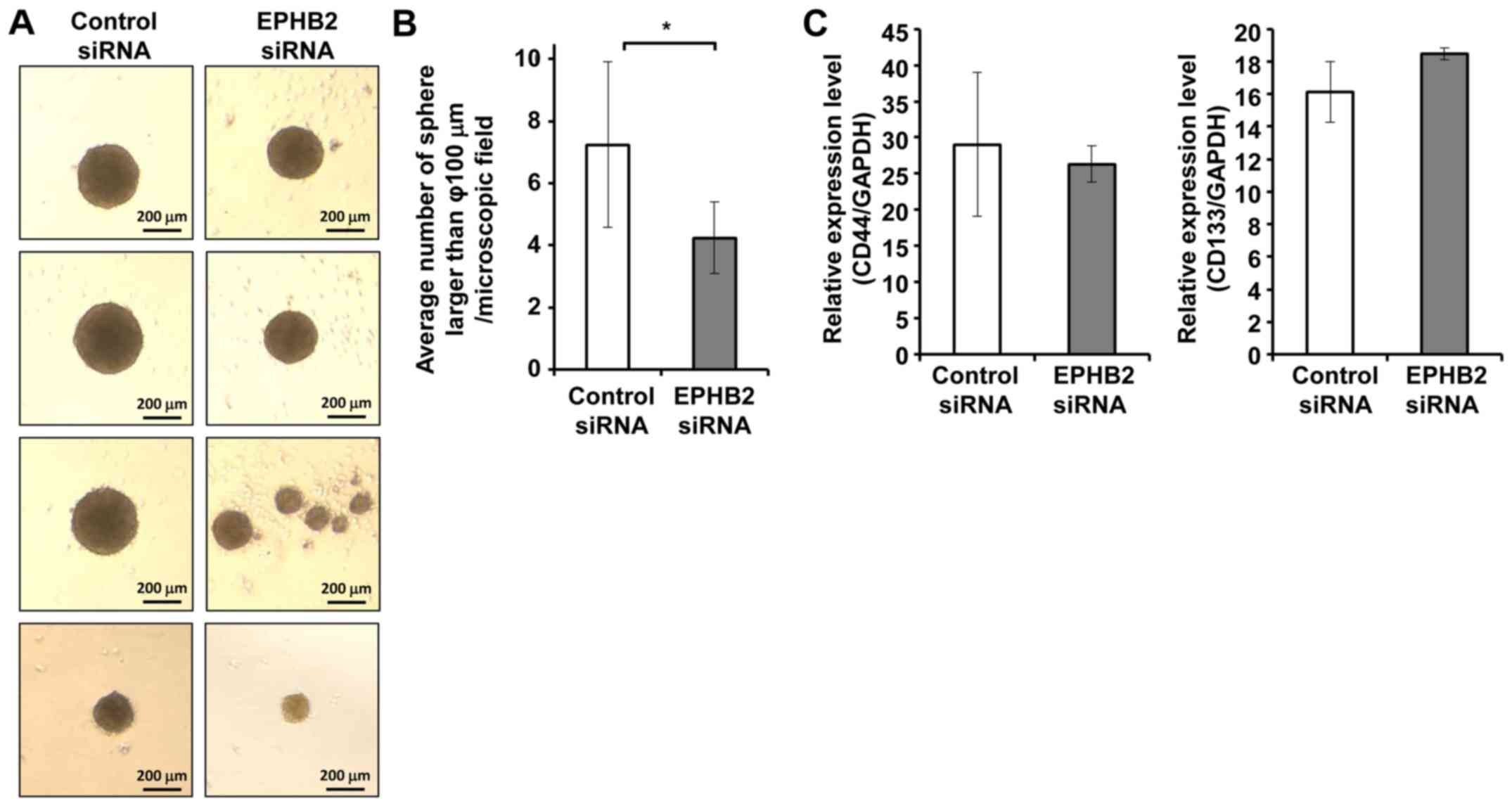
Knockdown of EPHB2 resulted in reduced
tumor sphere formation
Accumulating evidence suggests that EMT is involved
in the acquisition of cancer stem cell properties by tumor cells
(20). To investigate whether
silencing of EPHB2 could promote formation of cancer stem cells in
A431 cells, sphere forming efficacy was analyzed. Cells transfected
with EPHB2 siRNA or control siRNA were grown in suspension culture
with serum-free sphere medium for 5 days, and the number of spheres
>100 µm in diameter was counted. In contrast to our prediction,
the number of spheres formed in EPHB2 silenced cells was
significantly lower than that in control cells (Fig. 3A and B). On the other hand, no clear
differences were observed in the expression levels of cell stemness
markers between EPHB2 silenced cells and control cells (Fig. 3C).
Tumor spheres showed increased expression of EPHB2
and reduced expression of EMT markers compared to cells cultured in
adherent form. To elucidate the roles of EPHB2 in tumor sphere
formation and EMT, the gene expression patterns of tumor spheres
and adherent cells were also analyzed. As shown in Fig. 4, the expression level of EPHB2 was
significantly higher in tumor spheres compared to cells cultured in
adherent form. Consistent with this observation, the expression
patterns of all EMT markers except SNAI1 indicated suppression of
EMT in tumor spheres compared to adherent cells (Fig. 4).
 | Figure 4.Spheres formed using A431 cells
exhibit upregulated EPHB2 expression and reduced
epithelial-mesenchymal transition-specific gene expression. Cells
were cultured under normal adherent conditions or sphere forming
conditions for 5 days. Total RNA was extracted from AD and SP, and
analyzed by reverse transcription-quantitative polymerase chain
reaction. Data are presented as the mean ± standard deviation of
triplicate experiments. *P<0.05, **P<0.01 and ***P<0.001,
as indicated. AD, adherent cells; SP, tumor spheres; EPHB2,
erythropoietin-producing hepatocellular B2; CDH1, E-cadherin; FN,
fibronectin; SNAI1, snail family transcriptional repressor 1; ZEB,
zinc finger E-box binding homeobox; TWIST, Twist family
basic-helix-loop-helix transcription factor 1. |
Discussion
The results of the present study indicated that
silencing of EPHB2 induced EMT-like morphological changes
accompanied by the up- and the down-regulation of the mesenchymal
and the epithelial marker gene expression, respectively. In
addition, EPHB2 silencing resulted in reduced efficiency of tumor
sphere formation. Accordingly, tumor spheres derived from A431
showed increased EPHB2 expression level, along with decreased
expression of genes characteristic of cells undergoing EMT.
As activation of EMT has been implicated in the
acquisition of stem cell properties by cancer cells (20), and this makes cancer cells anoikis
resistant and enables them to survive under non-adherent conditions
(21,22), we expected that anchorage-independent
cell growth would be enhanced in EPHB2 knockdown cells. In contrast
to our prediction, however, EPHB2 knockdown cells formed
significantly fewer tumor spheres than control cells. The lack of
significant differences in expression levels of cell stemness
markers between tumor spheres and adherent cells indicated that the
increased number of tumor spheres in EPHB2 knockdown cells might
not be due to an increased stemness. Alternatively, it might be
attributed to their decreased cell-cell adhesion along with the
promotion of EMT. This hypothesis was supported by the observations
that EPHB2 expression level was increased and the expression
profiles characteristic of EMT were suppressed in tumor
spheres.
Previously, it has been reported that expression
levels of EPHB2 were increased in both mouse cSCC (13,14) and
human cSCC (14). Farshchian et
al (14) demonstrated that
depletion of EPHB2 in cSCC cells reduces their
proliferation, invasion and migration abilities in association with
the down-regulation of genes implicated in these oncogenic
processes. The present findings indicated that EPHB2 could promote
anchorage-independent cell growth by increasing cell-cell adhesion
through the suppression of EMT processes. Taken together, these
findings strongly suggest that EPHB2 has oncogenic functions,
promoting the growth and acquisition of malignant phenotypes in
cSCC.
Among the EMT markers examined, expression pattern
of SNAI1 was opposite to the other markers in EPHB2 knockdown cells
and spheres. Similar to ZEB1/2 and TWIST, it has been well-known
that SNAI1 contributes to the induction of EMT mainly through the
repression of CDH1 transcription. However, the regulatory
mechanisms behind the transcription of these EMT inducers are
dependent on cellular context and signaling. For example, it has
been reported that Insulin-like growth factor receptor signals
induces SNAI1 and ZEB1 via NF-kB activation, whereas this signal
augments Twist and ZEB1 via Ras/Raf/ERK signal (23). Under our experimental conditions,
therefore, knockdown of EPHB2 might potentially activate certain
signal pathway(s) linked to the transcriptional activation of
ZEB1/2 and Twist, but to SNAI1.
In contrast to our current findings, it has been
reported that EPHB2 activates EMT in cervical cancer cells
(6) and breast cancer cells
(24). In breast cancer cells,
activation of wild-type p53 inhibited basal and TGF-β3-induced EMT
by reducing the expression level of EPHB2 (24). This discrepancy between our results
and those reported previously may be due to the variety of
mutations in the molecules involved in EMT processes. Indeed, A431
cells carry a missense mutation in p53 gene at exon 8 (25). As observations in breast cancer
indicated that there is cross-talk among EPHB2, TGF-β, and p53
(24), the mutated p53 may affect
the roles of EPHB2 in induction of EMT in A431 cells. In addition,
although EMT has been implicated in promotion of invasion and
migration activity of tumor cells, the present results indicated
that silencing of EPHB2 induced EMT-like phenotypes but did not
affect the invasion and migration abilities of the cells. This
result may also have been due to genetic variation of genes related
to cell functions, including EMT, among cells. To adequately
address this issue, further analyses using many types of cell lines
harboring different types of genetic variations are required.
In conclusion, the present study showed that
knockdown of EPHB2 induces EMT-like phenotypes in A431 cells, and
that spheres formed by A431 showed increased expression of EPHB2
and suppressed gene expression patterns characteristic of cells
undergoing EMT. These findings indicated that EPHB2 is involved at
least partially in suppression of EMT processes in A431 cells.
Acknowledgements
The authors would like to thank Ms. A. Oguni for
their technical support and Ms. K. Tagata for their secretarial
assistance (both Division of General Medicine, Department of
Medicine, Nihon University School of Medicine Tokyo, Japan).
Funding
The present study was supported in part by JSPS
KAKENHI (grant no. 15K09791) and MEXT-Supported Program for the
Strategic Research Foundation at Private Universities (grant no.
2011-2015).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contribution
KF and HN conceived and designed the study. KF, YI,
TT, MY, DW, JH and TO performed the experiments. NF performed the
statistical analysis. MS made substantial contributions to the
analysis and interpretation of data. KF and MS wrote the paper. TO,
HN, MS and NF critically revised the paper for important
intellectual content and gave final approval of the paper to be
published. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pasquale EB: Eph-ephrin bidirectional
signaling in physiology and disease. Cell. 133:38–52. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gale NW, Holland SJ, Valenzuela DM,
Flenniken A, Pan L, Ryan TE, Henkemeyer M, Strebhardt K, Hirai H,
Wilkinson DG, et al: Eph receptors and ligands comprise two major
specificity subclasses and are reciprocally compartmentalized
during embryogenesis. Neuron. 17:9–19. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wijeratne DT, Rodger J, Wood FM and Fear
MW: The role of Eph receptors and Ephrins in the skin. Int J
Dermatol. 55:3–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pasquale EB: Eph receptor signalling casts
a wide net on cell behaviour. Nat Rev Mol Cell Biol. 6:462–475.
2005. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pasquale EB: Eph receptors and ephrins in
cancer: Bidirectional signalling and beyond. Nat Rev Cancer.
10:165–180. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao Q, Liu W, Cai J, Li M, Gao Y, Lin W
and Li Z: EphB2 promotes cervical cancer progression by inducing
epithelial-mesenchymal transition. Hum Pathol. 45:372–381. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kumar SR, Scehnet JS, Ley EJ, Singh J,
Krasnoperov V, Liu R, Manchanda PK, Ladner RD, Hawes D, Weaver FA,
et al: Preferential induction of EphB4 over EphB2 and its
implication in colorectal cancer progression. Cancer Res.
69:3736–3745. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kumar SR, Masood R, Spannuth WA, Singh J,
Scehnet J, Kleiber G, Jennings N, Deavers M, Krasnoperov V, Dubeau
L, et al: The receptor tyrosine kinase EphB4 is overexpressed in
ovarian cancer, provides survival signals and predicts poor
outcome. Br J Cancer. 96:1083–1091. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hafner C, Becker B, Landthaler M and Vogt
T: Expression profile of Eph receptors and ephrin ligands in human
skin and downregulation of EphA1 in nonmelanoma skin cancer. Mod
Pathol. 19:1369–1377. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Herath NI, Doecke J, Spanevello MD,
Leggett BA and Boyd AW: Epigenetic silencing of EphA1 expression in
colorectal cancer is correlated with poor survival. Br J Cancer.
100:1095–1102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Batlle E, Bacani J, Begthel H, Jonkheer S,
Gregorieff A, van de Born M, Malats N, Sancho E, Boon E, Pawson T,
et al: EphB receptor activity suppresses colorectal cancer
progression. Nature. 435:1126–1130. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li JJ, Liu DP, Liu GT and Xie D: EphrinA5
acts as a tumor suppressor in glioma by negative regulation of
epidermal growth factor receptor. Oncogene. 28:1759–1768. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fujiwara K, Ghosh S, Liang P, Morien E,
Soma M and Nagase H: Genome-wide screening of aberrant DNA
methylation which associated with gene expression in mouse skin
cancers. Mol Carcinog. 54:178–188. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Farshchian M, Nissinen L, Siljamäki E,
Riihilä P, Toriseva M, Kivisaari A, Ala-Aho R, Kallajoki M,
Veräjänkorva E, Honkanen HK, et al: EphB2 promotes progression of
cutaneous squamous cell carcinoma. J Invest Dermatol.
135:1882–1892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li RX, Chen ZH and Chen ZK: The role of
EPH receptors in cancer-related epithelial-mesenchymal transition.
Chin J Cancer. 33:231–240. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Savagner P: The epithelial-mesenchymal
transition (EMT) phenomenon. Ann Oncol. 21 (Suppl 7):vii89–vii92.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yuan W and Chen Z, Wu S, Ge J, Chang S,
Wang X, Chen J and Chen Z: Expression of EphA2 and E-cadherin in
gastric cancer: Correlated with tumor progression and lymphogenous
metastasis. Pathol Oncol Res. 15:473–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saito T, Masuda N, Miyazaki T, Kanoh K,
Suzuki H, Shimura T, Asao T and Kuwano H: Expression of EphA2 and
E-cadherin in colorectal cancer: Correlation with cancer
metastasis. Oncol Rep. 11:605–611. 2004.PubMed/NCBI
|
|
19
|
Schauer MC, Stoecklein NH, Theisen J,
Kropil F, Baldus S, Hoelscher A, Feith M, Bolke E, Matuschek C,
Budach W and Knoefel WT: The simultaneous expression of both ephrin
B3 receptor and E-cadherin in Barrett's adenocarcinoma is
associated with favorable clinical staging. Eur J Med Res.
17:102012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou P, Li B, Liu F, Zhang M, Wang Q, Liu
Y, Yao Y and Li D: The epithelial to mesenchymal transition (EMT)
and cancer stem cells: implication for treatment resistance in
pancreatic cancer. Mol Cancer. 16:522017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Izumiya M, Kabashima A, Higuchi H,
Igarashi T, Sakai G, Iizuka H, Nakamura S, Adachi M, Hamamoto Y,
Funakoshi S, et al: Chemoresistance is associated with cancer stem
cell-like properties and epithelial-to-mesenchymal transition in
pancreatic cancer cells. Anticancer Res. 32:3847–3853.
2012.PubMed/NCBI
|
|
22
|
Guadamillas MC, Cerezo A and Del Pozo MA:
Overcoming anoikis-pathways to anchorage-independent growth in
cancer. J Cell Sci. 124:3189–3197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Malaguarnera R and Belfiore A: The
emerging role of insulin and insulin-like growth factor signaling
in cancer stem cells. Front Endocrinol (Lausanne). 5:102014.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lam S, Wiercinska E, Teunisse AF, Lodder
K, ten Dijke P and Jochemsen AG: Wild-type p53 inhibits
pro-invasive properties of TGF-β3 in breast cancer, in part through
regulation of EPHB2, a new TGF-β target gene. Breast Cancer Res
Treat. 148:7–18. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Harlow E, Williamson NM, Ralston R,
Helfman DM and Adams TE: Molecular cloning and in vitro expression
of a cDNA clone for human cellular tumor antigen p53. Mol Cell
Biol. 5:1601–1610. 1985. View Article : Google Scholar : PubMed/NCBI
|