Introduction
Diffuse intrinsic pontine glioma (DIPG) is one of
the most fatal malignant pediatric brain tumors (1) and is a major cause of cancer-associated
mortality in children (2). The mean
overall survival of patients with DIPG is 8–14 months, and the only
effective treatment modality is radiation (3). However, clinical outcomes are
disappointing, and a combination of various adjuvant chemotherapies
has failed to prolong overall survival compared with radiation
alone (4). In general, routine tumor
biopsy is avoided due to the location of the tumor, and the paucity
of tumor tissues has limited active research on the biology and
pathogenesis of DIPG. Although several clinical trials for
molecular targeted agents are underway (4–6), the
focus is on canonical oncogenic signaling pathways, such as the
epidermal growth factor receptor and platelet-derived growth factor
receptor (PDGFR) signaling pathways. Furthermore, these therapeutic
trials are problematic since they are not based on the unique
molecular genomic features of DIPG (4).
A number of genetic and epigenetic alterations in
DIPG have recently been discovered, revealing a mutation signature
that is completely different from that of high-grade gliomas of
other regions of the brain. Specifically, the lysine-to-methionine
mutation at position 27 of the H3 histone variant (H3K27M),
H3.1K27M or H3.3K27M, was identified in 78% of DIPGs and was
accompanied by activating mutations in PDGFRA or activin A receptor
type I (ACVR1) (7–9). The fact that DIPG is detected
exclusively in the ventral pons of children aged between 6 and 7
years (10,11) may explain the very different genomic
and molecular features compared with other gliomas. Furthermore,
the specific peak age and anatomical location suggest abnormalities
in postnatal neurodevelopmental mechanisms. Indeed, aberrancy in
one of the signaling pathways associated with the differentiation
of neural stem cells (NSCs) may cause an imbalance in the number
and viability of local cells, and may contribute to growth and
neoplastic behavior (12).
Signal transducer and activator of transcription 3
(STAT3) is a member of the STAT family and has extensive functions,
including neural differentiation, in a number of organisms
(13,14). STAT3 was originally identified as a
mediator of interleukin-6 (IL-6) receptor signaling (15). The receptors for IL-6 utilize gp130,
which activates STAT3 via phosphorylation; phosphorylated STAT3
dimerizes and translocates to the nucleus, where it binds to a
specific DNA sequence to activate the transcription of genes
involved in glial differentiation (16). Furthermore, a previous study
demonstrated that STAT3 is essential for astrocyte differentiation
(17).
Additionally, STAT3 acts as an oncogene in numerous
types of cancer, including breast (18), lung (19) and pancreatic cancer (20). Constitutive activation of STAT3 was
first observed to be associated with oncogenic transformation by
the viral Src oncoprotein (21).
Similarly, other oncogenic tyrosine kinases activate STAT3,
resulting in oncogenic transformation (22–24). The
contribution of STAT3 to a given tumor phenotype depends on the
tissue context. Overall, the role of STAT3 has been studied more in
glioma compared with other types of cancer (25–27). In
adult glioma, a gain-of-function mutation in STAT3 induces
angiogenesis, immunosuppression, invasion and temozolomide
resistance (28,29). However, to the best of our knowledge,
the association between STAT3 and DIPG has not yet been
elucidated.
In the present study, mRNA expression levels of
various genes associated with the differentiation of NSCs (30) were compared between DIPG tissues and
normal brain tissues using open public data. All of the screened
genes, including Notch receptor 1 (NOTCH1), ACVR1 and STAT3, were
significantly upregulated in DIPG tissues compared with in normal
brain tissues. Based on its function in gliogenesis during
neurodevelopment and in the oncogenesis of malignant tumors,
including gliomas, the possible role of STAT3 in the tumor biology
of DIPG was investigated using the human DIPG SF8628 cell line.
STAT3 activation was modulated by treatment with the STAT3
inhibitor AG490 and transfection using STAT3 short hairpin (sh)RNA.
Cell viability assays were performed to assess cell viability after
modifying STAT3 activation. Protein expression was analyzed via
western blotting and RNA expression was evaluated via reverse
transcription-semi-quantitative PCR (RT-semi-qPCR). To investigate
the effect of STAT3 inhibition on the therapeutic effect of
irradiation, SF8628 cells were treated with a combination of STAT3
inhibition and irradiation.
Materials and methods
In silico R2 analysis
Comparison of mRNA expression levels of genes
associated with NSC differentiation [namely NOTCH1, inhibitor of
DNA binding 1, ACVR1, Hes family bHLH transcription factor 1
(HES1), SMAD family member 1, E1A binding protein p300, LIF
receptor subunit α and STAT3] between normal brain tissues [n=172;
Gene Expression Omnibus (GEO) ID: GSE11882 (31)] and DIPG tumor tissues [n=27, GEO ID:
GSE26576 (32)] was performed using
R2 and the Megasampler module (33):
Genomics Analysis and Visualization Platform (http://r2.amc.nl), a microarray analysis and
visualization platform. One-way ANOVA was used to compare the
expression levels of STAT3 mRNA between the normal brain tissue and
DIPG GEO datasets.
Cell culture
The patient-derived DIPG SF8628 cell line (Merck
KGaA) harboring the histone H3.3 Lys 27-to-methionine (K27M)
mutation was used in the present study. The cells were cultured in
DMEM with high glucose supplemented with 10% FBS (both from
Biowest) and 1% penicillin/streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) at 37°C with 95% humidity and 5%
CO2.
STAT3 inhibitor treatment
To inhibit endogenous STAT3 activity, SF8628 cells
were treated with 20 µM of the STAT3 inhibitor AG490 (Cell
Signaling Technology, Inc.) dissolved in DMSO.
Lentivirus-mediated shRNA silencing of
STAT3 expression
Lentiviral shRNA particles were purchased from
Sigma-Aldrich (Merck KGaA). Viral shRNA transfections using 10 µl
(1×107 titer units/0.1 ml) STAT3-targeting shRNA or
non-targeting shRNA were performed by incubating SF8628 cells in
culture medium containing lentiviral particles for 12 h in the
presence of 0.8 µg/ml polybrene (EMD Millipore). Subsequently,
Puromycin (Thermo Fisher Scientific, Inc.) was added to the medium
of the transfected cells to select shRNA-expressing cells.
RT-semi-qPCR
RT-semi-qPCR was performed to determine the
transcript level of STAT3 in SF8628 cells, and the amplification of
β-actin transcripts was used as the control to normalize the
transcript levels of molecules. Total RNA was isolated from cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
and RT was performed to synthesize cDNA in a 20 µl reaction mixture
(SuperScript One-Step kit; Invitrogen; Thermo Fisher Scientific,
Inc.) containing 2.5 µl of each 10 pmol gene-specific primer, 1 µg
RNA, 2X reaction buffer, 0.4 µl Taq polymerase, 0.4 mM of each dNTP
and 1.2 mM MgCl2, for 30 min at 50°C. After an initial
denaturation step of cDNA for 2 min at 94°C, the cDNA of STAT3
transcripts was amplified for 25 cycles of 30 sec at 94°C, 30 sec
at 58°C and 30 sec at 72°C, while the cDNA of β-actin transcripts
was amplified for 18 cycles of 30 sec at 94°C, 30 sec at 52°C and
30 sec at 70°C. The PCR cycling numbers had been optimized to avoid
amplification saturation. A total of 5 µl qPCR product was
separated on 1% agarose gels, which were subsequently stained with
RedSafe™ Nucleic Acid Staining solution (Intron
Biotechnology, Inc.). Primer sequences were as follows: STAT3
forward, 5′-ACCCAACAGCCGCCGTAG-3′ and reverse,
5′-CAGATGGTTGTTTCCATTCAGAT-3′; and β-actin forward,
5′-ACACCTTCTACAATGAGCTG-3′ and reverse,
5′-CATGATGGAGTTGAAGGTAG-3′.
Western blotting
Lysates of subconfluent (60-80% confluent) cells
were obtained using SDS lysis buffer [125 mM Tris-HCl (pH 6.8), 4%
SDS, 0.004% bromophenol blue and 20% glycerol]. The Pierce BCA
Protein assay kit (Thermo Fisher Scientific, Inc.) was used to
determine protein concentration. The cell lysates (30 µg
protein/lane) were separated by 10% SDS-PAGE and transferred to
polyvinylidene difluoride membranes (EMD Millipore) that were
blocked with 5% skim dry milk in Tween-20 (0.05%)-TBS (TTBS) for 1
h at room temperature. Subsequently, the membranes were incubated
overnight at 4°C with primary antibodies (1:1,000) against one of
the following: Cleaved caspase 3 (cat. no. ab2302; Abcam), cleaved
poly (ADP-ribose) polymerase (cat. no. 9542; Cell Signaling
Technology, Inc.), phosphorylated-STAT3 (pSTAT3; cat. no. 9131;
Cell Signaling Technology, Inc.), STAT3 (cat. no. 9139; Cell
Signaling Technology, Inc.), cyclin D1 (cat. no. MA5-16356; Thermo
Fisher Scientific, Inc.), E-cadherin (cat. no. 610181; BD
Biosciences), N-cadherin (cat. no. C3865; Sigma-Aldrich; Merck
KGaA;), vimentin (cat. no. 3390S; Cell Signaling Technology, Inc.),
Twist (cat. no. SC-15393; Santa Cruz Biotechnology, Inc.), Snail
(cat. no. SC-28199; Santa Cruz Biotechnology, Inc.) and matrix
metallopeptidase 9 (MMP-9; cat. no. RB-1539-P; NeoMarkers, Inc.).
Horseradish peroxidase-conjugated anti-rabbit IgG (cat. no.
bs-0295G-HRP) or anti-mouse IgG (cat. no. bs-0296G-HRP) (both
1:4,000; BIOSS) were used as secondary antibodies, and the
membranes were incubated for 2 h at room temperature. Enhanced
chemiluminescence (Invitrogen; Thermo Fisher Scientific, Inc.) was
used to detect the immunoreactive proteins. β-actin (primary
antibody, 1:1,000; cat. no. SC-47778; Santa Cruz Biotechnology,
Inc.) served as the loading control.
Cell viability assay
Cell viability was assessed using a Cell Counting
Kit (CCK)-8 assay (Dojindo Molecular Technologies, Inc.) according
to the manufacturer's protocol. Briefly, 5×103 cells
were seeded in each well of a 96-well plate and incubated for 48 h
at 37°C. The CCK-8 reagent was added to each well 1 h before the
incubation endpoint. Optical density values at 420 nm were
determined using an ELISA plate reader (Bio-Rad Laboratories,
Inc.).
Transwell migration and invasion
assays
A 24-well Transwell Insert system with an 8-µm pore
size polyethylene terephthalate membrane was purchased from BD
Biosciences. The upper chambers were coated with or without
Matrigel for 1 h at 37°C for invasion or migration, respectively.
Medium containing 10% FBS was placed in the lower chambers and
served as a chemoattractant. SF8628 cells (1×104
cells/insert) in 300 µl 1% FBS-containing medium were seeded in the
upper chamber of each Transwell insert and allowed to migrate for
48 h at 37°C. Non-migrated cells were removed from the top of each
insert with a cotton swab. Migrated cells on the bottom surface of
the insert were stained with 0.2% crystal violet in 20% methanol
for 30 min at room temperature and visualized with an inverted
light microscope (magnification, ×40). The stained cells were lysed
in 10% SDS for 30 min, and the absorbance was measured at 562 nm
using an ELISA plate reader.
F-actin assay
To examine whether STAT3 inhibition causes
cytoskeletal reorganization, filamentous actin (F-actin) was
visualized. Cells were incubated with 165 nmol/l Alexa
Fluor-633-conjugated phalloidin (cat. no. A22284; Invitrogen;
Thermo Fisher Scientific, Inc.) for 10 min at room temperature,
followed by 4′6′-diamidio-2-phenoylindole (DAPI) staining for 10
min at room temperature. Immunofluorescence was observed by
fluorescence microscopy (magnification, ×400).
Combination of STAT3 inhibition and
irradiation
Control cells and STAT3-inhibited cells (by shRNA
transfection or AG490 treatment) were cultured in 96-well plates at
a density of 5×103/well for 24 h at 37°C with 95%
humidity and 5% CO2. The cells were irradiated with 4 Gy
using a 6-MV photon beam in a linear accelerator (21EX-S; Varian
Medical Systems) and incubated for 24 h at 37°C. Subsequently, a
CCK-8 assay was used as aforementioned to measure cell viability.
All treatments were tested in triplicate.
Phosphorylated H2A X variant histone
(γH2AX) assay
Cells were cultured on a 4-well chamber slide
(3×104 cells/chamber). Following irradiation as
aforementioned, cells were incubated for 1, 4 or 24 h, fixed in 4%
paraformaldehyde for 10 min and permeabilized using 0.5% Triton
X-100 for 5 min, both at room temperature. After blocking with 5%
skim milk diluted in 0.05% Triton X-100 for 30 min at room
temperature, the slides were washed for 5 min in TTBS and incubated
with a primary rabbit anti-γH2AX antibody (clone 2577; cat. no.
2577; Cell Signaling Technology, Inc.) at a 1:200 dilution in TTBS
for 2 h at room temperature in a humidified chamber. Subsequently,
the slides were washed three times in TTBS for 5 min each and
incubated with an Alexa Fluor 488 goat anti-rabbit secondary IgG
antibody (cat. no. A11008; Invitrogen; Thermo Fisher Scientific,
Inc.) diluted 1:500 in 1% BSA for 1 h at room temperature in a
humidified chamber. The slides were washed twice in TTBS and
mounted with DAPI-containing mounting medium. Images of γH2AX were
acquired with a fluorescence microscope (magnification, ×400; BX51;
Olympus Corporation).
Statistical analysis
Associations between STAT3 activation and either the
cell viability index, invasion or migration were analyzed using a
two-tailed unpaired Student's t-test (for differences between two
groups) using Excel 2016 (Microsoft Corporation) or one-way ANOVA
(for multiple comparisons) followed by Tukey's post hoc test using
SPSS v23 (IBM Corp.). Data are presented as the mean ± SD (n≥3).
P<0.05 was considered to indicate a statistically significant
difference.
Results
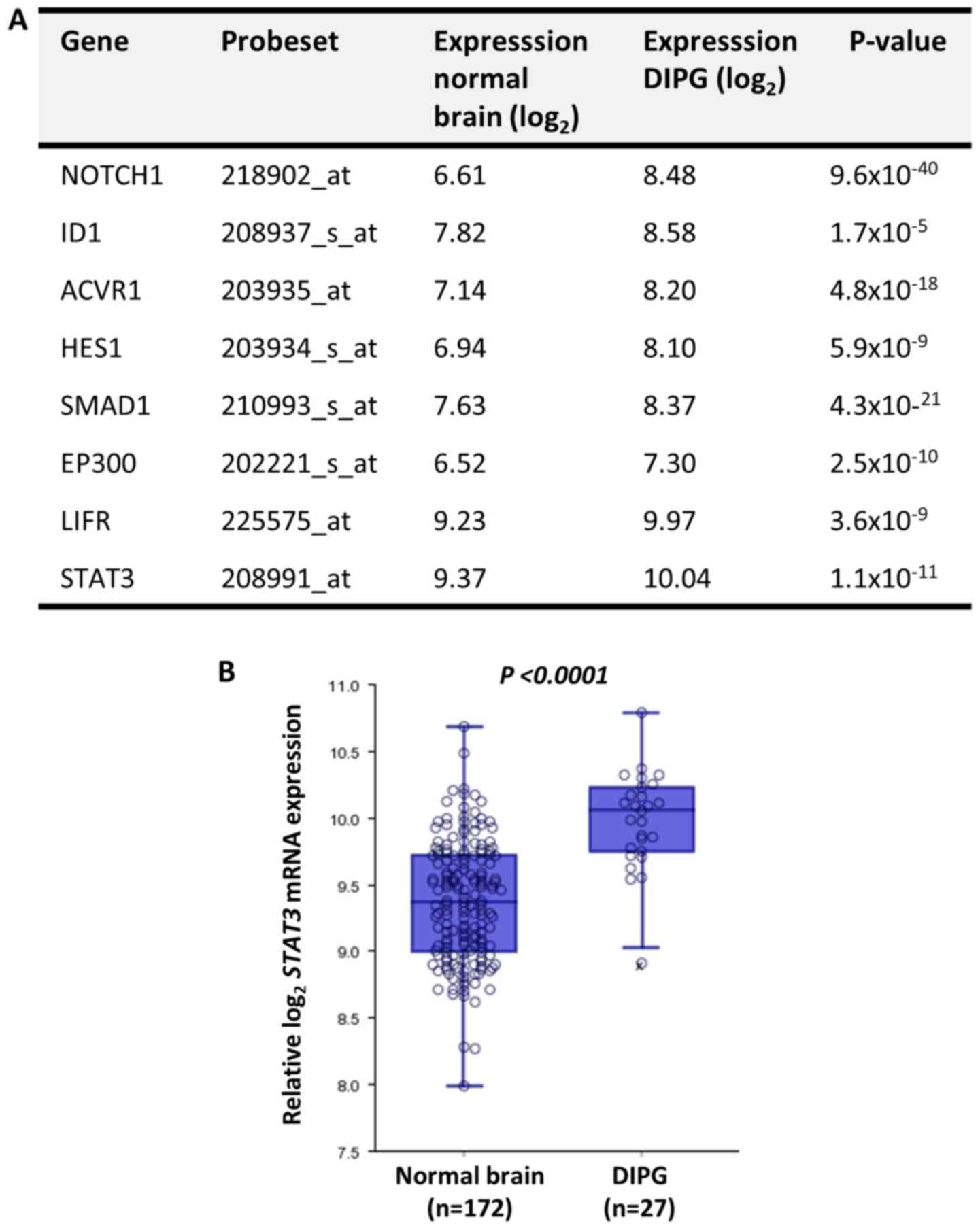
In silico analysis identifies STAT3 as
a potential oncogene in DIPG
The expression levels of astrogliogenesis-associated
genes (30) between normal brain and
DIPG tissues were compared using the publicly available microarray
analysis platform R2 and the Megasampler module, previously
described by Kumar et al (33). According to the expression analysis,
all of these molecules were significantly upregulated in DIPG
compared with in normal brain tissues (Fig. 1). Among the analyzed molecules, HES1
and STAT3 are transcription factors that regulate hallmarks of
cancer (34,35). Based on the results of a previous
study (36) on the radiosensitizing
effect of STAT3 inhibition in glioma, STAT3 was further
investigated as a potential target to inhibit the oncogenic
phenotype of DIPG cells.
STAT3 activation is associated with
DIPG cell viability
To determine the oncogenic role of STAT3, the effect
of STAT3 inactivation on the viability of SF8628 cells was examined
via treatment with the STAT3 inhibitor AG490 or via STAT3 shRNA
transfection. The transfections with shRNAs were confirmed by
RT-semi-qPCR and gel electrophoresis (Fig. 2A). SF8628 DIPG cells were treated
with various concentrations of AG490. Western blotting revealed
that treatment of SF8628 cells with various concentrations of AG490
resulted in a substantial decrease in the protein expression of the
active form of STAT3 (pSTAT3) in a dose-dependent manner, whereas
the protein expression of total STAT3 was not changed (data not
shown). In SF8628 cells treated with 30 µM AG490, cell viability
was significantly reduced compared with cells treated vehicle
control (DMSO), and was similar to the viability of cells treated
with 20 µM AG490 (Fig. 2B).
Therefore, 20 µM AG490 was used in the following experiments. The
CCK-8 assay revealed that the viability of AG490-treated SF8628
cells after 48 h was decreased compared with that of control
vehicle-treated cells (Fig. 2C).
Similar results were observed for cells expressing STAT3 shRNA
(Fig. 2D). Since AG490 treatment did
not change the status of cell apoptosis manifested by cleaved
caspase 3 and cleaved poly (ADP-ribose) polymerase (data not shown)
in SF8628 cells, it was hypothesized that decreased cell viability
by STAT3 inactivation was not a result from increased cell
apoptosis. To further examine the role of STAT3 in the viability of
DIPG cells, the effect of STAT3 inhibition on the expression of a
representative viability marker, cyclin D1, was analyzed. Western
blotting revealed that cyclin D1 expression decreased after STAT3
inhibition using AG490 or STAT3 shRNA (Fig. 2E).
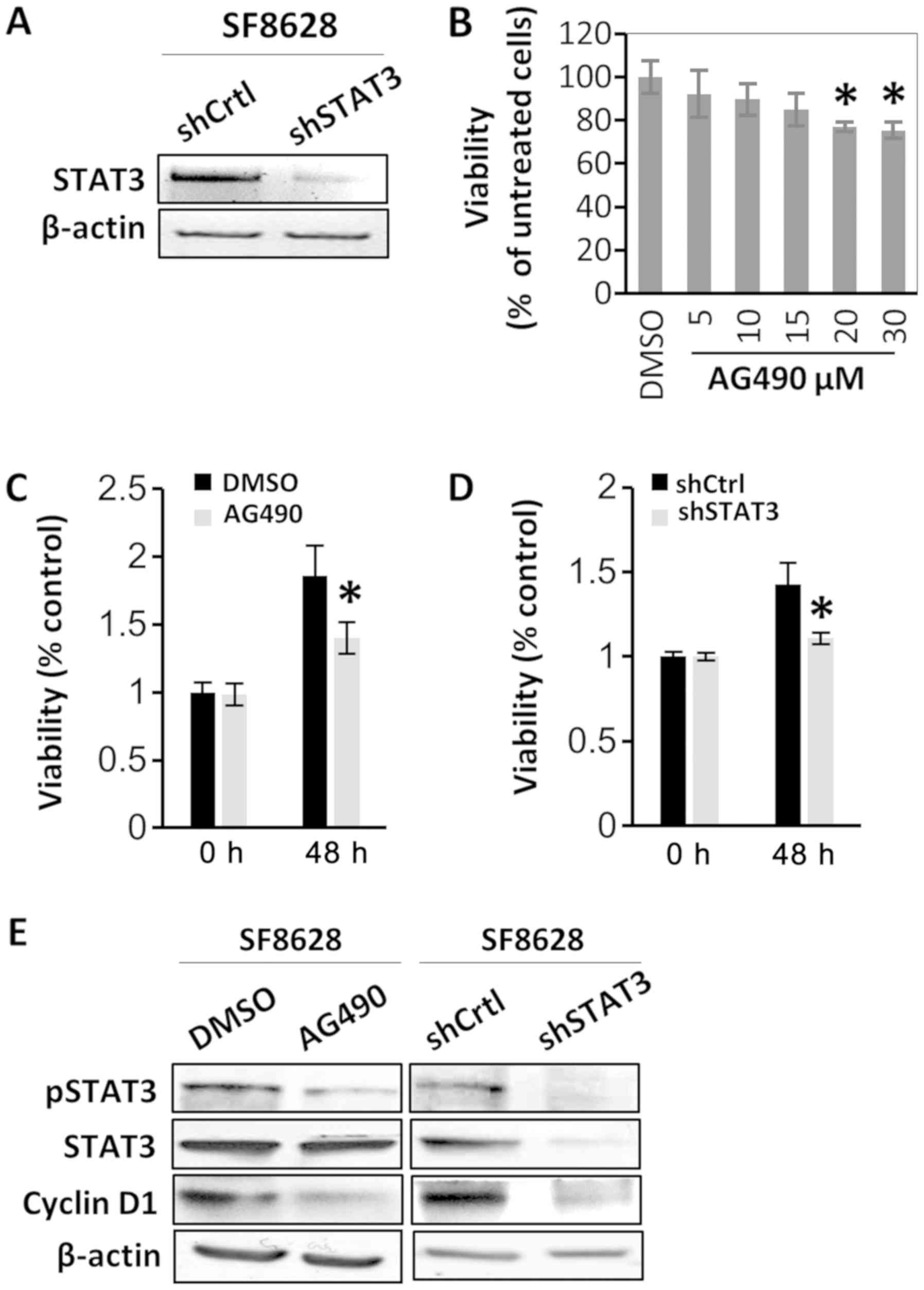 | Figure 2.STAT3 inhibition suppresses human
diffuse intrinsic pontine glioma cell viability. SF8628 cells were
treated with vehicle control (DMSO) or AG490. SF8628 cells were
transfected shCtrl or shSTAT3. (A) mRNA STAT3 expression was
determined via reverse transcription-semi-quantitative PCR. β-actin
mRNA was used as the loading control. (B) Cell viability data of
AG490-treated cells. Cells were treated with various concentrations
of AG490. Cell viability was analyzed using a CCK-8 assay, and
absorbance was measured at 420 nm. *P<0.05 vs. DMSO-treated
cells using one-way ANOVA. Data are presented as the mean ± SD. (C
and D) CCK-8 assay of control cells and STAT3-inhibited cells. Cell
viability was analyzed using a CCK-8 assay, and absorbance was
measured at 420 nm. *P<0.05 vs. DMSO-treated cells or
shCtrl-transfected cells at 48 h using a two-tailed unpaired
Student's t-test. Data are presented as the mean ± SD. (E) Western
blot analysis of pSTAT3, total STAT3 and cyclin D1. Protein samples
were extracted from SF8628 control cells and STAT3-inhibited cells;
β-actin was used as the loading control. Exposure time was 3 min
for pSTAT3, 1 min for STAT3, 30 sec for cyclin D1 and 15 sec for
β-actin. STAT3, signal transducer and activator of transcription 3;
pSTAT3, phosphorylated STAT3; shCtrl, control short hairpin RNA;
shSTAT3, STAT3 short hairpin RNA; CCK-8, Cell Counting Kit-8. |
STAT3 regulates EMT, as well as the
migration and invasion of DIPG cells
As the main oncogenic mechanism of STAT3 is the
promotion of EMT (37), the effect
of STAT3 inactivation in SF8628 cells on EMT, motility and
invasiveness was investigated. Western blotting revealed that STAT3
inactivation increased E-cadherin (epithelial cell marker)
expression, but decreased the expression levels of N-cadherin,
vimentin, Twist, Snail and MMP-9 (mesenchymal cell markers)
(Fig. 3A). Therefore, the present
data suggested a regulatory role of STAT3 in the mesenchymal
transition in DIPG cells. To confirm that STAT3 may promote the
mesenchymal transition in DIPG cells, actin organization was
examined. Control SF8628 cells produced numerous filopodia-like
extensions containing actin-rich bundles, which were absent or less
visible in STAT3-inhibited cells (Fig.
3B). This suggested that the control cells exhibited more
mesenchymal characteristics than the STAT3-inactivated cells.
Migration and invasion assays were performed to assess the direct
effect of STAT3 inhibition on cell motility and invasiveness. STAT3
inhibition significantly decreased cell migration and invasion
(Fig. 3C and D, respectively), which
further confirmed the potential effect of STAT3 on EMT induction in
DIPG cells.
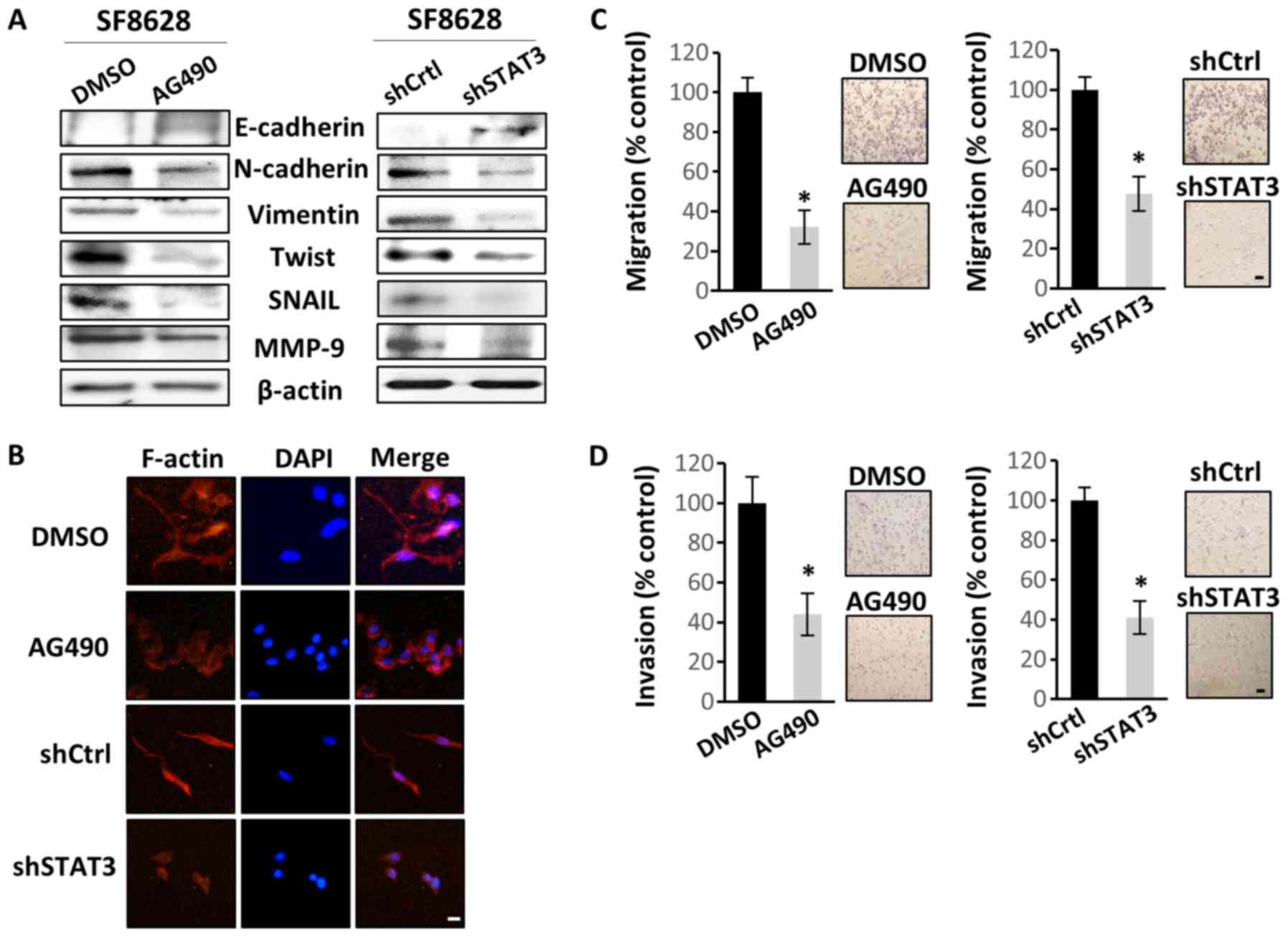 | Figure 3.STAT3 inhibition suppresses human
diffuse intrinsic pontine glioma cell migration and invasion by
regulating EMT. SF8628 cells were treated with vehicle control
(DMSO) or AG490. SF8628 cells were transfected with shCtrl or
shSTAT3. (A) Western blot analysis of EMT markers. Protein samples
were extracted from SF8628 control cells and STAT3-inhibited cells.
Protein samples were tested for E-cadherin, N-cadherin, Vimentin,
Twist, Snail, MMP-9 and β-actin (loading control) expression.
Exposure time was 5 min for E-cadherin, 1 min for N-cadherin, Twist
and Snail, 10 sec for Vimentin, 3 min for MMP-9 and 15 sec for
β-actin. (B) Organization of the actin cytoskeleton was determined
by immunofluorescence staining. Alexa Fluor 633-conjugated
phalloidin was used to visualize F-actin (red), and DAPI staining
(blue) was used for visualization of cell nuclei. Original
magnification, ×400. Scale bar, 2 µm. (C) Cells were seeded in the
upper chamber of Transwell inserts, and the cell migration ability
was assessed 48 h after cell plating. Magnification, ×40. Scale
bar, 100 µm. (D) Cells were seeded in the upper chamber of
Transwell inserts coated with Matrigel, and the cell invasion
ability was measured 48 h after cell plating. Magnification, ×40.
Scale bar, 100 µm. The results were calculated as percentages
relative to control cells. Representative images of invasive cells
are shown next to each bar graph. Data are presented as the mean ±
SD. *P<0.05 using a two-tailed unpaired Student's t-test. STAT3,
signal transducer and activator of transcription 3; shCtrl, control
short hairpin RNA; shSTAT3, STAT3 short hairpin RNA; EMT,
epithelial-mesenchymal transition; MMP-9, matrix
metallopeptidase-9; DAPI, 4′6′-diamidio-2-phenoylindole. |
STAT3 inactivation increases the
therapeutic effect of radiation
As STAT3 inhibitors have been reported to enhance
radiation efficacy in various types of cancer (38–40), the
present study investigated whether STAT3 inactivation was able to
enhance the sensitivity of DIPG cells to radiation. SF8628 cells
were exposed to 4 Gy of radiation 8 h after treatment with AG490 or
DMSO. CCK-8 assays 24 h after radiation indicated that viability
was significantly decreased in cells treated with AG490 compared
with that in cells treated with DMSO (Fig. 4A). Similar results were obtained when
SF8628 cells expressing control shRNA or STAT3 shRNA were exposed
to irradiation, which further suggests that STAT3 inhibition
increases the therapeutic effect of radiation in DIPG cells
(Fig. 4B).
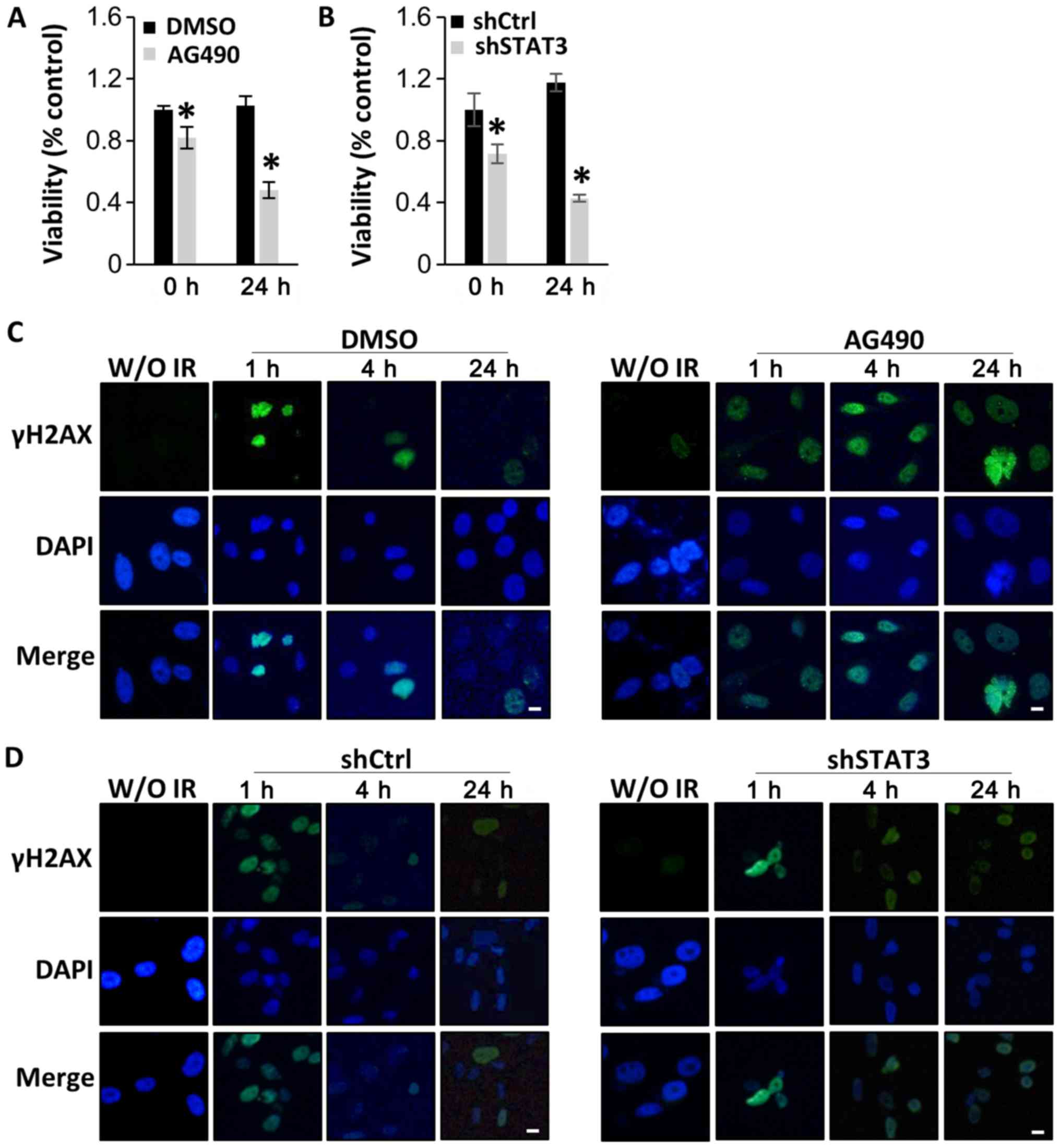 | Figure 4.STAT3 inhibition sensitizes human
diffuse intrinsic pontine glioma cells to radiation by interfering
with DNA damage repair. SF8628 cells were treated with control
vehicle (DMSO) or AG490. SF8628 cells were transfected with shCtrl
or shSTAT3. (A and B) CCK-8 assays of control cells and
STAT3-inhibited cells at 0 and 24 h after radiation treatment. Cell
viability was analyzed using a CCK-8 assay, and absorbance was
measured at 420 nm. *P<0.05 using one-way ANOVA. Data are
presented as the mean ± SD. (C and D) Analysis of DNA damage repair
after treatment with 4 Gy radiation by visualizing the
double-strand marker γH2AX. The panel shows representative images
of γH2AX (green), DAPI-stained nuclei (blue) and merged images of
SF8628 cells without irradiation and at 1, 4 and 24 h after
radiation. Scale bar, 1 µm. STAT3, signal transducer and activator
of transcription 3; shCtrl, control short hairpin RNA; shSTAT3,
STAT3 short hairpin RNA; CCK-8, Cell Counting Kit-8; γH2AX,
phosphorylated H2A X variant histone; DAPI,
4′6′-diamidio-2-phenoylindole. |
Subsequently, the effect of STAT3 inhibition on the
repair of radiation-induced DNA damage was examined using the DNA
double strand break marker γH2AX at 1, 4 and 24 h after treatment
with 4 Gy radiation. As shown in Fig.
4C, γH2AX was localized in the nucleus in both AG490-treated
and control cells after 1 h; these results indicated that DNA
damage occurred early in both AG490-treated and control cells after
radiation treatment. However, decreased expression of γH2AX was
observed over time in cells treated with DMSO and radiation, but
not in those treated with AG490 and radiation, indicating that
repair of DNA damage predominantly occurred in control cells
treated with radiation (Fig. 4C).
Similar results were observed in cells transfected with STAT3
shRNA. Control shRNA-expressing cells treated with radiation
exhibited lower γH2AX positivity than STAT3 shRNA-expressing cells
at 24 h after radiation treatment (Fig.
4D). The present results suggest that STAT3 inhibition enhances
the radiation sensitivity of DIPG cells.
Discussion
Recent technical advances have allowed the
investigation of the concept that DIPG occurrence is associated
with neural differentiation (41).
As STAT3 is often upregulated in numerous types of cancer and
serves a role in astrocyte differentiation, the present study
investigated the effect of STAT3 inhibition on the oncogenic
behavior of the DIPG SF8628 cell line. First, it was observed that
STAT3 expression was upregulated in DIPG tissues compared with
normal brain tissues. Second, the effect of STAT3 upregulation was
assessed by inhibition of STAT3 using shRNA or the STAT3 inhibitor
AG490, and it was revealed that cell viability, migration and
invasion were decreased as STAT3 was inhibited. Additionally, STAT3
inhibition induced changes opposite to those observed in EMT, such
as an upregulated E-cadherin expression and downregulated Snail and
MMP9 expression. Furthermore, the degree of cellular actin
remodeling was decreased, suggesting the possibility that STAT3 may
contribute to EMT in DIPG cells. Finally, the therapeutic potential
of STAT3 inhibition was assessed in combination with radiation,
which is the current mode of treatment. Inhibition of STAT3 in DIPG
cells decreased their viability, as well as their DNA repair
capacity. Although the current study contains the limitation of
using a single DIPG cell line, the present results are consistent
with previous reports on the role of STAT3 in cell viability,
migration, prognosis and therapeutic resistance in glioma (28,29).
STAT3 is one of the major potential targets that may
be applied in molecular targeted therapy due to its known
regulatory effects on oncogenic proteins. For example, STAT3
transcriptionally regulates cyclin D1 (42) and mesenchymal cell markers (37), such as Slug, Snail and Twist, which
drive transformation and ultimately metastasis. In the present
study, STAT3 inactivation decreased cyclin D1 expression and SF8628
cell viability. However, the current data did not reveal the
transcriptional mechanism underlying cyclin D1 regulation by STAT3;
nonetheless, based on previous studies (42,43), it
is hypothesized that STAT3 may act as a transactivator of cyclin D1
transcription in DIPG cells.
EMT confers migration and invasion abilities, and
induces molecular phenotype changes in cancer cells (44). In addition, various types of cancer
that are resistant to cytotoxic therapy are prone to displaying a
mesenchymal phenotype (45). For
example, chemoresistance has been attributed to EMT in adult
glioblastoma (46,47). Since EMT is one of the primary
changes induced by STAT3 in cancer, the present study evaluated the
effect of STAT3 inhibition in DIPG cells in association with this
process. STAT3 inhibition decreased the migration and invasiveness
of DIPG cells. Furthermore, STAT3 inhibition upregulated the
expression levels of E-cadherin, an epithelial cell marker, and
downregulated the expression levels of mesenchymal cell markers,
indicating a change toward an epithelial phenotype. Therefore, the
present results suggest that STAT3 contributes to the mesenchymal
phenotype of DIPG through EMT, consistent with evidence supporting
the possible role of the JAK/STAT signaling pathway in the
mesenchymal transition in pediatric glioma (48). Although the JAK/STAT3/TWIST signaling
pathway is involved in neurogenesis and astrogliogenesis under
normal conditions (49), aberrant
activation of this pathway may induce EMT in cancer (50,51).
The STAT3 signaling pathway is known to serve a role
in radioresistance in numerous types of cancer, including squamous
cell carcinoma and head and neck carcinoma (52), and similar results have been reported
for high-grade gliomas (53,54). A previous in vitro and in
vivo study has demonstrated that a STAT3 inhibitor potentiates
the radiosensitizing effect of temozolomide in glioblastoma
(55). In particular, previous
studies (56,57) have reported that STAT3-induced EMT
mediates radioresistance in esophageal squamous carcinoma and
glioblastoma. In esophageal squamous carcinoma, IL6/STAT3/TWIST
signaling pathway-mediated EMT confers radioresistance both in
vitro and in vivo (56).
Additionally, the STAT3/Slug axis induces the EMT phenotype and
radioresistance (57). A possible
mechanism underlying STAT3/EMT/radioresistance is hypothesized to
be associated with the role of STAT3 in DNA damage repair. Several
studies have indicated that radiation-induced ataxia telangiectasia
mutated, which is a key molecule initiating DNA damage repair upon
radiation (58), positively
regulates STAT3 via an unknown mechanism (59,60).
Therefore, STAT3 may serve a role in DNA damage repair mechanisms
and mediate radiation resistance. In the present study, the
combination of STAT3 inhibition and radiation resulted in
significant combinatorial efficacy in DIPG cells. Therefore, the
present data support STAT3 inhibition as a potential treatment in
clinical studies of DIPG, but further investigations are required
to clarify the precise mechanism underlying radiation
resistance.
The role of STAT3 in neural development may reflect
its participation in the oncogenesis of DIPG. It has been
demonstrated that activation of STAT signaling via gp130 in
cooperation with bone morphogenetic protein (BMP) signaling leads
to the formation of the STAT3/SMAD/p300 complex on the
astrogliogenic gene promoter (61)
and inhibits oligodendrocyte progenitor cell differentiation into
mature oligodendrocytes (62).
Notably, ~25% of DIPGs display activating mutations in ACVR1, which
encodes the activin receptor-like kinase-2 in the BMP signaling
pathway. Furthermore, the present study revealed that expression
levels of principal astrogliogenesis-inducing proteins were mostly
increased in DIPG compared with in normal brain. Overall, it can be
hypothesized that increased STAT3 expression, in combination with
the overexpression of BMP signaling pathway components caused by
ACVR1 mutations, may cause excessive astrogliogenesis. Accordingly,
malfunction of the BMP signaling pathway, including its key
molecules ACVR1 and STAT3, may be a key driver in the development
of DIPG with an astrocytic phenotype.
In conclusion, the present study revealed that STAT3
was upregulated in human DIPG samples compared with in normal brain
samples, and that STAT3 inhibition decreased the oncogenic
phenotype of DIPG cells. Furthermore, STAT3 inhibition enhanced the
therapeutic effects of radiation therapy in DIPG cells. Therefore,
STAT3 may serve as a therapeutic target for DIPG.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Research Foundation of Korea Grant funded by the Korean Government
(grant no. NRF-2018R1A5A2025964), the Bio & Medical Technology
Development Program of the National Research Foundation of Korea
funded by the Ministry of Science & Information and
Communications Technology (grant no. NRF-2018-M3A9H3021707) and the
Seoul National University Hospital Research Fund (grant no.
03-2016-0350).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The datasets GSE11882 (https://www.ncbi.nlm.nih.gov/nucleotide?cmd=search&term=GSE11882)
and GSE26576 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE26576)
were analyzed using the Genomics Analysis and Visualization
platform (http://r2.amc.nl).
Authors' contributions
JP and JYL designed the study and wrote the original
draft. JP was in charge of all experiments. SY, SPK, WL, KHK, JIK,
SKK and KCW interpreted the data and revised the manuscript. All
authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Warren KE: Diffuse intrinsic pontine
glioma: Poised for progress. Front Oncol. 2:2052012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Khatua S, Sadighi ZS, Pearlman ML, Bochare
S and Vats TS: Brain tumors in children-current therapies and newer
directions. Indian J Pediatr. 79:922–927. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Vanan MI and Eisenstat DD: DIPG in
Children-what can we learn from the past? Front Oncol. 5:2372015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jansen MH, Van Vuurden DG, Vandertop WP
and Kaspers GJ: Diffuse intrinsic pontine gliomas: A systematic
update on clinical trials and biology. Cancer Treat Rev. 38:27–35.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pollack IF, Jakacki RI, Blaney SM, Hancock
ML, Kieran MW, Phillips P, Kun LE, Friedman H, Packer R, Banerjee
A, et al: Phase I trial of imatinib in children with newly
diagnosed brainstem and recurrent malignant gliomas: A pediatric
brain tumor consortium report. Neuro Oncol. 9:145–160. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Geoerger B, Hargrave D, Thomas F, Ndiaye
A, Frappaz D, Andreiuolo F, Varlet P, Aerts I, Riccardi R, Jaspan
T, et al: Innovative therapies for Children with cancer pediatric
phase I study of erlotinib in brainstem glioma and
relapsing/refractory brain tumors. Neuro Oncol. 13:109–118. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B,
Li Y, Zhu X, Qu C, Chen X, Zhang J, et al: The genomic landscape of
diffuse intrinsic pontine glioma and pediatric non-brainstem
high-grade glioma. Nat Genet. 46:444–450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hall A, Giese NA and Richardson WD: Spinal
cord oligodendrocytes develop from ventrally derived progenitor
cells that express PDGF alpha-receptors. Development.
122:4085–4094. 1996.PubMed/NCBI
|
|
9
|
Verschueren K, Dewulf N, Goumans MJ,
Lonnoy O, Feijen A, Grimsby S, Vandi Spiegle K, ten Dijke P, Morén
A, Vanscheeuwijck P, et al: Expression of type I and type IB
receptors for activin in midgestation mouse embryos suggests
distinct functions in organogenesis. Mech Dev. 52:109–123. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Buczkowicz P, Bartels U, Bouffet E, Becher
O and Hawkins C: Histopathological spectrum of paediatric diffuse
intrinsic pontine glioma: Diagnostic and therapeutic implications.
Acta Neuropathol. 128:573–581. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Saratsis AM, Kambhampati M, Snyder K,
Yadavilli S, Devaney JM, Harmon B, Hall J, Raabe EH, An P, Weingart
M, et al: Comparative multidimensional molecular analyses of
pediatric diffuse intrinsic pontine glioma reveals distinct
molecular subtypes. Acta Neuropathol. 127:881–895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Achanta P, Sedora Roman NI and
Quiñones-Hinojosa A: Gliomagenesis and the use of neural stem cells
in brain tumor treatment. Anticancer Agents Med Chem. 10:121–130.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Levy DE and Lee CK: What does Stat3 do? J
Clin Invest. 109:1143–1148. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Duncan SA, Zhong Z, Wen Z and Darnell JE
Jr: STAT signaling is active during early mammalian development.
Dev Dyn. 208:190–198. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wegenka UM, Buschmann J, Lütticken C,
Heinrich PC and Horn F: Acute-phase response factor, a nuclear
factor binding to acute-phase response elements, is rapidly
activated by interleukin-6 at the posttranslational level. Mol Cell
Biol. 13:276–288. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levy DE and Darnell JE Jr: Stats:
Transcriptional control and biological impact. Nat Rev Mol Cell
Biol. 3:651–662. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fan G, Martinowich K, Chin MH, He F, Fouse
SD, Hutnick L, Hattori D, Ge W, Shen Y, Wu H, et al: DNA
methylation controls the timing of astrogliogenesis through
regulation of JAK-STAT signaling. Development. 132:3345–3356. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ma JH, Qin L and Li X: Role of STAT3
signaling pathway in breast cancer. Cell Commun Signal. 18:332020.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Dutta P, Sabri N, Li J and Li WX: Role of
STAT3 in lung cancer. JAKSTAT. 3:e9995032014.PubMed/NCBI
|
|
20
|
Corcoran RB, Contino G, Deshpande V,
Tzatsos A, Conrad C, Benes CH, Levy DE, Settleman J, Engelman JA
and Bardeesy N: STAT3 plays a critical role in KRAS-induced
pancreatic tumorigenesis. Cancer Res. 71:5020–5029. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu CL, Meyer DJ, Campbell GS, Larner AC,
Carter-Su C, Schwartz J and Jove R: Enhanced DNA-binding activity
of a Stat3-related protein in cells transformed by the Src
oncoprotein. Science. 269:81–83. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Garcia R, Yu CL, Hudnall A, Catlett R,
Nelson KL, Smithgall T, Fujita DJ, Ethier SP and Jove R:
Constitutive activation of Stat3 in fibroblasts transformed by
diverse oncoproteins and in breast carcinoma cells. Cell Growth
Differ. 8:1267–1276. 1997.PubMed/NCBI
|
|
23
|
Lund TC, Prator PC, Medveczky MM and
Medveczky PG: The Lck binding domain of herpesvirus saimiri tip-484
constitutively activates Lck and STAT3 in T cells. J Virol.
73:1689–1694. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wen X, Lin HH, Shih HM, Kung HJ and Ann
DK: Kinase activation of the non-receptor tyrosine kinase Etk/BMX
alone is sufficient to transactivate STAT-mediated gene expression
in salivary and lung epithelial cells. J Biol Chem.
274:38204–38210. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
West AJ, Tsui V, Stylli SS, Nguyen HPT,
Morokoff AP, Kaye AH and Luwor RB: The role of interleukin-6-STAT3
signalling in glioblastoma. Oncol Lett. 16:4095–4104.
2018.PubMed/NCBI
|
|
26
|
Ganguly D, Fan M, Yang CH, Zbytek B,
Finkelstein D, Roussel MF and Pfeffer LM: The critical role that
STAT3 plays in glioma-initiating cells: STAT3 addiction in glioma.
Oncotarget. 9:22095–22112. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim JE, Patel M, Ruzevick J, Jackson CM
and Lim M: STAT3 Activation in glioblastoma: Biochemical and
therapeutic implications. Cancers (Basel). 6:376–395. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ouédraogo ZG, Biau J, Kemeny JL, Morel L,
Verrelle P and Chautard E: Role of STAT3 in genesis and progression
of human malignant gliomas. Mol Neurobiol. 54:5780–5797. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chang N, Ahn SH, Kong DS, Lee HW and Nam
DH: The role of STAT3 in glioblastoma progression through dual
influences on tumor cells and the immune microenvironment. Mol Cell
Endocrinol. 451:53–65. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chuang JH, Tung LC and Lin Y: Neural
differentiation from embryonic stem cells in vitro: An overview of
the signaling pathways. World J Stem Cells. 7:437–447. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Berchtold NC, Cribbs DH, Coleman PD,
Rogers J, Head E, Kim R, Beach T, Miller C, Troncoso J, Trojanowski
JQ, et al: Gene expression changes in the course of normal brain
aging are sexually dimorphic. Proc Natl Acad Sci USA.
105:15605–15610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Paugh BS, Broniscer A, Qu C, Miller CP,
Zhang J, Tatevossian RG, Olson JM, Geyer JR, Chi SN, da Silva NS,
et al: Genome-wide analyses identify recurrent amplifications of
receptor tyrosine kinases and cell-cycle regulatory genes in
diffuse intrinsic pontine glioma. J Clin Oncol. 29:3999–4006. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kumar SS, Sengupta S, Lee K, Hura N,
Fuller C, DeWire M, Stevenson CB, Fouladi M and Drissi R: BMI-1 is
a potential therapeutic target in diffuse intrinsic pontine glioma.
Oncotarget. 8:62962–62975. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu ZH, Dai XM and Du B: Hes1: A key role
in stemness, metastasis and multidrug resistance. Cancer Biol Ther.
16:353–359. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kamran MZ, Patil P and Gude RP: Role of
STAT3 in cancer metastasis and translational advances. Biomed Res
Int. 2013:4218212013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang YP, Chang YL, Huang PI, Chiou GY,
Tseng LM, Chiou SH, Chen MH, Chen MT, Shih YH, Chang CH, et al:
Resveratrol suppresses tumorigenicity and enhances radiosensitivity
in primary glioblastoma tumor initiating cells by inhibiting the
STAT3 axis. J Cell Physiol. 227:976–993. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wendt MK, Balanis N, Carlin CR and
Schiemann WP: STAT3 and epithelial-mesenchymal transitions in
carcinomas. JAKSTAT. 3:e289752014.PubMed/NCBI
|
|
38
|
Lau J, Ilkhanizadeh S, Wang S,
Miroshnikova YA, Salvatierra NA, Wong RA, Schmidt C, Weaver VM,
Weiss WA and Persson AI: STAT3 blockade inhibits radiation-induced
malignant progression in glioma. Cancer Res. 75:4302–4311. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bharadwaj U, Eckols TK, Xu X, Kasembeli
MM, Chen Y, Adachi M, Song Y, Mo Q, Lai SY and Tweardy DJ:
Small-molecule inhibition of STAT3 in radioresistant head and neck
squamous cell carcinoma. Oncotarget. 7:26307–26330. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
You S, Li R, Park D, Xie M, Sica GL, Cao
Y, Xiao ZQ and Deng X: Disruption of STAT3 by niclosamide reverses
radioresistance of human lung cancer. Mol Cancer Ther. 13:606–616.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Puget S, Philippe C, Bax DA, Job B, Varlet
P, Junier MP, Andreiuolo F, Carvalho D, Reis R, Guerrini-Rousseau
L, et al: Mesenchymal transition and PDGFRA amplification/mutation
are key distinct oncogenic events in pediatric diffuse intrinsic
pontine gliomas. PLoS One. 7:e303132012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Leslie K, Lang C, Devgan G, Azare J,
Berishaj M, Gerald W, Kim YB, Paz K, Darnell JE, Albanese C, et al:
Cyclin D1 is transcriptionally regulated by and required for
transformation by activated signal transducer and activator of
transcription 3. Cancer Res. 66:2544–2552. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Gu J, Li G, Sun T, Su Y, Zhang X, Shen J,
Tian Z and Zhang J: Blockage of the STAT3 signaling pathway with a
decoy oligonucleotide suppresses growth of human malignant glioma
cells. J Neurooncol. 89:9–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Brabletz T, Kalluri R, Nieto MA and
Weinberg RA: EMT in cancer. Nat Rev Cancer. 18:128–134. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shintani Y, Okimura A, Sato K, Nakagiri T,
Kadota Y, Inoue M, Sawabata N, Minami M, Ikeda N, Kawahara K, et
al: Epithelial to mesenchymal transition is a determinant of
sensitivity to chemoradiotherapy in non-small cell lung cancer. Ann
Thorac Surg. 92:1794–1804. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Siebzehnrubl FA, Silver DJ, Tugertimur B,
Deleyrolle LP, Siebzehnrubl D, Sarkisian MR, Devers KG, Yachnis AT,
Kupper MD, Neal D, et al: The ZEB1 pathway links glioblastoma
initiation, invasion and chemoresistance. EMBO Mol Med.
5:1196–1212. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kahlert UD, Nikkhah G and Maciaczyk J:
Epithelial-to-mesenchymal(−like) transition as a relevant molecular
event in malignant gliomas. Cancer Lett. 331:131–138. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Meel MH, Schaper SA, Kaspers GJL and
Hulleman E: Signaling pathways and mesenchymal transition in
pediatric high-grade glioma. Cell Mol Life Sci. 75:871–887. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cao F, Hata R, Zhu P, Nakashiro K and
Sakanaka M: Conditional deletion of Stat3 promotes neurogenesis and
inhibits astrogliogenesis in neural stem cells. Biochem Biophys Res
Commun. 394:843–847. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Uddin N, Kim RK, Yoo KC, Kim YH, Cui YH,
Kim IG, Suh Y and Lee SJ: Persistent activation of STAT3 by
PIM2-driven positive feedback loop for epithelial-mesenchymal
transition in breast cancer. Cancer Sci. 106:718–725. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Xiong H, Hong J, Du W, Lin YW, Ren LL,
Wang YC, Su WY, Wang JL, Cui Y, Wang ZH and Fang JY: Roles of STAT3
and ZEB1 proteins in E-cadherin down-regulation and human
colorectal cancer epithelial-mesenchymal transition. J Biol Chem.
287:5819–5832. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Han X, Xue X, Zhou H and Zhang G: A
molecular view of the radioresistance of gliomas. Oncotarget.
8:100931–100941. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gao L, Li F, Dong B, Zhang J, Rao Y, Cong
Y, Mao B and Chen X: Inhibition of STAT3 and ErbB2 suppresses tumor
growth, enhances radiosensitivity, and induces
mitochondria-dependent apoptosis in glioma cells. Int J Radiat
Oncol Biol Phys. 77:1223–1231. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yuan X, Du J, Hua S, Zhang H, Gu C, Wang
J, Yang L, Huang J, Yu J and Liu F: Suppression of autophagy
augments the radiosensitizing effects of STAT3 inhibition on human
glioma cells. Exp Cell Res. 330:267–276. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han TJ, Cho BJ, Choi EJ, Kim DH, Song SH,
Paek SH and Kim IA: Inhibition of STAT3 enhances the
radiosensitizing effect of temozolomide in glioblastoma cells in
vitro and in vivo. J Neurooncol. 130:89–98. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zang C, Liu X, Li B, He Y, Jing S, He Y,
Wu W, Zhang B, Ma S, Dai W, et al: IL-6/STAT3/TWIST inhibition
reverses ionizing radiation-induced EMT and radioresistance in
esophageal squamous carcinoma. Oncotarget. 8:11228–11238. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lin JC, Tsai JT, Chao TY, Ma HI and Liu
WH: The STAT3/Slug axis enhances radiation-induced tumor invasion
and cancer stem-like properties in radioresistant glioblastoma.
Cancers (Basel). 10(pii): E5122018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Guleria A and Chandna S: ATM kinase: Much
more than a DNA damage responsive protein. DNA Repair (Amst).
39:1–20. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding
Q, Xu Z and Chen Y: Inhibition of ATM reverses EMT and decreases
metastatic potential of cisplatin-resistant lung cancer cells
through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 38:1492019.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zhang Y, Cho YY, Petersen BL, Bode AM, Zhu
F and Dong Z: Ataxia telangiectasia mutated proteins, MAPKs, and
RSK2 are involved in the phosphorylation of STAT3. J Biol Chem.
278:12650–12659. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Shimazaki T and Okano H: Heterochronic
microRNAs in temporal specification of neural stem cells:
Application toward rejuvenation. NPJ Aging Mech Dis. 2:150142016.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Samanta J and Kessler JA: Interactions
between ID and OLIG proteins mediate the inhibitory effects of BMP4
on oligodendroglial differentiation. Development. 131:4131–4142.
2004. View Article : Google Scholar : PubMed/NCBI
|