Introduction
The protein synthesis is a very complex process
involved in cell growth, differentiation and survival. Molecular
chaperones, such as heat shock proteins (HSPs), are key elements in
these biological processes, helping the polypeptide folding
involved in reaching functional conformation, as well as
facilitating the protein stability, trafficking and proteolytic
turnover (1).
HSPs are present in almost all living organisms and
their expression is increased in response to different physical,
chemical and biological insults, including temperature, presence of
heavy metals and oxidative stress (2). Most of the elements inducing cellular
stress might cause protein denaturation and must be reduced or
eliminated with the help of molecular chaperones. In the process of
cell senescence and in several diseases a defective chaperone
function was noted (1,2). If cellular stress proceeds unchecked by
the common mechanisms of action regulated by chaperones,
intracellular proteins become denatured and insoluble, and then
precipitates as aggregates. The development of inclusion bodies is,
for instance, a usual pathological process in several disorders,
even in the absence of cellular stress (2). Drugs able to modulate the heat shock
response and induce HSP expression might have a potential
therapeutic value. In neoplastic cells, HSPs (mainly HSP90) are
often overexpressed. As reported by Garcia-Carbonero and
collaborators, the oncogenic potential of cells is dependent on
their ability to survive despite endogenous (hypoxia, pH changes,
nutrient deprivation, dysregulated signaling pathways) and
exogenous (radiation or chemotherapy) insults (3). Moreover, increased HSP expression may
stabilize oncogenic proteins and promote independence of growth
factors, cancer-cell survival, proliferation, immortalization,
neo-vascularization and metastasis (3).
HSP90 is constitutively expressed in normal cells,
representing 1–2% of the total intracellular protein. Since in
neoplastic cells, its levels increase to 4–6% (4), HSP90 has thus emerged as significant
target in several diseases and, in particular, being a protein with
the ability to act as regulator on tumor cell cycle progression, it
is considered a remarkable therapeutic target for anticancer drug
development. Therefore, the interest for new possible HSP90
inhibitors is intensified (5).
With the identification of this interesting new
target, pure natural molecules isolated from plants and other
natural organisms have been found to be an important source of new
HSP90 inhibitors, with the potential of being of interest for the
future development of novel selective anticancer agents (6).
Geldanamycin and radicicol are natural products to
inhibit the HSP90 activity. In the past, studies on the natural
antibiotic geldanamycin demonstrated that this benzoquinone
ansamycin exhibits potent antitumor activity against human cancer
cells (7), but unfortunately its
therapeutic usage has been limited, due to its poor water
solubility and severe hepatotoxicity. Recently, the chemical
structure of geldanamycin has been modified with the aim to reduce
toxicity issues and to increase its water-solubility. The position
17 of the benzoquinone group represents the modified moiety to
generate new derivatives, including 17-allylamino-, 17-demethoxy
geldanamycin (17-AAG) and
17-N,N-dimethylaminoethylamino-17-dimethoxy geldanamycin (17-DMAG),
studied in phase II/III or I in clinical trials, respectively
(8).
Radicicol (9), the
second mentioned natural active agent, is a macrocyclic anti-fungal
antibiotic, also able to inhibit HSP90 activity by interacting with
the same site of action of geldanamycin (9,10).
However, due to its intrinsic chemical instability, it displayed
very limited in vivo effects.
Starting from these and other known natural
molecules, many synthetic efforts have been described in literature
to obtain other Hsp90 inhibitors with better features. Some of them
reported the 1,3-dihydroxybenzene (resorcinol ring, present also in
radicicol structure) bound to a pyrazole or isoxazole ring as an
important scaffold for very active molecules, such as the drug
candidate NVP-AUY922 (Luminespib) (11).
Some other synthetic compounds, including the
derivatives used in the present study and containing an isoxazole
nucleus, have recently shown potent and selective inhibition of
HSP90 (12,13). The presence of the heterocyclic
nucleus seems to exert an important role in the docking of these
derivatives to the ATP-binding site of HSP90 (14).
We (12,13) and other research groups (11,14) have
studied new resorcinol structurally related molecules. Our novel
synthetized inhibitors of HSP90 (compounds 1–8, Fig. 1) (12,13),
investigated in the present work, are characterized by
modifications on the isoxazole scaffold focusing mainly on the C-4
position, by introducing of a second amide group to ameliorate the
protein interaction, producing an extra interaction with Lys58, as
well as a concomitant reorientation of the aromatic portion. The
key interactions of the OH-resorcinol (1,3-dihydroxybenzene) groups
and the C-3 amide still remain identical in the series of
3,4-isoxazolediamides (12).
The other series here reported is represented by
4,5,6,7-tetrahydro-isoxazolo-[4,5-c]-pyridine analogues (compounds
9–13, Fig. 2), containing a
condensed bicyclic group. Also, in this series, the resorcinol
portion was maintained because of its importance and role in the
interaction with the HSP90 protein. However, structural alterations
and substitution of the resorcinol group were investigated
(13).
The first aim of the present study was to determine
whether these new derivatives exhibit antiproliferative effects on
the K562 human experimental cellular system (15). This model system was selected in
consideration of the fact that it has been proposed as very useful
for the screening of antitumor compounds, and that undergoes
terminal differentiation when exposed to some antitumor drugs
(16,17). Since inhibitory effects of tumor cell
growth might be associated to activation of early and late
apoptosis, the second and more general aim was to investigate the
possible pro-apoptotic effect of these compounds on K562 human
leukemia cells and on two additional cell lines representative of
solid tumors, the glioblastoma U251-MG and T98G cell lines. It
should be underlined that comparing glioblastoma cell lines which
respond (the U251-MG) or not (the T98G) to temozolomide (TMZ)
treatment is very important, since glioblastoma multiforme (VI
grade tumor) is one of the most aggressive solid tumors and TMZ
chemotherapy, while remaining the most commonly used clinical
treatment, cannot be proposed in TMZ resistant tumors (18).
Materials and methods
Chemical compounds
3,4-isoxazolediamides and
4,5,6,7-tetrahydro-isoxazolo-[4,5-c]-pyridine derivatives were
synthesized as reported in Baruchello et al (12,13).
Compounds 1–8 were synthesized as described in ref. 12. Compounds 9–11 were synthesized as
described in ref. 13. Compounds 12
and 13 were obtained similarly as described in ref. 13. For characterization of the compounds,
1H-NMR data were recorded at 200 MHz on a Bruker AC 200
spectrometer. Electrospray mass analyses were recorded on a double
focusing Finnigan MAT 95 instrument with BE geometry. Melting
points were determined on a Reichter-Kofler apparatus. HPLC
analysis were performed with a Jupiter column 100×4.6 mm, 5 µm
C18, 1 ml/min flow, H2O + 0.1%
TFA/Acetonitrile + 0.1% TFA, T0 95/5%, T25
0/100%, UV detector 220 nm. Combustion analysis of all the
compounds is compatible with a purity >95%. The compounds 1–13
were solubilized in dimethyl sulfoxide (DMSO) and further dilutions
(1 mM-1 µM) were made with ethanol (EtOH). The stock solutions
(10–50 mM) were stored at −20°C and thawed just before the
treatment.
Cell cultures
Human erythroleukemic K562 cells are a human
immortalized myelogenous leukemia cell line isolated and
characterized by Lozzio CB and Lozzio BB, from a patient with
chronic myelogenous leukemia (CML) in blast crisis (19,20).
U251-MG glioma cells (Sigma-Aldrich; Merck KGaA) are derived from a
male patient with malignant astrocytoma and they represent an
important biological model to study genetic aberrations and
molecular pathways in a complex tumor such as glioblastoma
(21). The T98G cell line
(Sigma-Aldrich; Merck KGaA) was derived from a glioblastoma
multiforme tumor of a Caucasian male (18). K562, U215 and T98G cells were
cultured in humidified atmosphere of 5% CO2, in
RPMI-1640 medium (Lonza) supplemented with 10% fetal bovine serum
filtered 0.2 µM (FBS; Biowest, Nuaillè, France), 50 U/ml penicillin
(Lonza) and 50 µg/ml streptomycin (Lonza). Cell viability of
control untreated cells was always >95%, based on trypan-blue
exclusion tests and on the FACS analyses presented in Figs. 3–6.
Antiproliferative activity
The cells are counted with the Beckman
Coulter® Z2 (Beckman Coulter, Inc.) to evaluate the
possible antiproliferative effect caused by the compounds 1–13
after 72 and 96 h from the treatment, when they are in the log
phase of growth. Preliminary tests were done to identify the
IC50 on K562 cells: 25,000 cells/ml were seeded in
24-well plates and treated with 3,4-isoxazolediamides and
4,5,6,7-tetrahydro-isoxazolo-[4,5-c]-pyridine compounds at
different concentrations. Preliminary tests were done using
large-scale concentrations (1 nM, 10 nM, 100 nM, 1 µM, 10 µM, 100
µM and 300 µM) in order to identify the IC50 values on
K562 cell line, and then experiments were made with selected
concentrations near the IC50 found concentrations. All
the experiments were repeated at least three times,
independently.
Pro-apoptotic assay
K562, U251-MG and T98G cell lines were treated with
two specific concentrations, both close to the IC50
values, previously determinated, in order to study the apoptotic
effects of the derivatives. Annexin V and Dead Cell assays on all
the used cell lines, untreated and treated with increasing doses of
the selected isoxazole derivatives 1–11, were performed with the
Muse cell analyzer (EMD Millipore), according to the instructions
supplied by the manufacturer. This procedure utilizes Annexin V to
detect phosphatidyl serine (PS) on the external membrane of
apoptotic cells. Four populations of cells can be distinguished
using this assay: live, early apoptotic, late apoptotic and dead
cells. The MUSE cell analyzer is able to discriminate ‘early’ from
‘late’ apoptosis since ‘early apoptotic cells’ are Annexin V
positive, and 7-AAD negative, while ‘late apoptotic cells’ are
positive to both Annexin V and 7-AAD. Cells were washed with
sterile 1X PBS, tripsinized, resuspended in the original medium and
diluted (1:2) with the one step addition of the Muse Annexin V
& Dead Cell reagent. After incubation of 20 min at room
temperature, samples were analyzed, using Triton X 0.01%, as
positive control (22). Triton X was
employed in order to obtain a complete destruction of the cell
membrane leading, following the MUSE analysis a very high death
pattern. Data are acquired and recorded utilizing the Annexin V and
Dead Cell Software Module (EMD Millipore). Caspase 3/7 activity was
also analyzed on T98G cell line with the Muse cell analyzer (EMD
Millipore), according to the instructions supplied by the
manufacturer. After treatment with isoxazoles, cells were
harvested, lysed and centrifuged. The Muse™ Caspase 3/7 is a
non-toxic cell membrane permeable reagent; it contains a DNA
binding dye that is linked to an amino acid sequence
Asp-Glu-Val-Asp (DEVD) substrate. Cleavage by active Caspase 3/7 of
the DEVD peptide substrate in the cell results in release of the
dye, translocation to the nucleus and binding of the dye to DNA
with high fluorescence emission. A dead cell marker, 7-AAD
(7-aminoactinomycin D), is also included in both the Annexin V and
Caspase 4/7 assays as an indicator of cell membrane structural
integrity and cell death. It is excluded from live, healthy cells,
as well as early apoptotic cells, but permeates later stage
apoptotic and dead cells. With this assay the percentage of cells
that are live, dead, in the early and late stages of apoptosis can
be detected. Control experiments demonstrated that no effects of
the solvents used was observed at the maximum concentrations used
(data not shown).
Induction of differentiation
K562 cells grow in culture as single,
undifferentiated, cells in suspension, with low basal production of
hemoglobin. As several antitumor compounds exert their action
through activation of differentiated functions (17), erythroid differentiation was assayed
on K562 cell cultures after 5–7 days of treatment (when K562 cells
are in the plateau phase of cell growth) by counting
benzidine/H2O2 positive cells in a solution
containing 0.2% benzidine in 5 M glacial acetic acid, 10%
H2O2 as described elsewhere (23,24).
Statistical analysis
In order to detect significance of the observed
effects, results have been expressed as mean ± standard errors
(SEM) and comparison among groups was made by using analysis of
variances (ANOVA) with Dunnett's test for comparison with a single
control, and with Tukey's test for complete post hoc test.
Statistical significance was defined as significant (*P<0.05)
and highly significant (**P<0.01).
Results
Antiproliferative effects of
3,4-isoxazolediamides and
4,5,6,7-tetrahydro-isoxazolo-[4,5-c]-pyridines derivatives on the
K562 cell line
The antiproliferative effects of the tested
compounds were first evaluated on the human myelogenous leukemia
K562 cell line. The cell count was performed after 72 and 96 h of
cell culture, when cells are in the log phase of growth. Table I indicates the antiproliferative
effects (IC50 values, i.e., the inhibitory concentration
required for obtaining 50% cell growth inhibition) of all the
tested compounds 1–13 (the experiments were repeated at least three
times in the presence of the same experimental condition).
 | Table I.Antiproliferative effects in terms of
IC50 of isoxazoles 1–13 on K562 cells. |
Table I.
Antiproliferative effects in terms of
IC50 of isoxazoles 1–13 on K562 cells.
| Isoxazole
derivative | IC50 of
K562 cells |
|---|
| 1 |
71.57±4.89 nM |
| 2 |
18.01±0.69 nM |
| 3 |
44.25±10.90 nM |
| 4 |
70.12±5.80 nM |
| 5 |
35.21±6.20 nM |
| 6 |
45.43±13.10 nM |
| 7 | 779.40±151.00
nM |
| 8 |
3.20±1.10 µM |
| 9 | 627.30±49.00
nM |
| 10 |
68.30±5.20 µM |
| 11 | 220.70±23.00
µM |
| 12 | >300 µM |
| 13 | >300 µM |
The obtained results indicate that, while two
compounds (i.e. 12 and 13) are inactive at the used concentrations
(IC50>300 µM), some derivatives show strong effects
in inhibiting the in vitro proliferation of K562 cells. This
is particularly remarkable for the compounds 1 (71.57±4.89 nM), 2
(18.01±0.69 nM), 3 (44.25±10.9 nM), 4 (70.1±5.8 nM), 5 (35.2±6.2
nM), 6 (45.43±13.1 nM) and 10 (68.3±5.2 µM) (for all of these
compounds P<0.05 with respect to control untreated cells).
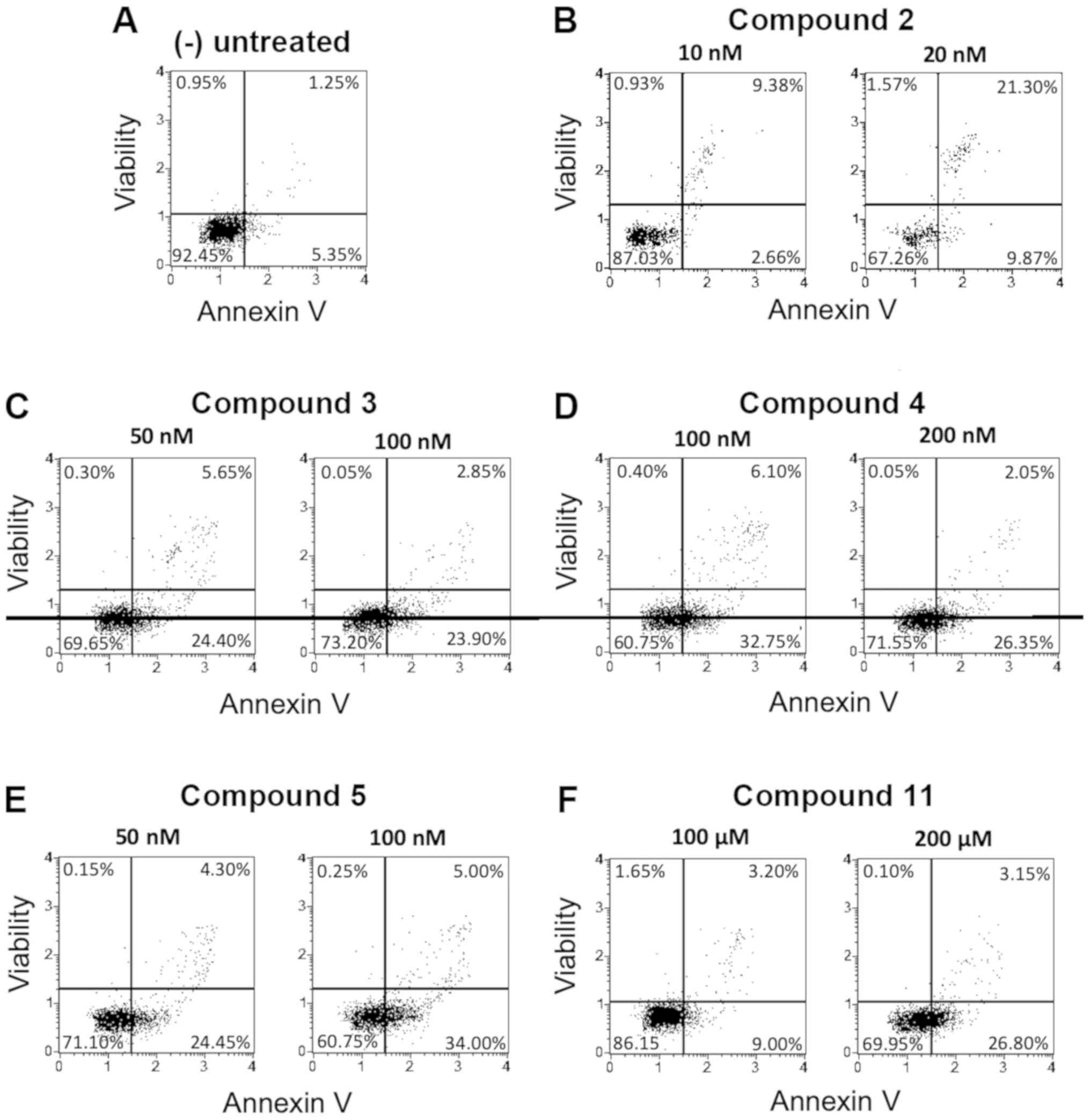
Apoptosis analysis on K562, U251-MG and T98G cell
lines was performed using compounds 1–11, selected on the basis of
their antiproliferative activity on K562 cells (Table I). Derivatives 12 and 13 were not
considered, being not active even when 300 µM concentration was
used (Table I). The pro-apoptosis
assays were conducted at two concentrations approaching the
IC50 values shown in Table
I for K562 cells and preliminarily verified for U251-MG and
T98G cells (data not shown). Representative data obtained from the
analysis of the corresponding Muse Analyzer plots after 72 h from
the treatment are shown in Figs.
3–5. The summaries of the data
obtained with all the selected 1–11 compounds are shown in Tables II–IV.
| Table II.Pro-apoptotic activity of isoxazole
derivatives 1–11 in K562 cells. |
Table II.
Pro-apoptotic activity of isoxazole
derivatives 1–11 in K562 cells.
| Sample | Early
apoptosis,% | Late
apoptosis,% | Total
apoptosis,% |
|---|
| Untreated cells
(−) | 0.10 | 0.10 | 0.20 |
| 1 (50 nM) | 14.00 | 27.21 | 41.21 |
| 1 (100 nM) | 16.15 | 30.30 | 46.45 |
| 2 (10 nM) | 4.15 | 2.15 | 6.30 |
| 2 (20 nM) | 20.46 | 10.26 | 30.72 |
| 3 (50 nM) | 29.45 | 6.70 | 36.15 |
| 3 (100 nM) | 28.75 | 10.00 | 38.75 |
| 4 (100 nM) | 25.75 | 7.80 | 33.55 |
| 4 (200 nM) | 64.90 | 15.20 | 80.10 |
| 5 (50 nM) | 23.60 | 6.60 | 30.20 |
| 5 (100 nM) | 31.90 | 11.95 | 43.85 |
| 6 (50 nM) | 30.30 | 6.40 | 36.70 |
| 6 (100 nM) | 25.85 | 16.00 | 41.85 |
| 7 (1 µM) | 36.40 | 10.40 | 46.80 |
| 7 (5 µM) | 37.55 | 24.80 | 62.35 |
| 8 (5 µM) | 30.30 | 15.55 | 45.85 |
| 8 (10 µM) | 54.05 | 36.55 | 90.60 |
| 9 (1 µM) | 44.90 | 13.25 | 58.15 |
| 9 (5 µM) | 39.85 | 24.45 | 64.30 |
| 10 (50 µM) | 15.40 | 2.70 | 18.10 |
| 10 (100 µM) | 23.75 | 16.70 | 40.45 |
| 11 (100 µM) | 39.40 | 17.30 | 56.70 |
| 11 (200 µM) | 63.45 | 25.05 | 88.50 |
| Table IV.Pro-apoptotic activity of isoxazole
derivatives 1–11 in T98G cells. |
Table IV.
Pro-apoptotic activity of isoxazole
derivatives 1–11 in T98G cells.
| Samples | Early
apoptosis,% | Late
apoptosis,% | Total
apoptosis,% |
|---|
| Untreated cells
(−) | 6.95 | 2.25 | 9.20 |
| 1 (50 nM) | 3.63 | 2.46 | 6.09 |
| 1 (100 nM) | 4.83 | 8.44 | 13.27 |
| 2 (10 nM) | 4.65 | 2.56 | 7.21 |
| 2 (20 nM) | 2.35 | 2.57 | 4.92 |
| 3 (50 nM) | 32.60 | 7.90 | 40.50 |
| 3 (100 nM) | 37.50 | 11.00 | 48.50 |
| 4 (100 nM) | 32.55 | 6.25 | 38.80 |
| 4 (200 nM) | 36.20 | 10.60 | 46.80 |
| 5 (50 nM) | 29.30 | 3.85 | 33.15 |
| 5 (100 nM) | 34.05 | 8.65 | 42.70 |
| 6 (50 nM) | 15.50 | 1.65 | 17.15 |
| 6 (100 nM) | 13.30 | 3.95 | 17.25 |
| 7 (1 µM) | 6.90 | 2.30 | 9.20 |
| 7 (5 µM) | 13.60 | 3.15 | 16.75 |
| 8 (5 µM) | 8.55 | 2.20 | 10.75 |
| 8 (10 µM) | 13.65 | 5.45 | 19.10 |
| 9 (1 µM) | 28.15 | 13.65 | 41.80 |
| 9 (5 µM) | 36.95 | 11.95 | 48.90 |
| 10 (50 µM) | 9.20 | 3.35 | 12.55 |
| 10 (100 µM) | 17.75 | 8.65 | 26.40 |
| 11 (100 µM) | 6.10 | 1.85 | 7.95 |
| 11 (200 µM) | 12.35 | 3.15 | 15.50 |
Pro-apoptotic activity of
3,4-isoxazolediamides and
4,5,6,7-tetrahydro-isoxazolo-[4,5-c]-pyridines derivatives on K562
cells
Analysis of apoptosis on K562 cells treated with the
studied isoxazoles was performed using the Annexin V release assay.
Representative results are shown in Fig.
3.
Table II shows the
complete set of results obtained studying the pro-apoptotic effects
of these compounds in early and late apoptosis stage, compared to
the untreated cells (negative control). These data were obtained
from the analysis of the Muse Analyzer plots, after 72 h from the
treatment (see Fig. 3 for
representative examples).
Among the tested compounds, all were found to
possess the ability to induce apoptosis on K562 cells, but the
highest effects were found using the isoxazoles 4, 7, 8, 9 and 11.
This remarkable biological activity is demonstrated by the fact
that induced apoptosis was found in >50% of the cells. In fact,
the derivatives 4 (100 nM), 8 (10 µM) and 11 (200 µM) displayed
high levels of apoptotic effect (80.10, 90.60 and 88.50%,
respectively) with a relevant dose-depending increase. Also
compound 2, 6 and 10 were found pro-apoptotic, but mainly at the
highest concentrations used.
Pro-apoptotic activity on U251-MG and
T98G glioma cells
Apoptosis analysis on both U251-MG and T98G cell
lines was obtained by performing treatments with the same compounds
previously analyzed on the K562 cell line. The following Figs. 4 and 5
(including representative experiments) and Tables III and IV (including the full sets of data) show
the results obtained, focusing on the effects of these isoxazoles
on the cells in early and late apoptosis stage.
| Table III.Pro-apoptotic activity of isoxazole
derivatives 1–11 in U251-MG cells. |
Table III.
Pro-apoptotic activity of isoxazole
derivatives 1–11 in U251-MG cells.
| Samples | Early
apoptosis,% | Late
apoptosis,% | Total
apoptosis,% |
|---|
| Untreated cells
(−) | 5.35 | 1.25 | 6.60 |
| 1 (50 nM) | 5.04 | 14.15 | 19.19 |
| 1 (100 nM) | 7.71 | 21.37 | 29.08 |
| 2 (10 nM) | 2.66 | 9.38 | 12.04 |
| 2 (20 nM) | 9.87 | 21.30 | 31.17 |
| 3 (50 nM) | 24.40 | 5.65 | 30.05 |
| 3 (100 nM) | 23.90 | 2.85 | 26.75 |
| 4 (100 nM) | 32.75 | 6.10 | 38.85 |
| 4 (200 nM) | 26.75 | 2.05 | 28.40 |
| 5 (50 nM) | 24.45 | 4.30 | 28.75 |
| 5 (100 nM) | 34.00 | 5.00 | 39.00 |
| 6 (50 nM) | 11.95 | 2.25 | 14.20 |
| 6 (100 nM) | 14.85 | 1.95 | 16.80 |
| 7 (1 µM) | 6.45 | 1.60 | 8.05 |
| 7 (5 µM) | 13.15 | 1.95 | 15.10 |
| 8 (5 µM) | 7.00 | 3.50 | 10.50 |
| 8 (10 µM) | 7.95 | 1.40 | 9.35 |
| 9 (1 µM) | 19.15 | 1.75 | 20.90 |
| 9 (5 µM) | 23.95 | 4.45 | 28.40 |
| 10 (50 µM) | 5.70 | 1.95 | 7.65 |
| 10 (100 µM) | 6.80 | 3.30 | 10.10 |
| 11 (100 µM) | 9.00 | 3.20 | 12.20 |
| 11 (200 µM) | 26.80 | 3.15 | 29.95 |
As we can observe in the Annexin V assay reported in
Figs. 4 and 5 and in Tables
III and IV, the isoxazoles
derivatives have been able to induce significant percentages of
apoptosis in both the glioblastoma cell lines, but with lower
levels of total apoptosis when comparison was done with the data
obtained using K562 cells. We reported in Fig. 4 the derivatives exhibiting the best
activity on U251-MG cells (the derivatives 2, 3, 4, 5 and 11,
showing total apoptosis values >30%). In Fig. 5 the effects of the most active
compounds on the TMZ-resistant T98G cells were reported (isoxazoles
3, 4, 5 and 9, showing total apoptosis values >40%). It should
be noted that in the TMZ-resistant T98G cell line the trend of
apoptosis induction is in general better than that observed using
the same compounds on U251-MG cells, since different derivatives
(for instance compounds 3, 4, 5, 9 and 10) showed higher activity
when used on the TMZ-resistant cell model. In particular, the
isoxazole 3 showed very high induction of apoptosis (48.50%) at the
concentration of 100 nM, while in U251-MG cells the obtained
activity was 25.75%. Furthermore, the compound 9 (used at 1 and 5
µM) induced the highest percentage of apoptosis in T98G cells
(41.80 and 48.90%, respectively), while in U251-MG cells the
apoptosis induction efficiency was 20.90 and 28.40%, respectively.
Compounds 6, 7 and 8 displayed low pro-apoptotic activities
(<25% at the highest concentration employed) on both glioma
cells lines.
Caspase-mediated pro-apoptotic effects
of compounds representative of the 3,4-isoxazolediamides and
4,5,6,7-tetrahydro-isoxazolo-[4,5-c]-pyridines structures on T98G
glioma cell line
The Caspase 3/7 assay was performed to further
confirm the pro-apoptotic effects (25) of isoxazoles derivatives 3–11 at the
concentration that produced the best results with the Annexin V
assay (Table IV). In particular,
the effects have been studied on the T98G glioma cells, since these
cells are resistant to induction of apoptosis caused by TMZ,
commonly used to treat glioblastoma patients) (10). The caspases 3 and 7 are effector
caspases of the enzymatic cascade of activation of the apoptotic
process. For this reason, they are considered biomarkers that
indicate cells in the stages of the programmed death process. The
data obtained are comparatively depicted in Fig. 6 as ‘percentage of apoptotic cells’
and include cells that are both in the early and late stages of the
apoptotic process.
In summary, the one-way ANOVA confirmed that all
compounds were able to induce an increase of apoptosis, being some
derivatives more effective (particularly the isoxazoles 3 and 9,
showing an apoptotic effect of 52.48±2.11 and 46.08 48±1.49%,
respectively). This effect was highly significant (Dunnett's test
P<0.0001, P<0.0001 and P<0.0083, respectively) for
isoxazole 3, 9 and 5 with respect to control untreated cells.
Differentiation induced by isoxazole
derivatives on the K562 cell line
When stimulated by various antitumor agents known
agents (such as mithramycin, MTH), K562 cells respond within few
days activating the terminal stages of differentiation, associated
with increased production of embryo-fetal hemoglobins (mainly Hb
Gower 1 and Hb Portland). As several antitumor compounds exert
their action through activation of differentiated functions
(26,27), erythroid differentiation was analyzed
in K562 cells treated with compounds exhibiting the highest
inhibitory effects on cell growth (the isoxazoles 1, 2, 3, 4, 5 and
6; Table I) and studied by
determining the proportion of blue benzidine-positive (hemoglobin
containing) cells. This enzymatic-colorimetric test was performed
after 7 days of cell culture. Blue cells were counted and the
obtained data expressed in percentage of benzidine positive cells
(Fig. 7). ANOVA confirmed that the
isoxazole derivatives 1–6 possessed a high capability in inducing
differentiation in the K562 cells (P<0.0001). For all the
compounds, very high significance was found when comparison was
performed with control untreated cells (Dunnett's test P<0.0001
for all compounds). Among the tested compounds, compound 2
exhibited the highest efficiency in inducing erythroid
differentiation in respect to the other compounds (Tukey's test
P-values were: P<0.0002 when comparison was performed for
compounds 1 and 3, and P=0.0046, P=0.0467 and P=0.0022 when
comparison was performed with 4, 5 and 6. The compounds 4, 5 and 6
possessed the same capability in inducing differentiation in the
K562 cells.
Discussion
Most types of human cancer share common hallmarks,
including self-sufficiency in growth signals, insensitivity to
growth-inhibitory mechanism leading to unrestricted cell
proliferation, evasion of programmed cell death, sustained
angiogenesis, tissue invasion and metastasis (28). Heat shock protein 90 (HSP90) is a
molecular chaperone protein involved in the conformational
maturation, stability and function of so-called ‘client’ proteins.
It has been proved that HSP90 is a key regulator protein of tumor
cell cycle progression where it is overexpressed (29,30).
This upregulation may be an adaptive response by cancer cells to
maintain protein homeostasis and promote cell survival in an
unfavorable environment as well as to stimulate cell proliferation
and inhibit cell death. The ability to attach carrier molecules
direct to specific tumors have a potential for the development into
selective anticancer agents avoiding toxic side effects on normal
healthy tissues. Thus, the interest of HSP90 as possible anticancer
targets and for throughput screening is getting high.
Natural compounds as geldanamycin and radicicol are
the pioneering compounds capable to inhibit this class of proteins,
nevertheless it was necessary to modify their chemical structure
because both of them exhibit several issues as poor solubility,
significant hepatotoxicity, intrinsic chemical instability altering
in vivo activity. For these reasons we generated novel
synthetic HSP90 inhibitors
(4,5,6,7-Tetrahydro-isoxazolo-[4,5-c]-pyridines and
3,4-Isoxazolediamide derivatives) in order to increase the
efficiency. We have elsewhere reported that these analogues are
very powerful in inhibiting HSP90, even when used at concentrations
lower than geldanamycin and radicicol; furthermore, geldanamycin
and radicicol are known to exhibit low solubility and cause in
vivo unwanted side effects, such as hepatotoxicity (31). Finally, this study envisages the
possible use of inhibitors of HSP-90 in searching for analogues
exhibiting high level of antiproliferative and pro-apoptotic
activities.
The general aim of this study was to determine the
biological activity of these new isoxazole compounds on tumor cell
lines.
An antiproliferative effect on K562 cells was
observed for the analogues 1–7 and 9, that exhibited cytotoxic
effect at nanomolar concentrations. Another compound that exhibited
lower but significant antiproliferative effect was compound 8,
characterized by a 3.2±1.1 µM IC50 value. Compounds 10
and 11 were able to induce antiproliferative effect on K562 cells
only when added at high concentrations (IC50 values
>60 µM), while 12 and 13 were completely inactive at the used
concentrations (IC50>300 µM). Further, the
differentiation was analyzed in K562 cells treated with the
compounds exhibiting the highest antiproliferative activity by
determining the proportion of benzidine-positive cells after 6 days
of treatment. Compounds 1–6 were found to stimulate erythroid
differentiation (especially compound 2) at very high level,
inducing very high percentages of benzidine-positive cells (98%, 50
nM, in the case of compound 2; 74–82% in the case of compounds 4, 5
and 8).
In this context, this study is limited, due to the
fact that few tumor cell lines were analyzed (the human
erythroleukemic K562 cell line, as a representative model for tumor
cell growing in suspension and the two glioma U251-MG and T98G cell
lines, growing attached to the flask). While other cancer cell
lines and further in vivo experiments should be considered,
the results of the present study about the in vitro cell
growth are encouraging. A second limitation is that in this study
the issue of combined antitumor therapy was not addressed. In any
case, considering the interesting inhibitory effects on the growth
of the TMZ-resistant T98G cells, the use of the molecules here
described could be combined with treatments based on drugs already
employed in conventional therapy (such as chemotherapy) of
glioblastoma, and might be of interest in the treatment of patients
not-responsive to TMZ.
A second conclusion of our study is that the
antiproliferative effects of the analyze compounds is the
activation of apoptosis. The possible pro-apoptotic effects of
these HSP90 inhibitors was investigated on K562 cells and on two
additional cell lines representative of neuronal solid tumors, i.e
the gliolastoma U251-MG and T98G cells. It should be noted that
glioblastoma multiforme (VI grade tumor) is one of the most
aggressive solid tumors and the TMZ-based chemotherapy, while
remaining the most commonly used treatment, cannot be considered
efficient in TMZ resistant gliomas. In this respect, the resistance
to antitumor compounds is the most important problem in the
clinical management of these patients, leading to low survival.
Therefore, in our investigation the effects on the U251-MG cell
line and on the TMZ-resistant T98G cell line were also
compared.
All the tested derivatives were used at two
concentrations that always included those demonstrated to cause 50%
inhibition of in vitro cell growth. Interestingly all
compounds were found to possess the ability to induce apoptosis on
K562 cells, especially the isoxazoles 4, 7, 8, 9 and 11, shown to
exhibit high levels pro-apoptotic activity (>50%). In fact, the
derivatives 4 (100 nM), 8 (10 µM) and 11 (200 µM) displayed very
high level of apoptotic effect (80.10, 90.60 and 88.50%,
respectively). More in detail, the common feature observed is that
the early apoptosis stage was always higher than the later one in
presence of each compound, with the exception of the derivative
1.
As far as the apoptosis analysis performed also on
U251-MG and T98G glioma cell lines using the compounds 1–11, the
derivatives 2, 3, 4, 5 and 11 exhibited the best activity, showing
total apoptosis values ≥30%; the top levels of apoptosis were
observed when the TMZ-resistant T98G cells were treated with the
isoxazoles 3, 4, 5 and 9 (total apoptosis >40%).
As far as apoptosis, this manuscript is limited to
the fact that only a FACS approach has been employed to investigate
apoptosis (i.e. the Annexin V and the Caspase 3/7 kit assays). A
number of assays (i.e. ELISA for histone/DNA fragment,
poly-ADP-ribose-polymerase cleavage assay, and terminal
deoxynucleotidyl transferase-mediated dUTP nick-end-labeling assay)
(32) should be also considered in
order to fully characterized the induction of the apoptotic
phenotype. Furthermore, the study of apoptosis-related molecular
markers (for instance, but not limited to, p53, BAX, Bcl2,
pro-apoptotic miRNAs) (33) will be
useful in the future to elucidate the activity of the investigated
isoxazole derivatives on specific targets.
In summary, despite the above-mentioned limitations,
this study supports the concept that isoxazole derivatives here
described are active in inhibiting tumor cell growth in
vitro, are able to induce apoptosis. Furthermore, all of them
are potent inducers of erythroid differentiation when the
erythroleukemic K562 cell system was employed. Therefore, the most
effective compound here described deserve further consideration in
testing their activity as antitumor drugs using in vivo
assays based on mouse tumor model systems.
Acknowledgements
The authors would like to thank Professor Giulio
Cabrini (Department of Neurosciences, Biomedicine and Movement,
University of Verona, Verona, Italy) for providing useful
discussion on the effects of the studied compounds on glioma cell
lines.
Funding
The present study was supported by Associazione
Italiana per la Ricerca sul Cancro (AIRC) (IG #13575; to RG) and by
Fondo per le Agevolazioni alla Ricerca (Lampronti-2018; to IL).
This study was also supported by the Interuniversity Consortium for
the Biotechnology (project no. 40/18), Italy.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
DS, RR and RB designed and synthesized the
compounds. IL, AF, MB, CT and RG designed and performed the
experiments, analyzed and the interpreted data. IL and RG wrote the
paper. CS applied statistical algorithms to the obtained results.
All authors agree to be accountable for all aspects of the research
in ensuring that the accuracy or integrity of any part of the work
are appropriately investigated and resolved. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
TMZ
|
temozolomide
|
|
HSPs
|
heat shock proteins
|
|
DMSO
|
dimethyl sulfoxide
|
|
EtOH
|
ethanol
|
|
PS
|
phosphatidyl serine
|
|
CML
|
chronic myelogenous leukemia
|
References
|
1
|
Macario AJ and Conway de Macario E: Sick
chaperones, cellular stress, and disease. N Engl J Med.
353:1489–1501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Powers MV and Workman P: Inhibitors of the
heat shock response: Biology and pharmacology. FEBS Lett.
581:3758–3769. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garcia-Carbonero R, Carnero A and Paz-Ares
L: Inhibition of HSP90 molecular chaperones: Moving into the
clinic. Lancet Oncol. 14:e358–e369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Welch WJ: The role of heat-shock proteins
as molecular chaperones. Curr Opin Cell Biol. 3:1033–1038. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rowlands MG, Newbatt YM, Prodromou C,
Pearl LH, Workman P and Aherne W: High-throughput screening assay
for inhibitors of heat-shock protein 90 ATPase activity. Anal
Biochem. 327:176–183. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cragg GM and Newman DJ: Plants as a source
of anti-cancer agents. J Ethnopharmacol. 100:72–79. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Whitesell L, Mimnaugh EG, De Costa B,
Myers CE and Neckers LM: Inhibition of heat shock protein
HSP90-pp60v-src heteroprotein complex formation by benzoquinone
ansamycins: Essential role for stress proteins in oncogenic
transformation. Proc Natl Acad Sci USA. 91:8324–8328. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sauville E: Heat shock protein-90 directed
therapeutics and target validation. Cancer Drugs Design and
Discovery. Elsevier; Amsterdam, Netherlands: pp. 336–350. 2008,
View Article : Google Scholar
|
|
9
|
Sharma SV, Agatsuma T and Nakano H:
Targeting of the protein chaperone, HSP90, by the transformation
suppressing agent, radicicol. Oncogene. 16:2639–2645. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lia Y, Zhanga D, Xu J, Shia J, Jiang L,
Yaoa N and Ye W: Discovery and development of natural heat shock
protein 90 inhibitors in cancer treatment. Acta Pharm Sin B.
2:238–245. 2012. View Article : Google Scholar
|
|
11
|
Brough PA, Aherne W, Barril X, Borgognoni
J, Boxall K, Cansfield JE, Cheung KM, Collins I, Davies NG,
Drysdale MJ, et al: 4,5-diarylisoxazole Hsp90 chaperone inhibitors:
Potential therapeutic agents for the treatment of cancer. J Med
Chem. 51:196–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Baruchello R, Simoni D, Grisolia G,
Barbato G, Marchetti P, Rondanin R, Mangiola S, Giannini G,
Brunetti T, Alloatti D, et al: Novel 3,4-isoxazolediamides as
potent inhibitors of chaperone heat shock protein 90. J Med Chem.
54:8592–8604. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baruchello R, Simoni D, Marchetti P,
Rondanin R, Mangiola S, Costantini C, Meli M, Giannini G, Vesci L,
Carollo V, et al: 4,5,6,7-Tetrahydro-isoxazolo-[4,5-c]-pyridines as
a new class of cytotoxic Hsp90 inhibitors. Eur J Med Chem.
76:53–60. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Biamonte MA, Van de Water R, Arndt JW,
Scannevin RH, Perret D and Lee WC: Heat shock protein 90:
Inhibitors in clinical trials. J Med Chem. 53:3–17. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Taldone T, Sun W and Chiosis G: Discovery
and development of heat shock protein 90 inhibitors. Bioorg Med
Chem. 17:2225–2235. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Khan MT, Lampronti I, Martello D, Bianchi
N, Jabbar S, Choudhuri MS, Datta BK and Gambari R: Identification
of pyrogallol as an antiproliferative compound present in extracts
from the medicinal plant Emblica officinalis: Effects on in
vitro cell growth of human tumor cell lines. Int J Oncol.
21:187–192. 2002.PubMed/NCBI
|
|
17
|
Bianchi N, Ongaro F, Chiarabelli C,
Gualandi L, Mischiati C, Bergamini P and Gambari R: Induction of
erythroid differentiation of human K562 cells by cisplatin analogs.
Biochem Pharmacol. 60:31–40. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brognara E, Fabbri E, Bazzoli E, Montagner
G, Ghimenton C, Eccher A, Cantù C, Manicardi A, Bianchi N, Finotti
A, et al: Uptake by human glioma cell lines and biological effects
of a peptide-nucleic acids targeting miR-221. J Neurooncol.
118:19–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lozzio CB and Lozzio BB: Human chronic
myelogenous leukemia cell-line with positive Philadelphia
chromosome. Blood. 45:321–334. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lampronti I, Khan MTH, Borgatti M, Bianchi
N and Gambari R: Inhibitory Effects of Bangladeshi Medicinal Plant
Extracts on Interactions between Transcription Factors and Target
DNA Sequences. Evid Based Complement Alternat Med. 5:303–312. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Brognara E, Fabbri E, Montagner G,
Gasparello J, Manicardi A, Corradini R, Bianchi N, Finotti A,
Breveglieri G, Borgatti M, et al: High levels of apoptosis are
induced in human glioma cell lines by co-administration of peptide
nucleic acids targeting miR-221 and miR-222. Int J Oncol.
48:1029–1038. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lampronti I, Borgatti M, Vertuani S,
Manfredini S and Gambari R: Modulation of the expression of the
pro-inflammatory IL-8 gene in cystic fibrosiscells by extracts
deriving from olive mill waste water. Evid Based Complement
Alternat Med. 2013:9606032013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Finotti A, Bianchi N, Fabbri E, Borgatti
M, Breveglieri G, Gasparello J and Gambari R: Erythroid induction
of K562 cells treated with mithramycin is associated with
inhibition of raptor gene transcription and mammalian target of
rapamycin complex 1 (mTORC1) functions. Pharmacol Res. 91:57–68.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fibach E, Prus E, Bianchi N, Zuccato C,
Breveglieri G, Salvatori F, Finotti A, Lipucci di Paola M, Brognara
E, Lampronti I, et al: Resveratrol: Antioxidant activity and
induction of fetal hemoglobin in erythroid cells from normal donors
and β-thalassemia patients. Int J Mol Med. 29:974–982.
2012.PubMed/NCBI
|
|
25
|
Scabini M, Stellari F, Cappella P,
Rizzitano S, Texido G and Pesenti E: In vivo imaging of early stage
apoptosis by measuring real-time caspase-3/7 activation. Apoptosis.
16:198–207. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bianchi N, Chiarabelli C, Borgatti M,
Mischiati C, Fibach E and Gambari R: Accumulation of gamma-globin
mRNA and induction of erythroid differentiation after treatment of
human leukaemic K562 cells with tallimustine. Br J Haematol.
113:951–961. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mischiati C, Sereni A, Lampronti I,
Bianchi N, Borgatti M, Prus E, Fibach E and Gambari R:
Rapamycin-mediated induction of gamma-globin mRNA accumulation in
human erythroid cells. Br J Haematol. 126:612–621. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Workman P: Pharmacogenomics in cancer drug
discovery and development: Inhibitors of the Hsp90 molecular
chaperone. Cancer Detect Prev. 26:405–410. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schopf FH, Biebl MM and Buchner J: The
HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 18:345–360. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
McDonald E, Workman P and Jones K:
Inhibitors of the HSP90 molecular chaperone: Attacking the master
regulator in cancer. Curr Top Med Chem. 6:1091–1107. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gorska M, Popowska U, Sielicka-Dudzin A,
Kuban-Jankowska A, Sawczuk W, Knap N, Cicero G and Wozniak F:
Geldanamycin and its derivatives as Hsp90 inhibitors. Front Biosci.
17:2269–2277. 2012. View
Article : Google Scholar
|
|
32
|
Ward TH, Cummings J, Dean E, Greystoke A,
Hou JM, Backen A, Ranson M and Dive C: Biomarkers of apoptosis. Br
J Cancer. 99:841–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gerl R and Vaux DL: Apoptosis in the
development and treatment of cancer. Carcinogenesis. 26:263–270.
2005. View Article : Google Scholar : PubMed/NCBI
|