Introduction
Non-small cell lung cancer (NSCLC) accounts for ~80%
of all types of lung cancer and was reported the leading cause of
cancer-associated mortality worldwide in 2016 (1,2).
Cisplatin (DDP)-based chemotherapy regimens are recommended as the
standard treatment modalities for NSCLC; however, only ~20% of
patients respond to the treatment (3,4).
Brother of Regulator of Imprinted Sites
(BORIS, also known as CCCTC-binding factor like, the
paralogue of CCCTC-binding factor (CTCF), is abnormally expressed
in the majority or different types of cancer and is therefore
considered as a potential therapeutic target for breast, lung and
cervical carcinoma (5–7). However, the functions of BORIS in
carcinoma remain unknown. In our previous study (8), it was revealed that BORIS suppressed
apoptosis and resisted fluorouracil (5-FU) treatment in colorectal
cancer. Since 5-FU or DDP induces DNA damage and apoptosis of
carcinoma cells (9–11), BORIS may induce resistance to DDP
therapy. Based on the prevalent expression of BORIS and the
high incidence of DDP resistance in NSCLC (4,12), it is
worth investigating whether BORIS contributes to DDP resistance in
NSCLC.
Studies on platinum resistance in different types of
cancer, such as ovarian cancer, lung cancer and breast cancer have
revealed multiple complex resistance mechanisms, including the
decreased accumulation of platinum, a decline in DDP-DNA adduct
levels and an increase in DNA damage repair (9,13,14). On
the other hand, cancer cells are also able to correct intrinsic
pathways, such as the DNA repair system (including DNA excision and
mismatch repair systems) and apoptosis to defend against
environmental toxins for survival (9,14). It
has been reported that aberrant activation of DNA repair pathways
of NSCLC contributes to DDP resistance (15–18).
The presents study aimed to investigate whether
BORIS promoted NSCLC cell proliferation and protected NSCLC cells
from being injured by DDP. BORIS was used as the candidate target
for NSCLC therapy to decrease DDP resistance.
Materials and methods
Cell culture and transfection
Cell lines K562, H1299, A549 and H460 were purchased
from the American Type Culture Collection. In the three lung cancer
cell lines, H1299 lacks the expression of p53 protein. A549 and
H460 cells are detectable for p53 (19). A549/DDP (DDP-resistant A549 cell
line) was purchased from Shanghai ExCell Biology, Inc. Cells were
cultured in RPMI 1640 medium containing 10% heat-inactivated fetal
bovine serum (FBS; Gemini Company, http://www.gembio.com/products) at 37°C and in a
humidified atmosphere containing 5% CO2 for 1 week.
Lipofectamine® 3000 reagent (Thermo
Fisher Scientific, Inc.) was used to transfect pBORIS plasmids
purchased from OriGene Technologies Inc.. The empty vector control
was constructed in our previous study (7). According to the requirement of
Lipofectamine® 3000, 70% confluent cells were prepared
for the transfection. Plasmid DNA (1 µg) was transfected per well
into6-well plates. Plasmid (50 ng) was transfected per well into
96-well plates. Lipofectamine RNAiMAX (Thermo Fisher Scientific,
Inc.) was used to knockdown genes according to the manufacturer's
instructions. Two siRNAs (siRNA-1 and siRNA-2) that targeted
BORIS mRNA were used to knockdown BORIS. All small
interfering RNAs (siRNA) were chemically synthesized by Xiang Yin
Biotechnology Co., Ltd. (Table SI).
The DDP treatment on transfected cells were performed one day after
transfection.
Reverse transcription-quantitative
(RT-q) PCR
Cells (5×105) were harvested from cell
culture plates. RNA was extracted using TRIzol® reagent
(Thermo Fisher Scientific, Inc.) and ethanol precipitation. Samples
were quantified using the NanoDrop 2000 system (Thermo Fisher
Scientific, Inc.) and proceeded to reverse transcription. The kit
(cat. no. CW0741) used for reverse transcription was purchased from
cwbiotech Beijing (https://www.cwbiotech.com/home.html). Subsequently,
gene expression levels were quantified using SYBR Green based qPCR
(cwbiotech Beijing). The thermocycling conditions were as follow:
Initial denaturation at 95°C for 3 min; 45 cycles of denaturation
at 95°C for 10 sec, annealing at 60°C for 10 sec, elongation at
72°C for 30 sec; and afinal extension at 72°C for 30 sec.
Amplification and signal capture were performed by Bio-Rad CFX
connect real-time system (Bio-Rad Laboratories). GAPDH and β-actin
were used as the internal control genes to evaluate the expression
levels of the candidate genes. The primer sequences used for qPCR
are presented in Table SII.
Western blotting
Total protein was extracted from collected samples
using RIPA buffer (Beyotime Institute of Biotechnology)
supplemented with phenylmethylsulfonyl fluoride (cat. no. CAS
329-98-6; Sigma-Aldrich; Merck KGaA) and protease inhibitor
cocktail (cat. no. 04693159001; Roche Applied Science). Total
protein was quantified using the bicinchoninic acid kit (cat. no.
P0012S; Beyotime Institute of Biotechnology) and 30 µg protein/lane
was separated via 10% SDS-PAGE gel. The separated proteins were
subsequently transferred onto nitrocellulose membranes and blocked
with 5% skimmed milk for 1 h at room temperature. The membranes
were incubated with BORIS polyclone primary antibody (cat. no.
Ab126778; Abcam) overnight at 4°C. The BORIS antibody was diluted
by 0.1% in 2% skimmed milk. Following the primary incubation,
membranes were incubated with secondary antibody conjugated with
HRP (cat. no. DW-0990; Dawen Biotechnology Co., Ltd., www.dawenbio.com) for 2 h at room temperature, which
was diluted by 0.05% in 2% skimmed milk and used for subsequent
experimentation. Protein blots were detected using the Advansta ECL
system (cat. no. K-12043-D10; Advansta Inc., http://advansta.com) and visualized using the Bio-Rad
chemidoc XRS+ system (Bio-Rad Laboratories, Inc.).
Cell viability analysis and colony
formation assay
For the cell viability assays, 2,000 cells were
seeded onto 96 well-plates/well. Cells were incubated with MTT (500
µg/ml, cat. no. M2128, Sigma-Aldrich; Merck KGaA) at 37°C for 4 h
in the dark. Following the MTT incubation, the purple formazan
crystals were dissolved using 100 µl dimethyl sulfoxide for 15 min
at room temperature, and viability was subsequently analyzed at a
wavelength of 490 nm, using a BioTeck Synergy 2 plate reader system
(http://www.mtxlsi.com/bio-tek-synergy-2.htm).
For the colony formation assays, treated cells were
seeded onto 6 well plates and fixed with 4% formaldehyde for 20 min
at 37°C, and subsequently rinsed twice with PBS. Cells were stained
with 0.5% crystal violet for 30 min at room temperature. Images
were capturedusing white light channel in Bio-Rad chemidoc XRS+
system (Bio-Rad Laboratories, Inc.).
Flow cytometry (FACS) assay
The Alexa Fluor 488 Annexin V/Dead Cell Apoptosis
kit (cat. no. V13241) was obtained from Thermo Fisher Scientific,
Inc. Following treatment with DDP, ~106 cells were
harvested and washed twice in cold PBS. According to the manual, 1X
annexin binding buffer was used to dilute the Annexin V and
propidium iodide (PI). Cells were then resuspended and incubated in
Annexin V/PI working buffer at room temperature for 15 min in the
dark. Subsequently, cells were stored in the dark and on ice. FACS
was performed using a FACSCalibur (BD Biosciences) to detect
apoptotic cells. Data analysis was performed using FlowJo software
(version no. 7.6.1; FlowJo LLC).
Immunofluorescence and TUNEL
assay
Cells mounted on coverslips were prepared for
treatments. After treatments cells were fixed with 4% formaldehyde
for 20 min at 37°C and subsequently permeabilized with 0.3% Triton
X-100 for 10 min at room temperature. Cells were subsequently
blocked with 1% bovine serum albumin (BSA; Sangon Biotech Co.,
Ltd.) for 30 min at room temperature. Cells were incubated with
primary antibody (1:200 in 1% BSA) overnight at 4°C. Cells were
washed three times with PBS and subsequently incubated with
secondary antibodies conjugated with tetramethyl rhodamine
isothiocyanate (TRITC) or fluorescein-5-isothiocyanate (FITC; 1:200
in 1% BSA) for 1 h at room temperature. Cell nuclei were stained
using DAPI (0.5 µg/ml) at room temperature for 5 min. Coverslips
were re-washed four times and visualized using a Leica fluorescence
microscope (magnification, ×20). BORIS primary antibody (cat. no.
Ab126778) was purchased from Abcam and γH2AX antibody (cat. no.
05-636) was purchased from EMD Millipore. Primary antibodies were
diluted 1:200 in 1% BSA. Flag antibody (cat. no. R1180B) was
purchased from OriGene Technologies, Inc. FITC conjugated rabbit
antibody (cat. no. DW-GAR001, www.dawenbio.com) and TRITC conjugated mouse antibody
(cat. no. DW-A0521, www.dawenbio.com) were purchased from Dawen
Biotechnology Co., Ltd..
TUNEL apoptosis detection kit (FITC) was bought from
Shanghai Yeasen Biotechnology Co., Ltd.. The TUNEL reaction system
was incubated at 37°C for 2 h in the dark. The cells were washed
twice with PBS for 5 min. DAPI (1:6,000) was used to stain the
nucleus in a dark room for 5 min in room temperature. After 2
washes with PBS, coverslips were sealed with glycerol and mounted
onto slides. A Leica fluorescence microscope was used to visualize
the cells and more than three scopes of images were captured by 20×
objective lens for further analysis.
RNA-sequencing (RNA-seq) analysis
RNA-seq data and the clinical information for
patients with NSCLC treated with DDP were downloaded from The
Cancer Genome Atlas (TGCA) data portal and manually curated
(11) (Table SIII). Clinical characteristics for
the lung cancer cases (n=156) include age, sex, ethnicity, BORIS
expression [fragments per kilobase of exon model per million reads
mapped (FPKM)], clinical-stage (Tumor-Node-Metastasis; TNM)
(3) and survival span. Treatment
outcomes were recorded for 23 patients (Table SIV). Cancer tissues from 16 of the
23 patients were collected and analyzed before DDP treatment, which
were shown to be ‘prospective’ in the present study. Cancer tissues
from 7 of the 23 patients were collected and analyzed after DDP
treatment, which were shown to be ‘retrospective’ in the present
study (Table I). Survival
probabilities of 156 NSCLC patients were evaluated when the
patients were divided into two groups using a cut-off value of
0.0035 (FPKM).
 | Table I.Characteristics of 156 patients with
NSCLC. |
Table I.
Characteristics of 156 patients with
NSCLC.
| Characteristic | High BORIS
expression, FPKM>0.0035, n | Low BORIS
expression FPKM<0.0035, n | P-value |
|---|
| Sex |
|
| 0.0011 |
|
Male | 57 | 37 |
|
|
Female | 21 | 41 |
|
| Race |
|
| 0.75 |
|
White | 63 | 59 |
|
|
Black | 5 | 6 |
|
|
Asian | 0 | 1 |
|
| Not
reported | 11 | 11 |
|
| Age, years |
|
| >0.999 |
|
>62 | 38 | 39 |
|
|
≤62 | 38 | 39 |
|
| Stage |
|
| 0.35 |
| I | 17 | 14 |
|
| II | 41 | 37 |
|
|
III | 16 | 23 |
|
| IV | 2 | 4 |
|
| Not
reported | 2 | 0 |
|
Statistical analysis
Lung cancer classification for stagingwas referenced
to worldwide standard (20).
BORIS expression differences between patient groups were
analyzed using the χ2 test (Table I). The survival probabilities of
investigated patients were analyzed via the Kaplan-Meier method and
compared using the log-rank test. BORIS expression
differences between NSCLC cell lines were analyzed by one-way ANOVA
followed Tukey post-hoc test. All experiments were performed in
triplicate. Statistical analysis was performed using SPSS 24.0
software (IBM Corp.) or GraphPad Prism software (version 5.0;
GraphPad Software, Inc.). Statistical differences were analyzed by
paired Student's t-test and presented as the mean ± standard
deviation. P<0.05 was considered to indicate a statistically
significant difference.
Results
Patients with NSCLC with higher BORIS
expression have a shorter survival span following DDP therapy
A total of 156 patients with NSCLC who received DDP
treatment were extracted from TCGA (http://www.tcga.org/) (Tables I, II
and SIII). The median of
BORIS expression among the patients was 0.0035 (FPKM), which
was used as a cut-off to study the association between BORIS
and the overall survival rate in the present study. A higher
BORIS expression level was significantly associated with a
short survival span (P=0.019; Fig.
1A). In addition, BORIS expression levels in female
patients were significantly lower than male patients. It is
suggested that BORIS may be induced at high levels in male
patients (Table I). BORIS
expression in 23 cases with records of treatment outcomes was
analyzed (Table SIV). BORIS
expression data were extracted from The Cancer Genome Atlas
database (http://www.tcga.org). Though no paired
tissues were collected from the same patient before and after DDP
therapy, BORIS levels declined from an average of 0.0071
FPKM in the prospective tissues to an average of 0.0027 FPKM in
retrospective tissues (Fig. 1B).
These data suggested that BORIS expression was associated
with the mortality rate of NSCLC upon DDP chemotherapy.
 | Figure 1.BORIS expression is associated
with overall survival rate in patients with NSCLC who received DDP
chemotherapy. (A) Overall survival rate curves in patients with
NSCLC who received DDP chemotherapy. The data were analyzed using
SPSS Kaplan-Meier. Statistical difference was tested using the
Log-rank (MANTEL-COX) test. (B) BORIS expression in 23
patients, who had records of treatment outcomes in The Cancer
Genome Atlas. The patients were featured according to the outcomes
following DDP treatment: Stable disease after DDP chemotherapy,
patients completely remitted or responded to DDP chemotherapy,
patients with progressive disease after DDP chemotherapy and a
patient who responded partly to DDP chemotherapy. (C) BORIS
expressions were compared among four NSCLC cell lines, one normal
pulmonary alveolar epithelial cell line (HPAEpiC) and one leukemia
cell line which was reported to express BORIS and was used
as the positive control for testing BORIS. The statistical
differences of BORIS level between cell lines were
calculated and shown by P-value in the right panel. The statistical
analyses were performed by one-way ANOVA followed Tukey post-hoc
test. (D) Relative proliferation rates of 4 lung cancer cell lines
as compared with a normal human pulmonary alveolar epithelial cell
line (HPAEpiC) at different time points. A549/DDP, DDP resistant
A549 cell line; BORIS, Brother of Regulator of Imprinted Sites;
DDP, cisplatin; NSCLC, non-small cell lung cancer; OD, optical
density; FPKM, fragments per kilobase of exon model per million
reads mapped. |
| Table II.Logistic regression analysis of
2-year survival and predictors in patients with NSCLC. |
Table II.
Logistic regression analysis of
2-year survival and predictors in patients with NSCLC.
|
| Univariate
analysis | Multivariate
analysisa |
|---|
|
|
|
|
|---|
| Variable | OR | 95% CI | P-value | OR | 95% CI | P-value |
|---|
| Surgery method |
| No
surgery | Reference |
|
| Reference |
| <0.001 |
| Local
tumor destruction | 0.18 | 0.16–0.20 | <0.001 | 0.29 | 0.26–0.32 | <0.001 |
| Wedge
or segmental resection | 0.10 | 0.09–0.12 | <0.001 | 0.15 | 0.13–0.16 | <0.001 |
|
Lobectomy | 0.15 | 0.13–0.16 | <0.001 | 0.13 | 0.11–0.15 | <0.001 |
| Liver
transplantation | 0.05 | 0.05–0.06 | <0.001 | 0.08 | 0.07–0.10 | <0.001 |
| Tumor
size, mm | 1.02 | 1.02–1.02 | <0.001 | 1.01 | 1.01–1.01 | <0.001 |
| Age,
years | 1.02 | 1.02–1.02 | <0.001 | 1.01 | 1.01–1.02 | <0.001 |
|
Education level | 1.01 | 1.01–1.02 | <0.001 | 0.98 | 0.98–0.99 | <0.001 |
| Family
income | 0.99 | 0.99–0.99 | <0.001 | 0.99 | 0.99–0.99 | <0.001 |
| Historic stage |
|
Localized | Reference |
|
| Reference |
|
|
|
Regional | 5.78 | 5.34–6.28 | <0.001 | 2.32 | 1.97–2.73 | <0.001 |
|
Distant | 1.04 | 0.98–1.11 | 0.202 | 1.09 | 0.98–1.22 | 0.105 |
| ACJJ_T |
| T1 | Reference |
|
| Reference |
|
|
| T2 | 1.17 | 1.09–1.26 | <0.001 | 1.21 | 1.10–1.33 | <0.001 |
| T3 | 5.50 | 5.06–5.97 | <0.001 | 1.77 | 1.58–1.98 | <0.001 |
| T4 | 7.00 | 5.78–8.54 | <0.001 | 1.66 | 1.31–2.12 | <0.001 |
| Diagnosis year |
|
2004-2008 | Reference |
|
| Reference |
|
|
|
2009-2012 | 0.88 | 0.82–0.94 | <0.001 | 0.72 | 0.66–0.79 | <0.001 |
|
2013-2015 | 0.57 | 0.54–0.61 | <0.001 | 0.48 | 0.44–0.53 | <0.001 |
| Grade |
| I | Reference |
|
| Reference |
|
|
| II | 0.97 | 0.90–1.06 | <0.001 | 1.30 | 1.18–1.43 | <0.001 |
|
III | 2.22 | 2.01–2.45 | <0.001 | 2.44 | 2.15–2.76 | <0.001 |
| IV | 3.34 | 2.50–4.51 | <0.001 | 3.68 | 2.59–5.29 | <0.001 |
| ACJJ_M | 2.04 | 1.89–2.21 | <0.001 |
|
|
|
| M0 | Reference |
|
| Reference |
|
|
| M1 | 11.69 | 10.14–13.55 | <0.001 | 1.74 | 1.33–2.27 | <0.001 |
BORIS expression is positively
associated with NSCLC cell proliferation
Next, the proliferation rates of a panel of lung
cancer cell lines was studied, which revealed a positive
association between BORIS expression levels and cell
proliferation (Fig. 1C and D). All
cell lines tested demonstrated a statistically significant
difference (Fig. 1C). Among the cell
lines that were assessed, human pulmonary alveolar epithelial cell
line (HPAEpiC) was a normal cell line. HPAEpiC did not express
BORIS, suggesting that BORIS may just function in cancer
cells. In order to further investigate its effect on cell
proliferation, BORIS was silenced or overexpressed in NSCLC
cell lines. A549/DDP cells express a relative higher BORIS
compared with A549 (Fig. 1C). Next,
two siRNAs targeting BORIS generated a similar knockdown
efficiency with >50% decrease (Table
SI and Fig. 2D). The knockdown
efficiencies of siRNA-1 were all >70% in A549 and H460 cell
lines (Fig. S1); therefore, in the
present study, BORIS siRNA-1 was used for subsequent
experimentation. BORIS knockdown suppressed the colony
formation of NSCLC cells and induced the expression of p21,
a cyclin-dependent kinase inhibitor (Fig. 2A and B). It has previously been
demonstrated that elevated expression of p21 induces cell
cycle arrest (21–23). Likewise, overexpression of
BORIS promoted the proliferation of NSCLC cells and
inhibited the expression of p21 in H1299 and A549 cells
(Fig. 2B and C). Among the
investigated NSCLC cell lines in the present study, BORIS
was highly expressed in H1299 cells and expressed at low levels in
A549 cells. Thus, the regulations of p21 by BORIS was investigated
in these two cell lines, which express different BORIS
levels (Fig. 1C). As BORIS
knockdown inhibited cell cycles of NSCLC cells, the genome
stability was investigated by assessing γH2AX, which is sensitive
to DNA damage (24). BORIS
was silenced by either siRNA-1 or siRNA-2 and the knockdown
efficiencies were verified via western blotting and RT-qPCR
analyses. The results demonstrated that BORIS knockdown
induced γH2AX (Fig. 2D), suggesting
that BORIS deficiency induces DNA damage in NSCLC cells.
Collectively, these data demonstrated that BORIS expression
was associated with NSCLC cell proliferation, indicating that BORIS
may protect lung cancer cells from DNA damage.
BORIS resists DDP induced cell
suppression and apoptosis
In order to determine whether BORIS contributes to
DDP resistance, BORIS was first silenced or overexpressed in
H1299 cells, and subsequently treated by DDP at various
concentrations from 1–4 µg/ml. BORIS knockdown inhibited
H1299 cell proliferation. The supplement of 1 or 2 µg/ml DDP
synergized BORIS knockdown for cell proliferation
suppression (Fig. 3A). Conversely,
BORIS overexpression promoted cell proliferation.
BORIS overexpression resisted 1–4 µg/ml DDP treatment
(Fig. 3B). Subsequently, the extent
of apoptosis which was induced by 2 µg/ml DDP treatment was
investigated in BORIS overexpressed H1299 cells compared
with the negative control. The results demonstrated that
BORIS overexpression inhibited DDP induced apoptosis from
13.41 to 8.68%, and necrosis from 5.60 to 3.46% (Fig. 3C). Taken together, these results
suggest that BORIS protects NSCLC cells from injury by DDP
treatment, thus, confirm the importance of BORIS in DDP
resistance.
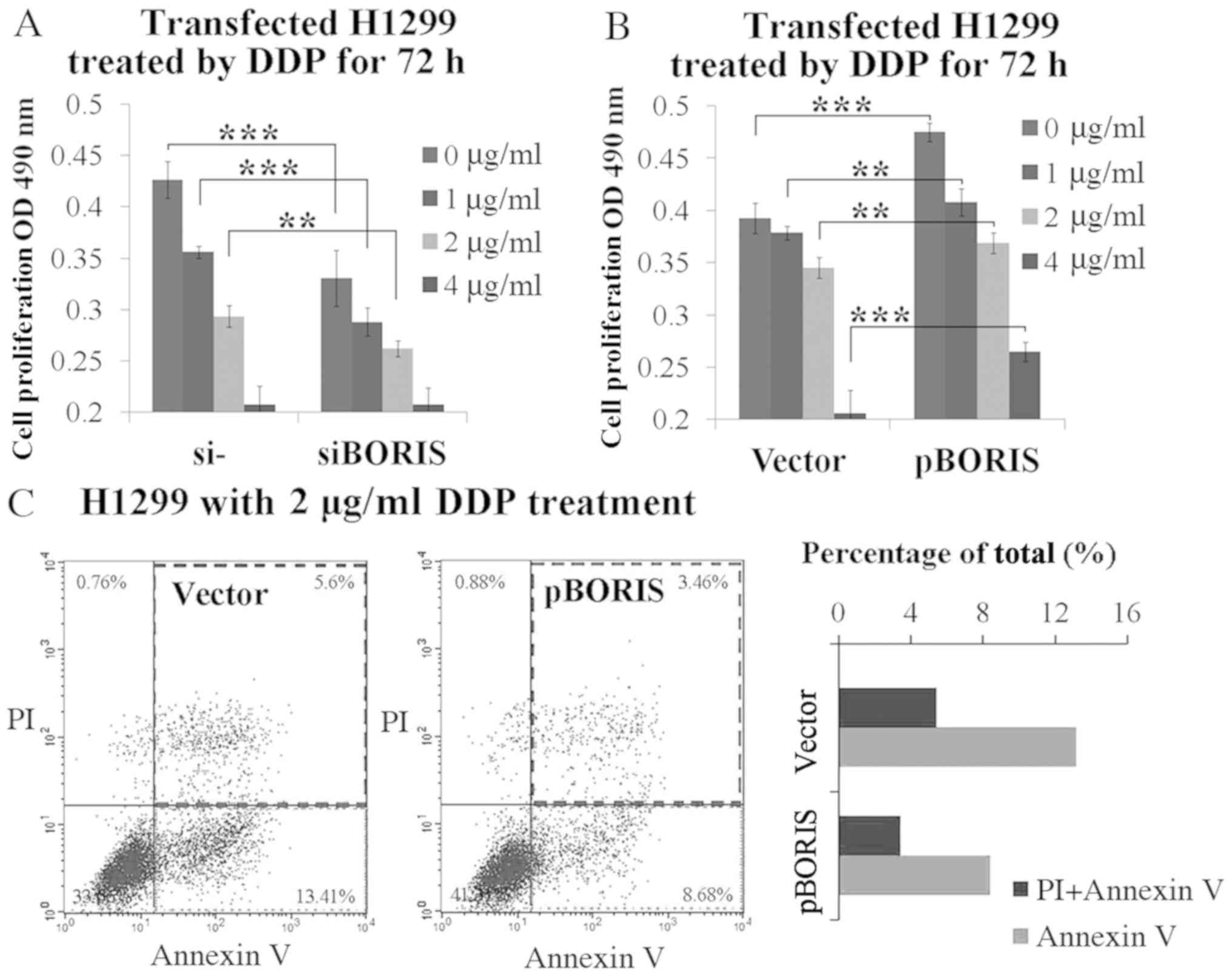 | Figure 3.BORIS resists DDP-induced tumor cell
suppression. (A) Cell proliferation study of H1299 cells
transfected by NC (negative siRNA control) or siBORIS (BORIS
siRNA-1) for 24 h and subsequently treated by DDP at different
doses (0, 1, 2 and 4 µg/ml) for 72 h. (B) Cell proliferation study
of H1299 cells transfected by the vector (empty plasmid) or pBORIS
(BORIS overexpression plasmid) for 24 h and subsequently treated by
DDP (0, 1, 2 and 4 µg/ml) for 72 h. (C) Cell apoptosis of H1299
cells with overexpression of BORIS after treatment using 2 µg/ml
DDP for 24 h. BORIS overexpression suppressed DDP-induced
apoptosis in H1299 cells. **P<0.01; ***P<0.001. BORIS,
Brother of Regulator of Imprinted Sites; NSCLC, non-small cell lung
cancer; OD, optical density; si, small interfering; PI, propidium
iodide; DDP, cisplatin. |
BORIS suppresses DNA damage and
activates the DNA repair system
As BORIS resisted DDP-induced NSCLC suppression
(Fig. 3), it was proposed that BORIS
may inhibit DDP-induced DNA damage. pBORIS plasmid
(BORIS-flag) was transiently transfected in A549 cells that
were treated subsequently by DDP to induce DNA damage. Cells with
overexpression of BORIS demonstrated a decreased γH2AX
signal (Fig. 4A and B), indicating
that BORIS attenuated DDP-induced DNA damage, thus promoting the
proliferation of NSCLC. DNA damage was detected by the TUNEL assay
in H1299 and A549/DDP when BORIS were silenced (Fig. 4C). BORIS may be beneficial for the
DNA repair system of NSCLC cells to sustain genome stability under
the treatment of DDP. The expression levels of few representative
genes of DNA repair system were investigated, such as BRCA1,
ERCC1, CMYC and MSH6 (Table
SII). BORIS was overexpressed by pBORIS or silenced by
siRNA-1 (siBORIS in Fig. 4D) in
H1299 and H460 cells. Fold changes of the investigated genes are
presented in Fig. 4D. MSH6
was regulated consistently and significantly by BORIS in H1299 and
H460 cells (Fig. 4D). Collectively,
the results of the present study suggest that BORIS may
enhance the mismatch repair (MMR) system in NSCLC cells to resist
DDP chemotherapy.
Discussion
The results of the present study demonstrated the
association between BORIS expression and DDP resistance in
NSCLC. As P53 mutants have frequently been detected in different
types of cancer and were reported to account for DDP resistance
(25), the present study
investigated the effects of BORIS knockdown in H1299 (lack
of p53) and A549 (express wild type p53) cells, respectively
(19). Suppression of cell viability
on BORIS knockdown in both H1299 and A549 cells suggested
that the function of BORIS was not associated with p53. BORIS may
promote DDP resistance by upregulating MSH6 expression.
However, the underlying molecular mechanism as to how BORIS
regulates MSH6 remains unknown. BORIS was reported to regulate
CMYC expression by demethylating the promoter (26). Demethylation may be a potential
mechanism for regulating MSH6 or other genes in the DNA
repair system by BORIS to resist DDP treatment. Furthermore,
unknown cellular environments may be constructed by BORIS in cancer
cells to resist chemotherapy. In neuroblastoma, BORIS was reported
to be upregulated along with the development of ALK inhibition
resistance (27). It is speculated
that BORIS expression may promote cancer cells to resist multiple
drug treatments, including DDP. It is well-known that EGFR mutants
cause tyrosine kinase inhibitors (RTKIs) resistance in NSCLC
(28,29), thus, it is worth investigating
whether BORIS knockdown attenuates RTKIs resistance.
BORIS knockdown induced DNA damage and
p21 expression in NSCLC cells. As BORIS is only
expressed in cancer cells and testes (30), BORIS may be a key protector for
cancer cell genome stability. BORIS is the paralogue of CTCF and is
speculated to compete with CTCF to induce the expression of the
oncogene (31–33). The spatial genome structure organized
by CTCF may be interfered by BORIS (30,34–36). DNA
and histone methylation modulated by BORIS may also promote genome
stability (26,37,38).
The overexpression of BORIS sustained
DDP-induced apoptosis and repaired DNA damage in NSCLC cells. DDP
resistance is usually associated with the increased repair of DNA
damage recognized by the mismatch repair system (13,14,39).
MutSa (composed of MSH2 and MSH6) recognized DNA lesions formed by
DDP and subsequently recruited downstream MMR proteins, including
MutLα (MLH1-PMS2), Exonuclease I, DNA polymerase δ and DNA ligase
(14,40). The elevated expression of MSH6
induced by BORIS overexpression in the present study could
facilitate the recognition of DNA lesions and attenuate the
recruitment of the phosphorylated form of γH2AX to sites of DNA
damage (24).
In conclusion, BORIS is required for genome
stability of NSCLC cells and is prospective therapeutic target for
decreasing DDP resistance. Considering that RTKIs resistance is
frequent in lung cancer cells, further study of targeted therapy on
BORIS will be expected in RTKIs resistant lung cancer cells.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Mr. Xiaoliang Zheng,
Ms Dongmei Yan, Mr. Jing Jia and Ms Jie Yuat the Center for
Molecular Medicine (Hangzhou, China) for their technical assistance
and discussion surrounding the data. The authors would also like to
thank Professor Tianhui Chenfrom Zhejiang Cancer Hospital
(Hangzhou, China) for revising the manuscript.
Funding
The present study was funded by grants from the
National Natural Science Foundation of China (grant no. 31871393);
Zhejiang Provincial Natural Science Foundation of China (grant nos.
LQY18H300001 and LQ18C070002); Youth Foundation of Zhejiang Academy
of Medical Sciences (grant nos. 2019Y006 and 2019Y003); and the
Medical and Health Science and Technology Project of Zhejiang
Province (grant nos. 2017KY308 and 2019RC030).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author upon reasonable
request.
Authors' contributions
YS and YZ designed the experiments. YS and CL
performed the cell culture, drug treatment experiments and
collected the data. JR performed extraction and gene expression
analysis. MF performed the plasmid construction. YS and JF
performed the statistical analyses. YZ wrote the manuscript. YZ and
XW analyzed the data and edited the manuscript. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
5-FU
|
fluorouracil
|
|
A549/DDP
|
DDP resistant A549 cell line
|
|
BORIS
|
Brother of Regulator of Imprinted
Sites
|
|
CTCF
|
CCCTC-binding factor
|
|
DDP
|
cisplatin
|
|
NSCLC
|
non-small-cell lung cancer
|
|
FACS
|
flow cytometry
|
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tanaka F and Yoneda K: Adjuvant therapy
following surgery in non-small cell lung cancer (NSCLC). Surg
Today. 46:25–37. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chevalier TL and Sori JC: Status and
trends of chemotherapy for advanced NSCLC. Eur J Cancer Suppl.
2:26–33. 2004. View Article : Google Scholar
|
|
4
|
Giaccone G: Clinical perspectives on
platinum resistance. Drugs. 59 (Suppl 4):S9–S17, S37-S38. 2000.
View Article : Google Scholar
|
|
5
|
Martin-Kleiner I: BORIS in human cancers-a
review. Eur J Cancer. 48:929–935. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dougherty CJ, Ichim TE, Liu L, Reznik G,
Min W, Ghochikyan A, Agadjanyan MG and Reznik BN: Selective
apoptosis of breast cancer cells by siRNA targeting of BORIS.
Biochem Biophys Res Commun. 370:109–112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Asano T, Hirohashi Y, Torigoe T, Mariya T,
Horibe R, Kuroda T, Tabuchi Y, Saijo H, Yasuda K, Mizuuchi M, et
al: Brother of the regulator of the imprinted site (BORIS) variant
subfamily 6 is involved in cervical cancer stemness and can be a
target of immunotherapy. Oncotarget. 7:11223–11237. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Fang M, Song Y, Ren J, Fang J and
Wang X: Brother of regulator of imprinted sites (BORIS) suppresses
apoptosis in colorectal cancer. Sci Rep. 7:407862017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Siddik ZH: Cisplatin: Mode of cytotoxic
action and molecular basis of resistance. Oncogene. 22:7265–7279.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Longley DB, Harkin DP and Johnston PG:
5-Fluorouracil: Mechanisms of action and clinical strategies. Nat
Rev Cancer. 3:330–338. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lewin F, Ringborg U, Skog S and Tribukait
B: The effect of 5-fluorouracil on cisplatin induced DNA
interstrand cross-linking in a mouse ascites tumor growing in vivo.
Anticancer Drugs. 6:465–470. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoon SL, Roh YG, Chu IS, Heo J, Kim SI,
Chang H, Kang TH, Chung JW, Koh SS, Larionov V and Leem SH: A
polymorphic minisatellite region of BORIS regulates gene expression
and its rare variants correlate with lung cancer susceptibility.
Exp Mol Med. 48:e2462016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Almeida GM, Duarte TL, Farmer PB, Steward
WP and Jones GD: Multiple end-point analysis reveals cisplatin
damage tolerance to be a chemoresistance mechanism in a NSCLC
model: Implications for predictive testing. Int J Cancer.
122:1810–1819. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shen D, Pouliot LM, Hall MD and Gottesman
MM: Cisplatin resistance: A cellular self-defense mechanism
resulting from multiple epigenetic and genetic changes. Pharmacol
Rev. 64:706–721. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rosell R, Lord RVN, Taron M and Reguart N:
DNA repair and cisplatin resistance in non-small-cell lung cancer.
Lung Cancer. 38:217–227. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cohen SM and Lippard SJ: Cisplatin: From
DNA damage to cancer chemotherapy. Prog Nucleic Acid Res Mol Biol.
67:93–130. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lippard SJ: Platinum, Gold, and Other
Metal Chemotherapeutic Agents. Chemistry and Biochemistry. 209.
American Chemical Society; 1983
|
|
18
|
Basu A and Krishnamurthy S: Cellular
responses to cisplatin-induced DNA damage. J Nucleic Acids.
2010:2013672010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gomyo Y, Sasaki JI, Branch CD, Roth JA and
Mukhopadhyay T: 5-aza-2′-deoxycytidine upregulates caspase-9
expression cooperating with p53-induced apoptosis in human lung
cancer cells. Oncogene. 23:6779–6787. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Detterbeck FC, Boffa DJ, Kim AW and Tanoue
LT: The eighth edition lung cancer stage classification. Chest.
151:193–203. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bunz F, Dutriaux A, Lengauer C, Waldman T,
Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B:
Requirement for p53 and p21 to sustain G2 arrest after DNA damage.
Science. 282:1497–1501. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Waldman T, Kinzler KW and Vogelstein B:
p21 is necessary for the p53-mediated G1 arrest in human cancer
cells. Cancer Res. 55:5187–1590. 1995.PubMed/NCBI
|
|
23
|
el-Deiry WS, Tokino T, Velculescu VE, Levy
DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and
Vogelstein B: WAF1, a potential mediator of p53 tumor suppression.
Cell. 75:817–825. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mah L, Elosta A and Karagiannis TC:
GammaH2AX: A sensitive molecular marker of DNA damage and repair.
Leukemia. 24:679–686. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chee JL, Saidin S, Lane DP, Leong SM, Noll
JE, Neilsen PM, Phua YT, Gabra H and Lim TM: Wild-type and mutant
p53 mediate cisplatin resistance through interaction and inhibition
of active caspase-9. Cell Cycle. 12:278–88. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nguyen P, Barsela G, Sun L, Bisht KS, Cui
H, Kohn EC, Feinberg AP and Gius D: BAT3 and SET1A form a complex
with CTCFL/BORIS to modulate H3K4 histone dimethylation and gene
expression. Mol Cell Biol. 28:6720–6729. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Debruyne DN, Dries R, Sengupta S, Seruggia
D, Gao Y, Sharma B, Huang H, Moreau L, McLane M, Day DS, et al:
BORIS promotes chromatin regulatory interactions in
treatment-resistant cancer cells. Nature. 572:676–680. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kobayashi S, Boggon TJ, Dayaram T, Janne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lin Y, Wang X and Jin H: EGFR-TKI
resistance in NSCLC patients: Mechanisms and strategies. Am J
Cancer Res. 4:411–435. 2014.PubMed/NCBI
|
|
30
|
Sleutels F, Soochit W, Bartkuhn M, Heath
H, Dienstbach S, Bergmaier P, Franke V, Rosa-Garrido M, van de
Nobelen S, Caesar L, et al: The male germ cell gene regulator CTCFL
is functionally different from CTCF and binds CTCF-like consensus
sites in a nucleosome composition-dependent manner. Epigenetics
Chromatin. 5:82012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Loukinov DI, Pugacheva E, Vatolin S, Pack
SD, Moon H, Chernukhin I, Mannan P, Larsson E, Kanduri C, Vostrov
AA, et al: BORIS, a novel male germ-line-specific protein
associated with epigenetic reprogramming events, shares the same
11-zinc-finger domain with CTCF, the insulator protein involved in
reading imprinting marks in the soma. Proc Natl Acad Sci USA.
99:6806–6811. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pugacheva EM, Teplyakov E, Wu Q, Li J,
Chen C, Meng C, Liu J, Robinson S, Loukinov D, Boukaba A, et al:
The cancer-associated CTCFL/BORIS protein targets multiple classes
of genomic repeats, with a distinct binding and functional
preference for humanoid-specific SVA transposable elements.
Epigenetics Chromatin. 9:352016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bergmaier P, Weth O, Dienstbach S,
Boettger T, Galjart N, Mernberger M, Bartkuhn M and Renkawitz R:
Choice of binding sites for CTCFL compared to CTCF is driven by
chromatin and by sequence preference. Nucleic Acids Res.
46:7097–7107. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pugacheva EM, Rivero-Hinojosa S, Espinoza
CA, Méndez-Catalá CF, Kang S, Suzuki T, Kosaka-Suzuki N, Robinson
S, Nagarajan V, Ye Z, et al: Comparative analyses of CTCF and BORIS
occupancies uncover two distinct classes of CTCF binding genomic
regions. Genome Biol. 16:1612015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lobanenkov VV and Zentner GE: Discovering
a binary CTCF code with a little help from BORIS. Nucleus. 9:33–41.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hong JA, Kang Y, Abdullaev Z, Flanagan PT,
Pack SD, Fischette MR, Adnani MT, Loukinov DI, Vatolin S, Risinger
JI, et al: Reciprocal binding of CTCF and BORIS to the NY-ESO-1
promoter coincides with derepression of this cancer-testis gene in
lung cancer cells. Cancer Res. 65:7763–7774. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jelinic P, Stehle JC and Shaw P: The
testis-specific factor CTCFL cooperates with the protein
methyltransferase PRMT7 in H19 imprinting control region
methylation. PLoS Biol. 4:e3552006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sun L, Huang L, Nguyen P, Bisht KS,
Bar-Sela G, Ho AS, Bradbury CM, Yu W, Cui H, Lee S, et al: DNA
methyltransferase 1 and 3B activate BAG-1 expression via
recruitment of CTCFL/BORIS and modulation of promoter histone
methylation. Cancer Res. 68:2726–2735. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gautam A, Li ZR and Bepler G: RRM1-induced
metastasis suppression through PTEN-regulated pathways. Oncogene.
22:2135–1242. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Martin LP, Hamilton TC and Schilder RJ:
Platinum resistance: The role of DNA repair pathways. Clin Cancer
Res. 14:1291–1295. 2008. View Article : Google Scholar : PubMed/NCBI
|
















