Introduction
Ulcerative colitis (UC) poses a high risk of
developing colorectal cancer (CRC) (1), and the number of patients with
UC-associated CRC is increasing all over the world as that with UC
increases (2). The risk of
developing UC-associated CRC is affected by the degree and duration
of inflammation in UC and genetic predisposition (3). However, the detailed molecular
mechanism underlying transition from UC-associated inflammation to
carcinogenesis has not yet been elucidated.
Some animal models of sporadic and
colitis-associated CRC have been developed in rodents. Induction of
the most well-studied mouse model of chemical-induced
colitis-associated CRC requires a single intraperitoneal injection
of azoxymethane (AOM), a carcinogen of the colon, followed by
colitis induction through oral administration of dextran sodium
sulfate (DSS) (4,5). This AOM/DSS mouse model reproduces the
course of human colitis-associated CRC from inflammation to
dysplasia and carcinoma, producing severe colitis with weight loss,
bloody diarrhea, and multiple colon tumors (5,6).
There are other aspects to the mouse models of CRC,
including the AOM/DSS model. There are significant differences in
invasive and metastatic potentials of CRC between animal models and
human disease (6–8). At diagnosis, approximately 50% of CRC
patients have lymphatic metastases and 33% have hematogenous
metastases (6). In contrast, the
frequency of metastases is extremely low in AOM models of CRC
(8). In human CRC, the disease
progresses in order, and a usual route of hematogenous metastasis
first reaches the liver, and subsequently the lung. However,
metastatic liver tumors are rare in the AOM models of rodent CRC.
Although animal models of chemical-induced CRCs have provided much
information about human disease, many research approaches have not
been made available, and further research to establish animal
models of metastasis is needed.
Besides remodeling microenvironment to promote
metastasis, cancer cells actuate regulators of embryonic
morphogenesis to achieve epithelial-mesenchymal transition (EMT)
and stop the differentiation program. Thereby, cancer cells acquire
motility, invade, and gain stem-like characters (9,10).
Recently, it has been revealed that cancer stem cells (CSCs) and
EMT-type cells have common molecular features and play important
roles in tumor metastasis (11–13). The
development of CSCs and EMT can promote the formation of metastatic
tumors in CRC, but the molecular mechanisms of metastasis involving
CSCs and EMT remain unclear (14).
Furthermore, because CSCs are comparatively resistant to therapies
created to eradicate cancer cells with non-CSC characters, analyses
of CSCs, which are small in number and are multi-drug resistant,
form the bases for the development of new therapeutic agents
targeting CSCs, and open the door to new cancer treatments
(11).
Smad proteins are core mediators that transmit
signals from transforming growth factor (TGF)-β superfamily
receptors to the nuclei. They are regulatory proteins consisting of
conserved Mad homology (MH)1, intermediate linker, and MH2 domains
(15,16).
TGF-β type I receptor (TβRI) with catalytic activity
phosphorylates COOH-terminal serine (Ser) residues of
receptor-activated Smads, including Smad2 and Smad3 (17). Certain Ser or threonine (Thr)
residues in the linker are phosphorylated by Ras-related
(proline-directed) kinases, consisting of extracellular
signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and
cyclin-dependent kinase (CDK)4 (18–21).
TβRI and Ras-related kinases specifically phosphorylate Smad2 and
Smad3, creating several phosphoisoforms: Smad2/3 phosphorylated at
the COOH-terminal (pSmad2C and pSmad3C); Smad2/3 phosphorylated at
the linker (pSmad2L and pSmad3L); and Smad2/3 phosphorylated at
both the C-terminal and linker (pSmad2C/L and pSmad3C/L) (21–24).
Phosphorylated Smad2 and Smad3 promptly form oligomers with Smad4
and translocate to the nucleus. There, they control transcription
of the target genes (25).
In our previous study, we confirmed specific
expression of linker Thr-phosphorylated Smad2/3 (pSmad2/3L-Thr) in
mouse colon epithelial cells, and proposed that these cells are
colon epithelial stem-like cells (26). Subsequently, we investigated AOM/DSS
mouse model, and clarified that carcinogenic pSmad3L-Ser signaling
caused by chronic colitis is a significant early event of
colitis-associated CRC, by investigating the Smad2/3
phosphorylation profiles. Furthermore, the study has supported the
theory that pSmad2/3L-Thr immunostaining-positive cells are CSCs
(27). AOM/DSS mice were sacrificed
10 or 20 weeks after AOM administration in that study, and most
colon tumors showed the characteristics of intramucosal
adenocarcinoma. Around 40 weeks after AOM administration, the
number of AOM/DSS mice dying had markedly increased
pre-experimentally.
Therefore, in the present study, we extended our
observations until 30 weeks after AOM administration to study a
colitis-associated advanced CRC mouse model, and explore if
pSmad2/3L-Thr immunostaining-positive cells are involved in a
progressive course of colitis-associated CRC as CSCs.
Materials and methods
Mice
We purchased 5-week-old male Crl:CD-1 (ICR) mice
from Charles River Laboratories (Charles River Laboratories Japan,
Inc.). All mice were kept in the animal facility of Kansai Medical
University under specific pathogen-free environment. Mice were fed
commercial food pellets (F2; Funabashi Farm) and tap water. All
experimental protocols were approved by the Ethics Committee for
the Use of Experimental Animals of Kansai Medical University
(approval no. 19-006).
Chemicals
We purchased AOM, a colon carcinogen, from
Sigma-Aldrich Japan K.K. and DSS having a molecular weight of
36,000-50,000 from MP Biomedicals. DSS was diluted with water to
form 2% solution to induce colitis.
Experimental design
A single intraperitoneal injection of AOM (10 mg/kg
body weight) was administered to ICR mice, and one week after the
AOM administration, the mice were given 2% DSS in their drinking
water for 7 days. Mice administered with AOM/DSS (AOM/DSS mice)
were sacrificed by cervical dislocation 10 (week 10; n=25), 20
(week 20; n=25) or 30 (week 30; n=25) weeks after AOM
administration (Fig. 1) (5,27,28).
Colons were excised after flushing the lumens with
saline and cut open longitudinally. After several washes with
saline, they were cut and fixed in 10%-buffered formalin.
Paraffin-embedded sections were prepared by using standard
method.
Histopathological analysis
Histopathological changes were confirmed in
hematoxylin and eosin (H&E)-stained specimens. Colorectal
neoplasms were diagnosed based on the description of Ward (29). CRC infiltration into the submucosal
layer, and invasion into vessels could also be observed in these
sections. All cases of CRC invasion into vessels were re-confirmed
by immunohistochemical staining of the blood vessels or lymph
vessels as explained below in detail.
Domain-specific antibody against the
phosphorylated Smad2 and Smad3
Rabbit polyclonal anti-human pSmad2/3L-Thr (Smad2:
Thr 220; Smad3: Thr 179) sera were produced against the
phosphorylated linker region of Smad2 and Smad3 by immunizing
rabbits with synthetic peptides (23,30,31). The
antisera were subjected to antigen affinity purification using
phosphorylated peptides as previously described (32).
Immunohistochemistry
We performed immunohistochemical staining on
formalin-fixed paraffin-embedded sections as described previously
(26,27,33).
Briefly, paraffin-embedded sections were deparaffinized and
rehydrated by washing with xylene and ethanol. Non-enzymatic
antigen retrieval was performed by heating sections to 121°C for 10
min in 0.01 M sodium citrate buffer (pH 6.0). After cooling,
sections were immersed in Tris-buffered saline (TBS) and blocked
with 3% bovine serum albumin dissolved in TBS for 5 min. Primary
antibodies (Abs) were diluted with TBS containing 0.1% Tween-20,
and incubated at 4°C in a humidity chamber. The primary Abs used in
this study were as follows: mouse monoclonal anti-human β-catenin
Ab (sc-7963; Santa Cruz Biotechnology, Inc.), rat monoclonal
anti-mouse CD34 Ab (ab8158; Abcam), rat monoclonal anti-mouse
podoplanin Ab (015-24111; Fujifilm Wako Pure Chemical Corp.),
rabbit polyclonal anti-human E-cadherin Ab (sc-7870, Santa Cruz
Biotechnology, Inc.), rat monoclonal anti-mouse Ki67 Ab (652402;
BioLegend), goat polyclonal anti-human B cell-specific Moloney
murine leukemia virus integration site 1 (Bmi1) Ab (ab115251;
Abcam), and rabbit polyclonal anti-human pSmad2/3L-Thr Ab. The
secondary Abs used were the appropriate species-specific AlexaFluor
(488 or 568)-conjugated Abs (Thermo Fisher Scientific, Inc.).
Slides were mounted in VECTASHIELD mounting medium containing
4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories) to stain
nuclei. Images were taken and captured using a fluorescence
microscope (Olympus). After immunofluorescent staining, the
specimen slides were immersed in distilled water and the cover
glasses were removed so as not to damage the tissues. After
sufficiently immersing them in TBS, H&E staining was performed
by a standard staining procedure. Then, we observed the same
sections under a light microscope.
Well-oriented lesions from the base to the surface
of CRC were selected for counting immunostaining-positive cells
using inForm software (PerkinElmer) according to the manufacturer's
instructions.
Statistical analysis
Comparing the presence (positives) or absence
(negatives) of CRC infiltration into the submucosal layer and
invasion into vessels (grouping variable: 0, negatives; 1,
positives), we analyzed the data with Kruskal-Wallis test followed
by Mann-Whitney U test with Bonferroni correction.
Comparing the percentages of immunostaining-positive
cells, we expressed the values as mean ± standard error of the mean
(SEM). We analyzed the data with paired t-test.
P<0.05 was considered to indicate a statistically
significant difference.
Results
Microscopic observations
As previously reported, we observed flat, nodular,
polypoid, or caterpillar-like colon tumors in the middle and distal
colons of all AOM/DSS mice at weeks 10, 20 and 30 (data not shown)
(5,27,28). The
size and number of colon tumors tended to increase with time after
the AOM administration.
Colon tumors of AOM/DSS mice at weeks 10 (Fig. 2A), 20 (Fig. 2B), and 30 (Fig. 2C and D) had the typical appearance of
adenocarcinoma. The nuclei were enlarged, round or ovoid, with
remarkable nucleoli; nuclear polarity was almost lost; there were a
lot of mitoses; and goblet cells had completely disappeared.
Tumor infiltrations into the submucosal layer were
occasionally observed in AOM/DSS mice at weeks 10 (24%: 6/25) and
20 (28%: 7/25). Alternatively, these findings were frequently
observed in AOM/DSS mice at week 30 (72%: 18/25) (Fig. 2D; arrowhead). Compared with AOM/DSS
mice at weeks 10 and 20, submucosal tumor infiltrations of AOM/DSS
mice significantly increased at week 30 (P=0.0021 and P=0.0051,
respectively). No significant difference was found between AOM/DSS
mice at weeks 10 and 20 (P=0.76).
Tumor invasions into vessels were scarcely observed
in AOM/DSS mice at weeks 10 (0%: 0/25) and 20 (4%: 1/25). However,
these findings were sometimes observed in AOM/DSS mice at week 30
(40%: 10/25) (Fig. 2D; arrow).
Compared with AOM/DSS mice at weeks 10 and 20, tumor invasions into
vessels of AOM/DSS mice significantly increased at week 30
(P=0.0005 and P=0.0024, respectively). No significant difference
was found between AOM/DSS mice at weeks 10 and 20 (P=0.32). These
results are summarized in Table
I.
 | Table I.Frequency of tumor infiltration into
the submucosal layer and invasion into vessels in
azoxymethane/dextran sodium sulfate mice. |
Table I.
Frequency of tumor infiltration into
the submucosal layer and invasion into vessels in
azoxymethane/dextran sodium sulfate mice.
| Weeks | Submucosal
infiltration, positive | Vessel invasion,
positive |
|---|
| Week 10 | 6/25
(24%)a,b | 0/25
(0%)a,c |
| Week 20 | 7/25
(28%)b | 1/25
(4%)b |
| Week 30 | 18/25 (72%) | 10/25 (40%) |
Double immunofluorescent staining for
β-catenin and markers of blood and lymph vessels at sites of tumor
invasions into vessels
After we confirmed tumor invasions into vessels by
H&E-stained sections, we performed double immunofluorescent
staining for β-catenin (green; Fig. 3A,
B, D and E) with CD34 (red; Fig. 3A
and D) and podoplanin (red; Fig. 3B
and E) to distinguish invasions into blood from lymph vessels
in these AOM/DSS mice at weeks 20 (n=1) and 30 (n=10), using DAPI
nuclear staining (blue).
 | Figure 3.Double immunofluorescent staining of
β-catenin (green) with CD34 (red) and podoplanin (red) in AOM/DSS
mice at week 30. DAPI (blue) was used for nuclear staining. (A-C)
In AOM/DSS mice that showed blood vessel invasion, (A and B)
β-catenin-positive cells were diffusely distributed throughout the
tumors in vessels. (A) Although immunofluorescent staining of CD34
revealed ring-shaped positivity around the vessel lumens, (B)
podoplanin-positive cells were not detected in these vessels. (D-F)
In AOM/DSS mice that showed lymph vessel invasion, (D and E)
β-catenin-positive tumor cells were similarly distributed in
vessels. (D) Contrarily, CD34-positive cells were not detected in
the vessels, and (E) immunofluorescent staining of podoplanin
demonstrated ring-shaped positivity around the vessel lumens. (C
and F) Following immunofluorescent staining, the same sections were
stained with hematoxylin and eosin, and observed for tumor invasion
into vessels by light microscopy. Original magnification, (A, B, D
and E) ×200 and (C and F) ×100. Scale bars, 100 µm. AOM,
azoxymethane; DSS, dextran sodium sulfate. |
In AOM/DSS mice that showed blood vessel invasion,
β-catenin-positive cells were diffusely distributed throughout the
tumors in vessels (Fig. 3A and B).
Although immunofluorescent staining of CD34 showed ring-shaped
positivity around the vessel lumens (Fig. 3A), podoplanin-positive cells were not
detected in these vessels (Fig. 3B).
In AOM/DSS mice that showed lymph vessel invasion,
β-catenin-positive tumor cells were similarly distributed in
vessels (Fig. 3D and E). In
contrast, CD34-positive cells were not detected in the vessels
(Fig. 3D), and immunofluorescent
staining of podoplanin showed ring-shaped positivity around the
vessel lumens (Fig. 3E).
Of the 11 AOM/DSS mice with vessel invasion, tumor
invasions into blood and lymph vessels were observed in 3 and 8
mice, respectively.
After immunofluorescent staining, the same sections
were stained with H&E and observed for tumor invasions into
vessels under a light microscope (Fig.
3C and F).
Double immunofluorescent staining for
β-catenin and E-cadherin
Immunofluorescent staining of β-catenin (green;
Fig. 4A and D) showed weak
positivity only in the cell membrane of non-tumorous mucosae in
AOM/DSS mice at weeks 10 (Fig. 4A)
and 30 (Fig. 4D), using DAPI nuclear
staining (blue). Strongly β-catenin-positive cells were distributed
throughout the tumors in AOM/DSS mice, and their expression was
predominantly observed in the cytoplasm and nucleus of tumor cells.
In the same sections, immunofluorescent staining of E-cadherin
(red; Fig. 4B and E) showed strong
positivity in the cell membrane of non-tumorous mucosae in AOM/DSS
mice at weeks 10 (Fig. 4B) and 30
(Fig. 4E). Positive levels of
E-cadherin in the cell membrane of colon tumors in AOM/DSS mice,
particularly in mice at week 30 (Fig.
4E), were clearly reduced as compared with those of
non-tumorous mucosae.
After immunofluorescent staining, the same sections
were stained with H&E, and CRCs were confirmed using a light
microscope (Fig. 4C and F).
Double immunofluorescent staining for
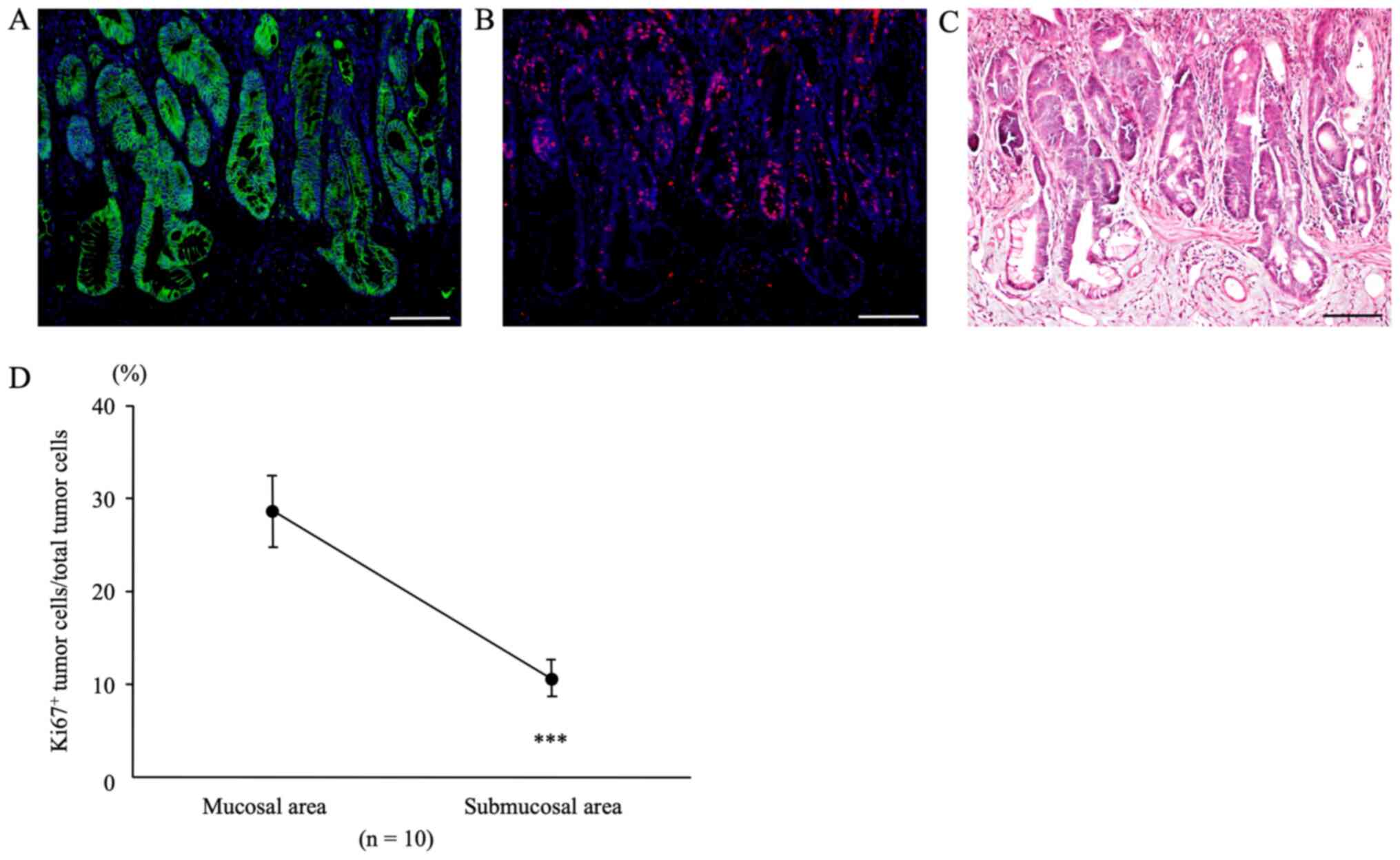
β-catenin and Ki67 at sites of submucosal infiltration
In colon tumors from AOM/DSS mice at week 30 that
showed tumor infiltration into the submucosal layer,
β-catenin-positive tumor cells (green; Fig. 5A) were diffusely distributed in both,
mucosal areas, and sites of submucosal infiltration, using DAPI
nuclear staining (blue). In the same sections, immunofluorescent
staining for Ki67 (red; Fig. 5B) was
performed. Subsequently, we calculated the Ki67-positive tumor cell
count/total tumor cell count separately for mucosal areas and sites
of submucosal infiltration using the software. The percentage of
Ki67-positive tumor cells in mucosal areas of AOM/DSS mice (28.66 ±
3.80%) was significantly higher than that in sites of submucosal
infiltration (10.66±1.97%) (Fig. 5D,
n=10, P=0.0001).
After immunofluorescent staining, the same sections
were stained with H&E, and CRCs and muscularis mucosae were
confirmed under a light microscope (Fig.
5C).
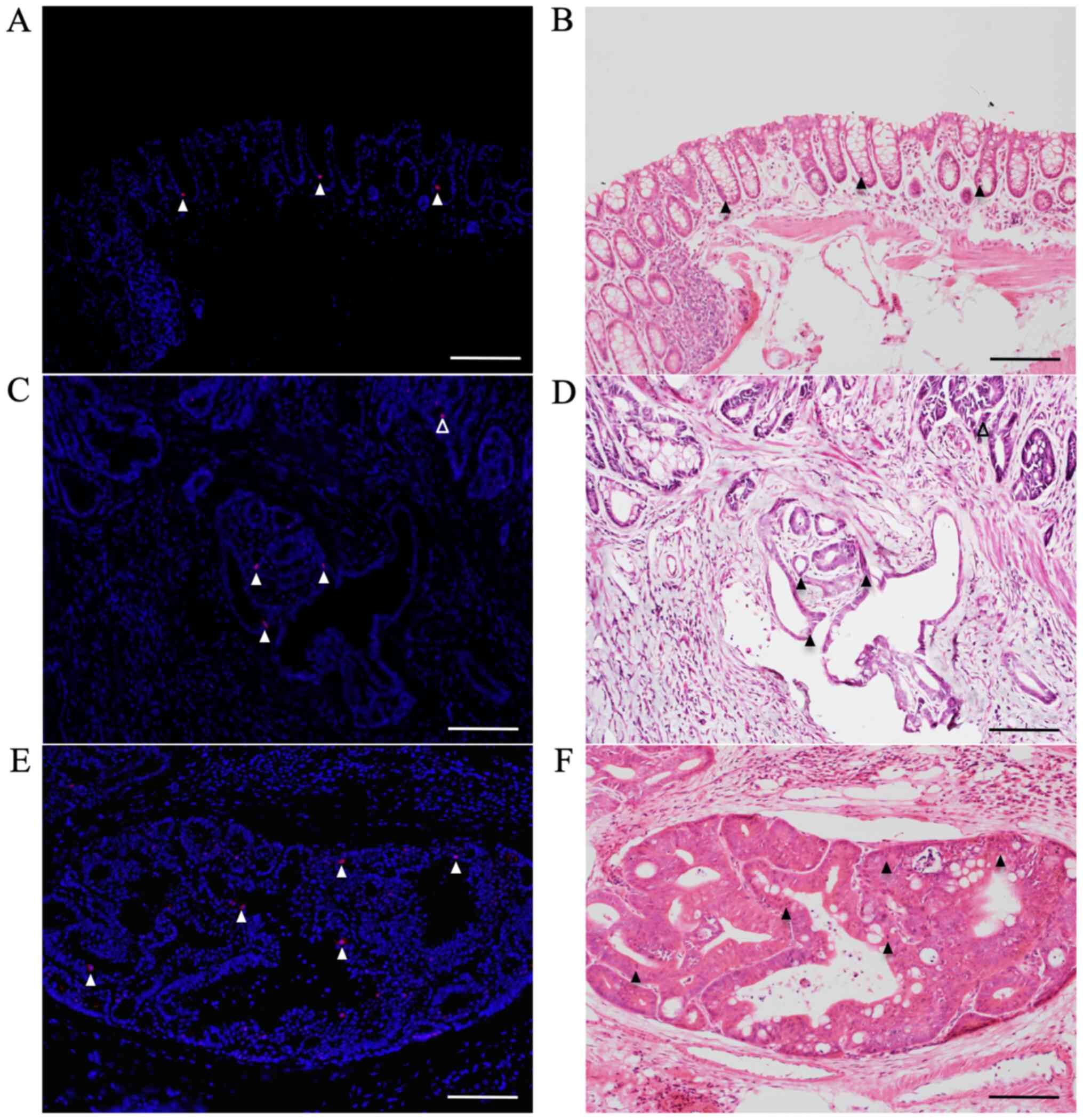
Immunofluorescent staining for
pSmad2/3L-Thr in CRCs with submucosal infiltration and vessel
invasion
pSmad2/3L-Thr-positive cells (red; arrowheads in
Fig. 6) were sparsely detected
around crypt bases in non-tumorous mucosae from AOM/DSS mice at
week 30 (Fig. 6A), using DAPI
nuclear staining (blue). In mucosal areas of colon tumors from
AOM/DSS mice, pSmad2/3L-Thr-positive cells were scattered within
tumor cells (open arrowhead; Fig.
6C). Furthermore, at both sites of submucosal infiltration
(filled arrowheads; Fig. 6C) and
vessel invasions (Fig. 6E) of these
tumors, pSmad2/3L-Thr-positive cells were also detected within
tumor cells.
After immunofluorescent staining, the same sections
were stained with H&E to confirm non-tumorous mucosae, CRCs,
and pSmad2/3L-Thr-positive cells (arrowheads) under light
microscopy (Fig. 6B, D and F).
Double immunofluorescent staining for
pSmad2/3L-Thr and β-catenin
In colon tumors from AOM/DSS mice at week 30 that
showed nuclear β-catenin expression in tumor cells, double
immunofluorescent staining for pSmad2/3L-Thr (red; arrowheads in
Fig. 7A) and β-catenin (green;
Fig. 7B) were performed and
analyzed, using DAPI nuclear staining (blue).
The percentage of pSmad2/3L-Thr-positive cells
within the nuclear β-catenin-positive tumor cells (9.98±1.82%) was
significantly higher than that within the cytoplasmic
β-catenin-positive tumor cells (3.67±0.77%) (Fig. 7D, n=20, P=0.0001).
After immunofluorescent staining, the same sections
were stained with H&E, and CRCs and pSmad2/3L-Thr-positive
cells (arrowheads) were confirmed using light microscopy (Fig. 7C).
Double immunofluorescent staining for
pSmad2/3L-Thr and Bmi1
In colon tumors from AOM/DSS mice at week 30, double
immunofluorescent staining for pSmad2/3L-Thr (red; arrowheads in
Fig. 8) and Bmi1 (green) were
performed, using DAPI nuclear staining (blue).
pSmad2/3L-Thr-positive cells were sparsely detected
around crypt bases in non-tumorous mucosae from AOM/DSS mice at
week 30 (Fig. 8A). In colon tumors
from AOM/DSS mice, pSmad2/3L-Thr-positive cells were scattered
within tumor cells (Fig. 8D). In
both non-neoplastic and neoplastic epithelial cells,
pSmad2/3L-Thr-positive cells showed immunohistochemical
co-localization with Bmi1 (Fig. 8B, C, E
and F).
Discussion
The prevalence of UC-associated CRCs has increased
with increasing numbers of UC patients (2). The risk for developing UC-associated
CRCs depends on the severity and duration of inflammation (3). The AOM/DSS mouse model manifests
pathological findings represented by severe colitis with subsequent
development of many colon tumors, and recapitulates the sequence of
colitis-associated CRC formation in humans (5,6).
Some mouse CRC models have been reported, but each
has certain limitations, such as the absence of spontaneous CRC and
the need for carcinogens to induce tumors. Also, in many mouse
models, there are differences between animals in the development of
intestinal tumors. Thus, there is an urgent need for models that
more closely reflect the biology and progression of human
cancer.
Mouse models of colitis-associated and sporadic
CRCs, including the AOM/DSS model, show a very low incidence of
invasion and metastases (8).
Although chemical-induced (autochthonous) mouse CRC models have
provided much information about human disease, there are still many
research approaches that have not been available. In our previous
study, we sacrificed AOM/DSS mice at weeks 10 or 20, and most colon
tumors were characterized by intramucosal adenocarcinoma (27). Therefore, we extended our
observations until 30 weeks after AOM administration, and have
developed a mouse (colitis-associated) model of advanced CRC, which
shows a high degree of authenticity and consistent results for the
pathology of disease progression found in human cancer patients.
AOM/DSS mice, at weeks 10 and 20, showed tumor infiltrations into
submucosa 24 and 28% of the times, respectively, whereas at week 30
it was 72%. AOM/DSS mice, at weeks 10 and 20, showed tumor
invasions into 0 and 4% of the vessels, respectively, whereas at
week 30 the value was 40%. Although no metastases to lymph nodes or
other organs were observed, AOM/DSS mice were considered to acquire
invasive and metastatic ability, especially between 20 and 30 weeks
after AOM administration.
Recent advances in cancer biology have further
emphasized the role of the immune system in creating a tumor
microenvironment that promotes cancer progression through tumor
growth, invasion, and metastasis. Potential uses of the model
include experiments with targeted therapies and conventional drugs
that affect the tumor microenvironment in the preoperative
environment. Research into the role of the tumor microenvironment
and the immune system, which may promote or inhibit CRC
progression, is important. Therefore, mouse models used in
preclinical studies must have complete immune responses to reveal
more detailed interactions with human cancer. The tumor
microenvironment is particularly important in the context of
metastatic disease, where new therapies are still in great need.
Because drugs aimed at regulating these cancer progression factors
are developed and evaluated preclinically, model systems in which
tumors have advanced will play major roles. In the future, if our
method can be improved in AOM/DSS mice to establish a CRC model
with metastasis to lymph nodes and organs, it is expected to
provide a mouse metastatic CRC model that is particularly useful in
preclinical studies for the development of drugs that target those
mechanisms.
The proteins involved in the regulation of
cytoskeletal apparatus function in conjunction with the junctional
proteins such as β-catenin and E-cadherin. Their dynamics influence
functional integrity that affects cell migration, invasion, and
polarity. β-catenin is a pivotal component of the
E-cadherin-mediated cell-cell adhesion system, and a key molecule
in the Wnt-APC signaling pathway (34). It controls the transcription of genes
involved in cell growth, development, and differentiation.
β-catenin is found in the cell membrane of non-tumorous colon
epithelial cells, but nuclear and cytoplasmic β-catenin
accumulation is associated with carcinogenesis in the colon
(35). Consistent with earlier
studies, we detected the nuclear and cytoplasmic β-catenin
accumulation in colon tumors of AOM/DSS mice, including vessel
invasions. Many studies have established the link between loss of
E-cadherin expression in cancer cells and transition to EMT
(36,37). Expression levels of E-cadherin differ
dramatically among human tumors, demonstrating that there is a
positive correlation between E-cadherin levels and patient survival
(38). In this regard, mutations in
the E-cadherin gene have been identified in cancer cells and are
thought to facilitate EMT development and cancer cell metastasis
(39,40). Consistent with these studies, we
detected strong E-cadherin positivity in the cell membrane of the
parts of non-tumorous mucosae in AOM/DSS mice, and levels of
E-cadherin positivity in the cell membrane were clearly attenuated
in the parts of colon tumors, especially in AOM/DSS mice at week
30, suggesting strong induction and promotion of EMT.
Ki67 antigen is a nuclear matrix protein expressed
in proliferating cells, but not in quiescent cells (41). In colon tumors from AOM/DSS mice at
week 30 that showed tumor infiltration into the submucosal layer,
the percentage of Ki67-positive tumor cells at the sites of
submucosal infiltration, the invasion front of CRCs, was
significantly lower than that in mucosal areas. This finding is
consistent with previous reports that the invasion front of human
CRCs shows low proliferative activity (42–44).
In our previous studies, we have confirmed
significant expression of pSmad2/3L-Thr in normal colon epithelial
cells of wild-type mice, and in tumorous colon epithelial cells of
AOM/DSS mice, suggesting that these cells are colon epithelial
stem-like cells and colorectal CSCs, respectively (26,27). In
addition, we have shown that pSmad2/3L-Thr-positive cells are BrdU
label-retaining, slow-cycling, and Ki67-negative quiescent cells in
the G0 phase, located adjacent to actively proliferating cells of
normal esophageal and colon epithelial cells (26,45). We
have consistently advocated that pSmad2/3L-Thr helps identify
normal epithelial stem-like cells in the esophagus, stomach, small
intestine, and colon and colorectal CSCs, immediately before they
re-enter the cell cycle from the dormant state of the G0 phase
(26,27,33,45).
Moreover, in the present study, pSmad2/3L-Thr-positive cells were
detected in both, the sites of submucosal infiltration as well as
vessel invasion of tumors in AOM/DSS mice at week 30. Additionally,
the percentage of pSmad2/3L-Thr-positive cells within the nuclear
β-catenin-positive tumor cells was significantly higher than that
within the cytoplasmic β-catenin-positive tumor cells. This result
is consistent with previous reports suggesting that nuclear
β-catenin accumulation is important for regulating intranuclear
CSC-related transcription factors and maintaining the CSC phenotype
(46,47). We performed double immunofluorescent
staining for pSmad2/3L-Thr and Bmi1, which is a representative
marker of slow-cycling (cancer) stem cells (48,49).
pSmad2/3L-Thr-positive cells showed immunohistochemical
co-localization with Bmi1 in non-tumorous mucosae and tumors in
AOM/DSS mice at week 30. We were able to re-confirm the results
supporting that pSmad2/3L-Thr is a biomarker of tissue and cancer
stem cells.
CSC theory suggests that a small number of
undifferentiated cancer cells that exhibit normal stem cell-like
characteristics promote tumor growth and spread. Although CSCs are
capable of self-renewal, they are relatively dormant and are
capable of proliferation, although not often cycling. They have
been shown to have significantly longer cell-cycle times when
compared to proliferating non-CSCs. It is presumed that this is due
to the arrest of CSCs in the G0 phase (50). Although CRC has been thoroughly
studied, little is known about the original cells of
carcinogenesis. In both non-neoplastic and neoplastic epithelial
cells, pSmad2/3L-Thr-positive cells consistently exhibit stem-like
properties. pSmad2/3L-Thr has a high probability of a stem-like
cell biomarker in other organs and CSCs. Future studies are needed
to further confirm that pSmad2/3L-Thr-positive cells are CSCs in
other neoplastic lesions.
In conclusion, in this study, we developed a mouse
(colitis-associated) advanced CRC model that showed tumor
infiltration into the submucosa and invasion into the vessels, with
the results of this study compelling us to re-acknowledge the
theory that pSmad2/3L-Thr immunostaining-positive cells are
CSCs.
Acknowledgements
Not applicable.
Funding
The present study was supported by a Grant-in-Aid
for Scientific Research (C) (grant nos. 16K09330 and 25460938) from
the Japan Society for the Promotion of Science.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
All authors have contributed to and agreed on the
content of the manuscript. YT carried out the experiments,
conducted data analyses and drafted the manuscript. TF conceived
the study, carried out data analyses, performed the statistical
analyses and helped to draft the manuscript. SH, YM, SM, RS, TTa,
TTo, TI, YA, AN and KO performed data analyses, and provided
significant advice and consultation. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Ethics Committee for the Use of Experimental Animals of Kansai
Medical University (approval no. 19-006).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
UC
|
ulcerative colitis
|
|
CRC
|
colorectal cancer
|
|
AOM
|
azoxymethane
|
|
DSS
|
dextran sodium sulfate
|
|
EMT
|
epithelial-mesenchymal transition
|
|
CSC
|
cancer stem cell
|
|
TGF-β
|
transforming growth factor-β
|
|
MH
|
Mad homology
|
|
TβRI
|
TGF-β type I receptor
|
|
Ser
|
serine
|
|
Thr
|
threonine
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
JNK
|
c-Jun NH2-terminal kinase
|
|
CDK
|
cyclin-dependent kinase
|
|
pSmad2/3L-Thr
|
linker threonine-phosphorylated
Smad2/3
|
|
ICR
|
Crl:CD-1
|
|
H&E
|
hematoxylin and eosin
|
|
TBS
|
Tris-buffered saline
|
|
Abs
|
antibodies
|
|
Bmi1
|
B cell-specific Moloney murine
leukemia virus integration site 1
|
|
DAPI
|
4′,6-diamidino-2-phenylindole
|
|
SEM
|
standard error of the mean
|
References
|
1
|
Eaden JA, Abrams KR and Mayberry JF: The
risk of colorectal cancer in ulcerative colitis: A meta-analysis.
Gut. 48:526–535. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ekbom A, Helmick C, Zack M and Adami HO:
Ulcerative colitis and colorectal cancer. A population-based study.
N Engl J Med. 323:1228–1233. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seril DN, Liao J, Yang GY and Yang CS:
Oxidative stress and ulcerative colitis-associated carcinogenesis:
Studies in humans and animal models. Carcinogenesis. 24:353–362.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanaka T: Development of an
inflammation-associated colorectal cancer model and its application
for research on carcinogenesis and chemoprevention. Int J Inflamm.
2012:6587862012.
|
|
5
|
Tanaka T, Kohno H, Suzuki R, Yamada Y,
Sugie S and Mori H: A novel inflammation-related mouse colon
carcinogenesis model induced by azoxymethane and dextran sodium
sulfate. Cancer Sci. 94:965–973. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tanaka T: Colorectal carcinogenesis:
Review of human and experimental animal studies. J Carcinog.
8:52009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boivin GP, Washington K, Yang K, Ward JM,
Pretlow TP, Russell R, Besselsen DG, Godfrey VL, Doetschman T, Dove
WF, et al: Pathology of mouse models of intestinal cancer:
Consensus report and recommendations. Gastroenterology.
124:762–777. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rosenberg DW, Giardina C and Tanaka T:
Mouse models for the study of colon carcinogenesis. Carcinogenesis.
30:183–196. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan
A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, et al: The
epithelial-mesenchymal transition generates cells with properties
of stem cells. Cell. 133:704–715. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kalluri R and Weinberg RA: The basics of
epithelial-mesenchymal transition. J Clin Invest. 119:1420–1428.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Clarke MF, Dick JE, Dirks PB, Eaves CJ,
Jamieson CH, Jones DL, Visvader J, Weissman IL and Wahl GM: Cancer
stem cells - perspectives on current status and future directions:
AACR Workshop on cancer stem cells. Cancer Res. 66:9339–9344. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Campbell LL and Polyak K: Breast tumor
heterogeneity: Cancer stem cells or clonal evolution? Cell Cycle.
6:2332–2338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sarkar FH, Li Y, Wang Z and Kong D:
Pancreatic cancer stem cells and EMT in drug resistance and
metastasis. Minerva Chir. 64:489–500. 2009.PubMed/NCBI
|
|
14
|
Brabletz T, Hlubek F, Spaderna S,
Schmalhofer O, Hiendlmeyer E, Jung A and Kirchner T: Invasion and
metastasis in colorectal cancer: Epithelial-mesenchymal transition,
mesenchymal-epithelial transition, stem cells and beta-catenin.
Cells Tissues Organs. 179:56–65. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Massagué J: TGF-beta signal transduction.
Annu Rev Biochem. 67:753–791. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wrana JL: Crossing Smads. Sci STKE.
2000:re12000.PubMed/NCBI
|
|
18
|
Kretzschmar M, Doody J, Timokhina I and
Massagué J: A mechanism of repression of TGFbeta/Smad signaling by
oncogenic Ras. Genes Dev. 13:804–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Matsuura I, Denissova NG, Wang G, He D,
Long J and Liu F: Cyclin-dependent kinases regulate the
antiproliferative function of Smads. Nature. 430:226–231. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mori S, Matsuzaki K, Yoshida K, Furukawa
F, Tahashi Y, Yamagata H, Sekimoto G, Seki T, Matsui H, Nishizawa
M, et al: TGF-beta and HGF transmit the signals through
JNK-dependent Smad2/3 phosphorylation at the linker regions.
Oncogene. 23:7416–7429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tarasewicz E and Jeruss JS:
Phospho-specific Smad3 signaling: Impact on breast oncogenesis.
Cell Cycle. 11:2443–2451. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matsuzaki K: Smad3 phosphoisoform-mediated
signaling during sporadic human colorectal carcinogenesis. Histol
Histopathol. 21:645–662. 2006.PubMed/NCBI
|
|
23
|
Matsuzaki K, Kitano C, Murata M, Sekimoto
G, Yoshida K, Uemura Y, Seki T, Taketani S, Fujisawa J and Okazaki
K: Smad2 and Smad3 phosphorylated at both linker and COOH-terminal
regions transmit malignant TGF-beta signal in later stages of human
colorectal cancer. Cancer Res. 69:5321–5330. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sapkota G, Knockaert M, Alarcón C,
Montalvo E, Brivanlou AH and Massagué J: Dephosphorylation of the
linker regions of Smad1 and Smad2/3 by small C-terminal domain
phosphatases has distinct outcomes for bone morphogenetic protein
and transforming growth factor-beta pathways. J Biol Chem.
281:40412–40419. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kishimoto M, Fukui T, Suzuki R, Takahashi
Y, Sumimoto K, Okazaki T, Sakao M, Sakaguchi Y, Yoshida K, Uchida
K, et al: Phosphorylation of Smad2/3 at specific linker threonine
indicates slow-cycling intestinal stem-like cells before reentry to
cell cycle. Dig Dis Sci. 60:362–374. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suzuki R, Fukui T, Kishimoto M, Miyamoto
S, Takahashi Y, Takeo M, Mitsuyama T, Sakaguchi Y, Uchida K, Nishio
A, et al: Smad2/3 linker phosphorylation is a possible marker of
cancer stem cells and correlates with carcinogenesis in a mouse
model of colitis-associated colorectal cancer. J Crohn's Colitis.
9:565–574. 2015. View Article : Google Scholar
|
|
28
|
Suzuki R, Kohno H, Sugie S and Tanaka T:
Sequential observations on the occurrence of preneoplastic and
neoplastic lesions in mouse colon treated with azoxymethane and
dextran sodium sulfate. Cancer Sci. 95:721–727. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ward JM: Morphogenesis of chemically
induced neoplasms of the colon and small intestine in rats. Lab
Invest. 30:505–513. 1974.PubMed/NCBI
|
|
30
|
Murata M, Matsuzaki K, Yoshida K, Sekimoto
G, Tahashi Y, Mori S, Uemura Y, Sakaida N, Fujisawa J, Seki T, et
al: Hepatitis B virus X protein shifts human hepatic transforming
growth factor (TGF)-beta signaling from tumor suppression to
oncogenesis in early chronic hepatitis B. Hepatology. 49:1203–1217.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sekimoto G, Matsuzaki K, Yoshida K, Mori
S, Murata M, Seki T, Matsui H, Fujisawa J and Okazaki K: Reversible
Smad-dependent signaling between tumor suppression and oncogenesis.
Cancer Res. 67:5090–5096. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Furukawa F, Matsuzaki K, Mori S, Tahashi
Y, Yoshida K, Sugano Y, Yamagata H, Matsushita M, Seki T, Inagaki
Y, et al: p38 MAPK mediates fibrogenic signal through Smad3
phosphorylation in rat myofibroblasts. Hepatology. 38:879–889.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fukui T, Kishimoto M, Nakajima A,
Yamashina M, Nakayama S, Kusuda T, Sakaguchi Y, Yoshida K, Uchida
K, Nishio A, et al: The specific linker phosphorylation of Smad2/3
indicates epithelial stem cells in stomach; particularly increasing
in mucosae of Helicobacter-associated gastritis. J Gastroenterol.
46:456–468. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miller JR and Moon RT: Signal transduction
through beta-catenin and specification of cell fate during
embryogenesis. Genes Dev. 10:2527–2539. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brabletz T, Jung A and Kirchner T:
Beta-catenin and the morphogenesis of colorectal cancer. Virchows
Arch. 441:1–11. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Edelman GM, Gallin WJ, Delouvée A,
Cunningham BA and Thiery JP: Early epochal maps of two different
cell adhesion molecules. Proc Natl Acad Sci USA. 80:4384–4388.
1983. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tepass U, Truong K, Godt D, Ikura M and
Peifer M: Cadherins in embryonic and neural morphogenesis. Nat Rev
Mol Cell Biol. 1:91–100. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hirohashi S: Inactivation of the
E-cadherin-mediated cell adhesion system in human cancers. Am J
Pathol. 153:333–339. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Muta H, Noguchi M, Kanai Y, Ochiai A,
Nawata H and Hirohashi S: E-cadherin gene mutations in signet ring
cell carcinoma of the stomach. Jpn J Cancer Res. 87:843–848. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saito A, Kanai Y, Maesawa C, Ochiai A,
Torii A and Hirohashi S: Disruption of E-cadherin-mediated cell
adhesion systems in gastric cancers in young patients. Jpn J Cancer
Res. 90:993–999. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Weidner N, Moore DH II and Vartanian R:
Correlation of Ki-67 antigen expression with mitotic figure index
and tumor grade in breast carcinomas using the novel
‘paraffin’-reactive MIB1 antibody. Hum Pathol. 25:337–342. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jung A, Schrauder M, Oswald U, Knoll C,
Sellberg P, Palmqvist R, Niedobitek G, Brabletz T and Kirchner T:
The invasion front of human colorectal adenocarcinomas shows
co-localization of nuclear beta-catenin, cyclin D1, and p16INK4A
and is a region of low proliferation. Am J Pathol. 159:1613–1617.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Palmqvist R, Rutegârd JN, Bozoky B,
Landberg G and Stenling R: Human colorectal cancers with an intact
p16/cyclin D1/pRb pathway have up-regulated p16 expression and
decreased proliferation in small invasive tumor clusters. Am J
Pathol. 157:1947–1953. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Palmqvist R, Sellberg P, Oberg A, Tavelin
B, Rutegård JN and Stenling R: Low tumour cell proliferation at the
invasive margin is associated with a poor prognosis in Dukes' stage
B colorectal cancers. Br J Cancer. 79:577–581. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Takahashi Y, Fukui T, Kishimoto M, Suzuki
R, Mitsuyama T, Sumimoto K, Okazaki T, Sakao M, Sakaguchi Y,
Yoshida K, et al: Phosphorylation of Smad2/3 at the specific linker
threonine residue indicates slow-cycling esophageal stem-like cells
before re-entry to the cell cycle. Dis Esophagus. 29:107–115. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Li XQ, Yang XL, Zhang G, Wu SP, Deng XB,
Xiao SJ, Liu QZ, Yao KT and Xiao GH: Nuclear β-catenin accumulation
is associated with increased expression of Nanog protein and
predicts poor prognosis of non-small cell lung cancer. J Transl
Med. 11:1142013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zhang G, Wang W, Yao C, Zhang S, Liang L,
Han M, Ren J, Qi X, Zhang X, Wang S, et al: Radiation-resistant
cancer stem-like cell properties are regulated by PTEN through the
activity of nuclear β-catenin in nasopharyngeal carcinoma.
Oncotarget. 8:74661–74672. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Espersen ML, Olsen J, Linnemann D, Høgdall
E and Troelsen JT: Clinical implications of intestinal stem cell
markers in colorectal cancer. Clin Colorectal Cancer. 14:63–71.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Srinivasan T, Walters J, Bu P, Than EB,
Tung KL, Chen KY, Panarelli N, Milsom J, Augenlicht L, Lipkin SM,
et al: NOTCH signaling regulates asymmetric cell fate of fast- and
slow-cycling colon cancer-initiating cells. Cancer Res.
76:3411–3421. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Boman BM, Fields JZ, Cavanaugh KL, Guetter
A and Runquist OA: How dysregulated colonic crypt dynamics cause
stem cell overpopulation and initiate colon cancer. Cancer Res.
68:3304–3313. 2008. View Article : Google Scholar : PubMed/NCBI
|