Introduction
Acute myeloid leukemia (AML) is the most common
acute leukemia in adults and one of the most common hematological
malignancies, with strong biological heterogeneity and clinical
heterogeneity (1). It accounted for
~20% of acute leukemia cases in children worldwide in 2008
(1). Due to chromosomal
abnormalities and genetic mutations, the normal hematopoietic
system undergoes malignant transformation at different stages,
which causes blockade of blood cell differentiation and results in
different subtypes of AML in the process of directional myeloid
differentiation (2). An important
pathogenic factor of AML is chromosomal abnormality, including
translocation, inversion, deletion and tandem duplication (3). There are varieties of recurrent genetic
abnormalities, such as t (8:21), inv (16) and t (15:17) (4). In addition to large chromosomal
abnormalities, genetic mutation is another feature for AML
(5). With the development and
widespread application of high-throughput sequencing technologies,
leukemia-associated mutant genes have been identified with
diagnostic and therapeutic values in AML. A few gene mutations,
including Fms related receptor tyrosine kinase 3 (FLT3),
nucleophosmin 1, KIT proto-oncogene, receptor tyrosine kinase,
CCAAT enhancer binding protein α, tet methylcytosine dioxygenase 2,
and DNA methyltransferase 3 α have been associated with AML
(6,7). At the individual level, more previously
unknown gene mutations associated with AML should be identified.
The present study reported two AML cases with kinship (a father-son
pair) in which genome-wide sequencing, whole-exome sequencing (WES)
and transcriptomic analysis was performed, and some novel mutant
genes and sites were identified.
Materials and methods
Patients
A father-son pair were both diagnosed with AML at
The General Hospital of Western Theater Command (Chengdu, China).
The son was diagnosed in April 2014 while the father was diagnosed
in May 2016. The father was aged 66 and the son aged 44 years.
According to National Comprehensive Cancer Network Guideline for
AML (8), both patients were
diagnosed with AML and vein blood samples were collected, and DNAs
were extracted for whole-genome sequencing (WGS) or WES using the
phenol-chloroform method. The Hospital Ethics Committee approved
the study and the two patients signed informed consent before
donating samples. For transcriptome sequencing, the RNA sample was
extracted from the blood of the son.
WGS
The DNA sample from the father was used for
genome-wide sequencing. DNA was extracted from peripheral white
blood cells using a DNA extraction kit (Qiagen GmbH) according to
the manufacturer's instructions. A total of 2 µl purified DNA was
used to measure the quality and quantity of the DNA (ng/µl)
(A260/A280) using an UV visible spectrophotometer (BioTek
Instruments, Inc.). Finally, 20 ng/µl DNA samples were stored at
20°C, until further use. DNA degradation and the molecular weight
was analyzed using 0.2% agarose gel electrophoresis (loading 5 µl
extracted DNA). DNA libraries were prepared following the
manufacturer's protocol (Illumina DNA Prep with Enrichment; cat.
no. 20025523; Illumina, Inc.). Briefly, for each sample, DNA was
firstly sheared into fragments of ~350 bp. The fragments were then
end-repaired, A-tailed, ligated to paired-end adaptors and PCR
amplified (according to the manufacturer's instructions) for
library construction. The final library concentration used was 6.2
pM. The resulting DNA libraries were subjected to 150 bp pair-end
sequencing on the Illumina HiSequencing PE150 platform (Illumina,
Inc.). The reference genome was downloaded from the University of
California, Santa Cruz (UCSC) database (GRCh38/hg38; http://genome.ucsc.edu). Low-quality reads were
discarded using the PRINSEQ software (v0.20.4) and the resulting
clean data were aligned to the human reference sequence (hg19) with
the Burrow-Wheeler Aligner (BWA) software (0.7.12-r1044) (9). Duplicate reads were removed using the
Picard software (http://sourceforge.net/projects/picard/). The aligned
reads were sorted with the Genome Analysis Toolkit (GATK) software
(v2.8.1; http://github.com/RRafiee/GenomeAnalysisToolkit).
SIFT (sift.jcvi.org) and PolyPhen2 (genetics.bwh.harvaed.edu) software were used to
predict the effects of amino acid mutations on protein structure
and function. All variants were annotated with ANNOVAR (10).
WES
The DNA sample from the son was used for WES. The
protocol of DNA extraction was identical to that of the father's
sample. DNA was randomly digested into 150–200 bp fragments using a
Bioruptor Pico ultrasound. Fragmented DNA was end-repaired,
A-tailed and then connected. Sample labeling and enrichment of DNA
were conducted using PCR amplification according to the
manufacturer's instructions. The DNA library with the specific
index was subjected to liquid phase hybridization with the
biotin-labeled RNA probe, and the target gene exon was obtained
using the streptavidin-labeled magnetic beads. Then, target exon
genes were enriched by PCR amplification. Sequencing was performed
using an Ilumina platform (AmpliSeq™ Focus Panel for Illumina; cat.
no. 20019164; 10 ng) and the HiSeq 3000/4000 PE (150 paired-end)
Cluster kit (both from Illumina, Inc.), according to the
manufacturer's instrustions. Clean reads were mapped to the
reference genome, GRCh37 using BWA. After removing duplications,
single nucleotide polymorphisms, and insertions and deletions
(Indel) were assigned and annotated using the (GATK) based on dbSNP
build 150.
Transcriptome sequencing
The total RNA from blood samples of the son was
extracted using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. RNA purity was determined using a NanoDrop 2000
spectrophotometer (NanoDrop Technologies; Thermo Fisher Scientific,
Inc.), and RNA integrity was verified using 1.5% agarose gels.
Magnetic beads conjugated with oligo (dT) were used to isolate
mRNA, which was fragmented into short lengths of ~200 bp using an
RNA Fragmentation kit (Illumina, Inc.). The short RNA fragments
were used as templates for first-strand DNA synthesis with random
primers according to the manufacturer's instruction. Next,
second-strand DNA was generated, purified using magnetic beads, end
repaired, and a single adenine (A) nucleotide was added to the 3′
ends. DNA degradation and the molecular weight were analyzed using
0.2% agarose gel electrophoresis (loading 5 µl extracted DNA).
RNA-Sequencing (RNA-Seq) libraries were prepared using the VAHTS
Total RNA-seq (H/M/R) Library Prep kit for Illumina (Vazyme Biotech
Co., Ltd.) following the manufacturer's instructions. The raw reads
were processed to remove the adapter and primer sequences using
SeqPrep software (v1.0; http://github.com/jstjohn/SeqPrep). Low-quality bases
at the 3′ ends were deleted using Sickle software (v1.33;
http://github.com/najoshi/sickle).
Fragments <30 bp in length were excluded from further analyses
and reads containing N >10% were removed. The obtained
high-quality sequencing sequence after quality control was aligned
with the designated Ensembl genome using TopHat (http://tophat.cbcb.umd.edu/). Saturation, duplicate
reads and coverage analysis were conducted software from Majorbio,
including SeQC-2.3.2 (http://code.google.com/p/rseqc/) and RSeQC-2.3.2
(http://code.google.com/p/rseqc/).
Predictionof novel transcripts in annotated genomes was performed
using Cufflink (http://cufflinks.cbcb.umd.edu/) (11). To identify the known long non-coding
(lnc)RNAs, the predicted novel transcript was aligned with a known
lncRNA using collection of databases, such as NONCODE (www.noncode.org), Ensembl (ensembl.org), NCBI (http://www.ncbi.nlm.nih.gov/), and UCSC (genome.ucsc.edu), LncRNAdb (http://www.lncrnadb.org/), GENCODE (https://www.gencodegenes.org/) and LncRNA Disease. The
gene expression levels and differentially expressed genes (DEGs)
were estimated using cufflinks (http://cole-trapnell-lab.github.io/cufflinks/)
(12–14). The default parameters for DEGs were
false discovery rate (FDR) ≤0.05 and log2 fold-change (FC) ≥1 or
≤-1.
Gene Ontology (GO) enrichment analysis was performed
using the software Goatools (https://github.com/tanghaibao/GOatools). The P-values
were corrected using four multiple test methods (Bonferroni's,
Holm, Sidak and FDR) to control the calculated false positive rate.
When the corrected P-value (or P-FDR) ≤0.05, it was considered
significant (15–17). Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway enrichment analysis was performed using
KOBAS (http://kobas.cbi.pku.edu.cn/home.do). A corrected
P≤0.05 was considered to indicate a significant difference
(18). Samtools (http://samtools.sourceforge.net/) and VarScan
version 2.2.7 (http://varscan.sourceforge.net/) software were used to
find candidate SNPs.
Results
WGS results
The WGS results for the father's blood sample were
as follows. A total of 638,386,172 raw reads were generated as
shown in Table I, achieving WGS mean
depth of 25. The coverage of the whole genome was 96.9%. All
properly pair-end mapped sequences were used for subsequent
variants detection. A total of 3,897,164 SNPs and 780,834 indels
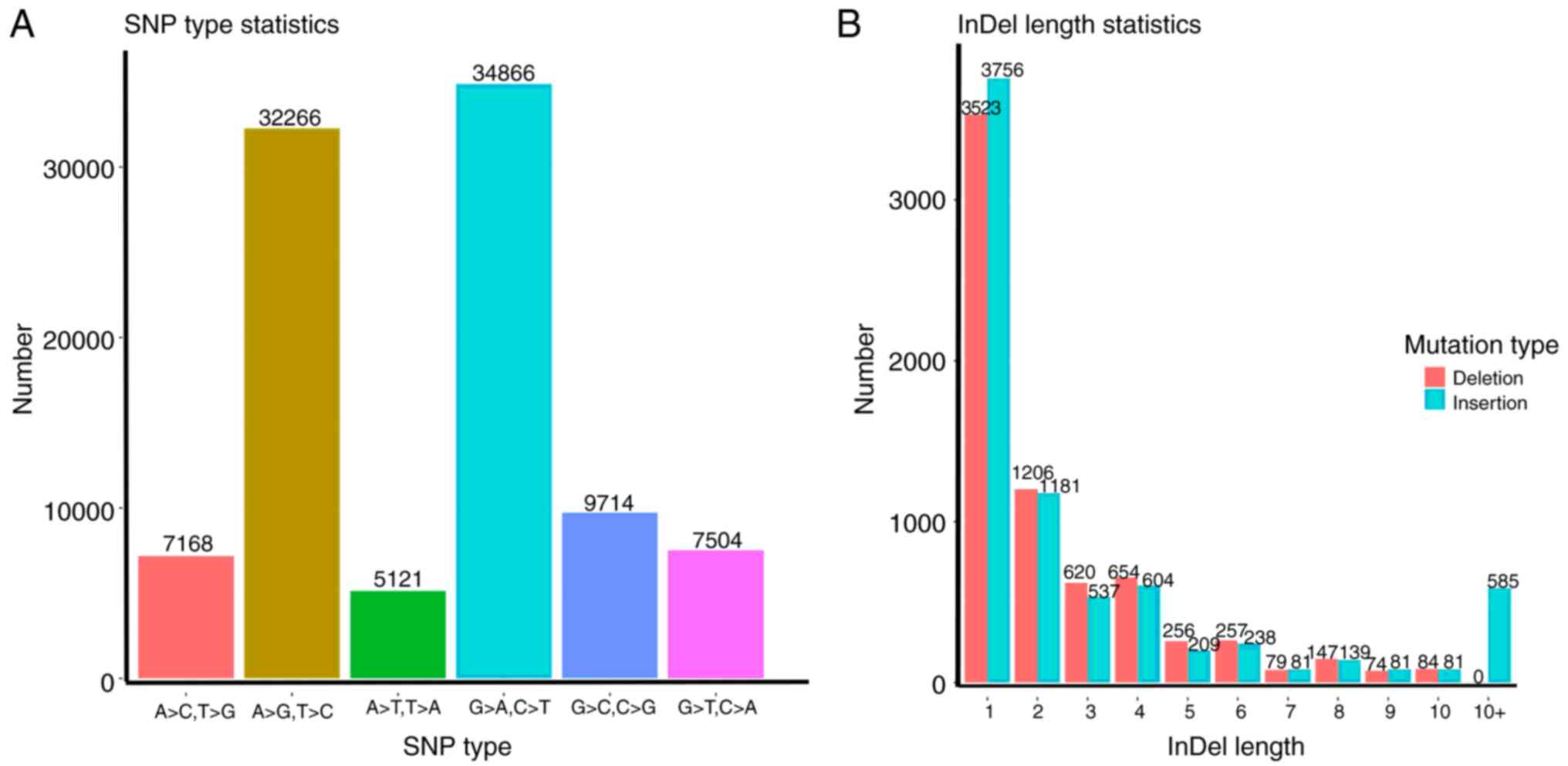
were identified (Fig. 1). The
distribution of each SNP locus was analyzed (Fig. 2A) (Table
II). The results showed that C>T and G>A were the most
common variations. The distribution of indel lengths is presented
in Fig. 2B. All SNP and indel
variants were annotated using the ANNOVAR program in conjunction
with the reference gene annotation information of the UCSC Genome
Browser. A total of 24,150 SNPs located in the exon region were
probed and 727 indel variants in the exon region were found. The
distribution of SNPs and indel variants in gene functional regions
shown in Table II.
 | Table I.Summary of the WGS and WES. |
Table I.
Summary of the WGS and WES.
| Index | WGS | WES |
|---|
| Raw reads | 638,386,172 | 638.39a |
| Raw data | 96,396,311,972 |
96,396.31b |
| Clean reads | 91,827,913,399 | 609.51a |
| Mean depth, x | 25 | 28.61 |
| Coverage, % | 96.90 | 99.91 |
| Table II.Acute myeloid leukemia-related
mutation genes in the exon region from father's whole-genome
sequencing results. |
Table II.
Acute myeloid leukemia-related
mutation genes in the exon region from father's whole-genome
sequencing results.
| Chrom | Position | Refs. | Alt | Gene | Transcript | Exon region | Variant
transcript | Variant
protein |
|---|
| 10 | 100219374 | T | A | HPSE2 | NM_001166245 | 10 | c.A1400T | p.Y467F |
|
|
|
|
|
| NM_001166244 | 11 | c.A1562T | p.Y521F |
|
|
|
|
|
| NM_021828 | 12 | c.A1736T | p.Y579F |
| 10 | 104934709 | T | C | NT5C2 |
NM_001134373/NM_012229 | 2/3 | c.A7G | p.T3A |
| 10 | 5136651 | C | G | AKR1C3 |
NM_001253909/NM_003739 | 1 | c.C15G | p.H5Q |
| 10 | 5139685 | G | A | AKR1C3 |
NM_001253908/NM_001253909/NM_003739 | 3 | c.G312A | p.K104K |
| 10 | 54531235 | C | T | MBL2 | NM_000242 | 1 | c.G161A | p.G54D |
| 10 | 70405855 | A | G | TET1 | NM_030625 | 4 | c.A3369G | p.I1123M |
| 10 | 88422116 | C | T | OPN4 | NM_033282 | 4 | c.C1181T | p.T394I |
|
|
|
|
|
| NM_001030015 | 9 | c.C1214T | p.T405I |
| 10 | 89623716 | G | A | PTEN | NM_001304717 | 1 | c.G10A | p.G4R |
| 10 | 89623901 | G | C | PTEN | NM_001304717 | 2 | c.G194C | p.C65S |
| 10 | 89624218 | C | G | PTEN | NM_001304717 | 2 | c.C511G | p.L171V |
| 11 | 102595492 | G | A | MMP8 | NM_002424 | 1 | c.C95T | p.T32I |
|
|
|
|
|
| NM_002424 | 2 | c.A259G | p.K87E |
|
|
|
|
|
|
NM_001304441/NM_001304442 | 3 | c.A190G | p.K64E |
| 11 | 35226155 | A | G | CD44 | NM_001001389 | 9 | c.A1121G | p.K374R |
|
|
|
|
|
| NM_000610 | 10 | c.A1250G | p.K417R |
| 11 | 35229673 | T | C | CD44 | NM_001001390 | 6 | c.T689C | p.I230T |
|
|
|
|
|
| NM_001001389 | 11 | c.T1307C | p.I436T |
|
|
|
|
|
| NM_000610 | 12 | c.T1436C | p.I479T |
| 13 | 28624294 | G | A | FLT3 | NM_004119 | 6 | c.C680T | p.T227M |
Regarding amino acid translation, there were 11,316
non-synonymous, 12 stop-loss, 12,033 synonymous, 92 stop-gain for
SNPs, 63 frameshift insertions, 73 frameshift deletions, 242
non-frameshift insertions, 248 non-frameshift deletions, 4
stop-gain, 2 stop-loss for indel variants (Table III).
| Table III.Summary of detected SNPs and indels
in all sequencing samples. |
Table III.
Summary of detected SNPs and indels
in all sequencing samples.
| A, SNPs |
|---|
|
|---|
| Index | WGS results | WES |
|---|
| Total SNPs in
exon | 24,150 | 22,656 |
| Synonymous SNP | 12,033 | 11,570 |
| Non-synonymous
SNP | 11,316 | 10,504 |
| Stop-gain | 92 | 87 |
| Stop-loss | 12 | 12 |
|
| B,
Indels |
|
| Index | WGS | WES |
|
| Total indels in
exon | 727 | 620 |
| Frameshift
insertion | 63 | 102 |
| Frameshift
deletion | 73 | 117 |
| Non-frameshift
insertion | 242 | 156 |
| Non-frameshift
deletion | 248 | 190 |
| Stop-gain | 4 | 3 |
| Stop-loss | 2 | 1 |
Non-synonymous SNPs can cause changes in the encoded
amino acids. Together, 2,436 mutations were destructive to protein
structure and function according to SIFT, and 1,154 mutations were
probably harmful according to PolyPhen2. Comparing with 121
AML-related genes identified in previous AML studies, 14 genes were
found in the father's exon region, including the following genes:
HPSE2 (19), NT5C2 (20), AKR1C3 (21), MBL2 (22), ARID5B (23), TET1 (24), WT1 (25), CD44 (26) and FLT3 (27). However, only 10 of these had
non-synonymous mutations changing encoded amino acids.
WES results
The WES results from the son achieved 96,396.31 Mb
raw data, generating a mean depth of 28.61. The coverage rate was
99.91%. All properly pair-end mapped sequences were used for
subsequent variants detection, as shown in Table II. Overall, 96,639 SNPs were
identified, including 10,504 non-synonymous SNPs. For the types of
base mutations in SNPs, C>T and G>A were the most commonly
observed in this sample. The indel results showed 15,033 mutations,
including 102 frameshift insertions, 117 frameshift deletions, 156
non-frameshift insertions, 190 non-frameshift deletions, three
stop-gain and one stop-loss (Table
III). Seven mutant genes were found in son's exon region
compared with 121 AML-related genes, including FLT3, GATA2
(28,29), MCM7 (30), PTEN (31), and RUNX1T1 (32) (Table
IV). Among them, six genes showed non-synonymous SNPs (Table IV).
| Table IV.Acute myeloid leukemia-related
mutation genes in the exon region from son's whole-exome sequencing
results. |
Table IV.
Acute myeloid leukemia-related
mutation genes in the exon region from son's whole-exome sequencing
results.
| Chrom | Position | Refs. | Alt | Gene | Transcript | Exon region | Variant
transcript | Variant
protein |
|---|
| 1 | 110882830 | C | T | RBM15 |
NM_001201545/NM_022768 | 1 | c.C803T | p.P268L |
| 3 | 128202797 | C | T | GATA2 |
NM_001145662/NM_032638 | 4 | c.G923A | p.R308Q |
| 7 | 99696316 | G | A | MCM7 |
NM_001278595/NM_182776 | 5 | c.C77T | p.P26L |
|
|
|
|
|
| NM_005916 | 6 | c.C605T | p.P202L |
| 8 | 93107611 | G | A | RUNX1T1 |
NM_001198634/NM_001198679 | 1 | c.C85T | p.R29W |
| 10 | 89623901 | G | C | PTEN | NM_001304717 | 2 | c.G194C | p.C65S |
| 13 | 28609806 | T | G | FLT3 | NM_004119 | 12 | c.A1423C | p.T475P |
| 13 | 28674628 | T | C | FLT3 | NM_004119 | 1 | c.A20G | p.D7G |
Then the mutant genes in the exon region were
compared between the father and son results, and some common ones
were observed. Notably, two missense mutations in the FLT3 gene
were identified. Internal tandem duplications in the FLT3 gene
(FLT3-ITD) were the earliest described molecular alterations in AML
(33). In the present results, the
alterations of FLT3 were as follows. In WGS data from the father:
Exon 6, c.C680T: p.T227M (NM_004119). In WES data from the son:
Exon 1, c.A20G: p.D7G (NM_004119) and exon 12, c.A1423C: p.T475P
(NM_004119). Another common gene was PTEN. As Tables III and IV show, in WGS data from the father, the
following mutations were identified: c.G10A: p.G4R (NM_001304717)
In exon 1, c.G194C: p.C65S(NM_001304717) in exon 2 and c.C511G:
p.L171V (NM_001304717) in exon 2. In WES data from the son the
c.G194C: p.C65S (NM_001304717) mutation was identified in exon
2.
Transcriptomic sequencing results
Transcriptomic sequencing generated 69,537,564 raw
reads containing 10,430,634,600 nucleotides for the son. Following
clean-up and quality filtering, 68,019,440 and 75,105,808 clean
reads were obtained, with a Q20 percentage (proportion of
nucleotides with quality value >20) >98.51%, which indicated
the RNA sequencing results were of high quality and suitable for
use in further analysis.
As Fig. 3 shows,
there were 54 differentially expressed mRNAs from transcriptomic
analysis, of which 31 were significantly upregulated in AML, and 23
were significantly downregulated. However, no common differentially
expressed mRNAs between WGS and WES were identified. As
aforementioned, FLT3 and PTEN might play an important role in AML
development. Compared with patients with AML, expression of FLT3
was decreased (P=0.035; FDR, 0.919; log2FC, 3.17). A lower
expression of PTEN were found in the patients with AML without
statistical significance (P=0.4135; FDR, 0.993; log2FC, −0.71).
GO annotations revealed that the aforementioned
genes were enriched in ‘single-organism signaling’, ‘response to
stimulus’, ‘biological regulation’ and ‘metabolic process’. The top
30 terms were all among the biological process class and are
presented in Fig. 4. KEGG pathway
analysis revealed 56 related pathways, while according to corrected
P-value, only three pathways were significantly enriched:
‘Influenza A’, ‘hematopoietic cell lineage’ and ‘cell adhesion
molecules’ (Fig. 5). Alternative
splicing is an important mechanism for regulating gene expression
and producing protein diversity, which can cause large differences
in genes and proteins (12). There
were 4,370 3′ end alternative splicing events, 2,575 3′
untranslated region (UTR) alternative splicing events, 4,927 5′ end
alternative splicing events, 1,783 5′ UTR alternative splicing
events, 2,468 exon spanning events, 336 intron retentions, and
2,761 other alternative splicing.
Discussion
Clinically, it is rare to identify AML cases similar
to familial aggregation. In the present study, the common molecular
mutation types of AML in the father were not consistent with those
of his son. Therefore, it was hypothesized that the father had some
molecular variation that had not been previously recognized, which
led to the occurrence of AML to the father, and it could be stably
and continuously expressed in his whole sexual maturity stage. The
molecular variation was passed on to his son through genetic
material and expressed at a certain developmental stage of the son,
leading to the disease. Therefore, the current study performed
genome-wide sequencing, WES and transcriptomic analyses of two
patients with AML patients with kinship (a father-son pair) in
order to investigate the profiling of AML development.
The father-son pair samples had a similar number of
SNPs and indel variants in the exon region (24,150 SNPs and 727
indel variants in father sample; 22,656 SNPs and 620 indel variants
in son sample), though different sequencing analysis technologies
were used, following the decision of the patient. Their SNPs and
indel variants were similar to previous studies of other types of
blood cancer (e.g. chronic myelomonocytic leukemias) (7,34).
However, transcriptomic analysis was also performed on the son
sample and 10,072 SNPs in the exon region were found.
In total, >200 common exon mutant genes were
identified in both the father and son, which suggested that AML may
be the result of a combination of multiple genes. In these genes,
an FLT3 mutation was identified in both father and son. They showed
different variations in the exon region. However, different
variations in FLT3 may cause the same result. FLT3-ITD has major
clinical implications and is associated with adverse outcomes in
recurrent somatic mutations in AML through multiple mechanisms
(35). Previous studies revealed
FLT3-ITD with different insertion sites spread over the whole
stretch of exons 14–15 (36,37). However, the present study observed
three mutations in exons 1, 6 and 12 and none of these were located
in exons 14 and 15. These mutations may cause changes in protein
structure or function, and the further study can investigate the
role of these exons. FLT3 is known to be overexpressed in
hematopoietic neoplasms (38). The
current transcriptomic sequencing results also showed increased
expression of FLT3 in AML. PTEN, a tumor suppressor gene (31), was another identified common gene. A
missense mutation in the PTEN gene was reported in concurrent germ
cell tumor (GCT)-associated AML (39). The mRNA and protein levels of PTEN in
newly diagnosed patients with AML and patients with relapsed AML
are significantly lower compared with those in the control and
remission groups, which was supports the present findings (39).
Through high-throughput analysis, the mutation
profiles of two patients with AML with a father-son relationship
were obtained at the genetic level, and the son showed a changed
mRNA profiles at the gene expression level. The study also
identified some new mutation sites in known AML-associated genes,
such as FLT3. The present case report may provide novel insight
into the molecular events governing the pathogenesis of AML.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the NCBI-SRA repository (https://www.ncbi.nlm.nih.gov/sra/). The BioSample
accession number is: SAMN16395638.
Authors' contributions
SHL and YCL designed the study and wrote the
manuscript. SZ, RD, YD and FYF performed all the experiments. SHL,
SZ and RD confirm the authenticity of all the raw data. All authors
approved the final version of the manuscript.
Ethics approval and consent to
participate
This study has been approved by The General Hospital
of Western Theater Command (Ethics approval number: 2016KY82).
Informed consent to participate was provided from both
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
De Kouchkovsky I and Abdul-Hay M: ‘Acute
myeloid leukemia: A comprehensive review and 2016 update’. Blood
Cancer J. 6:e4412016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tenen DG: Disruption of differentiation in
human cancer: AML shows the way. Nat Rev Cancer. 3:89–101. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rouli DD: Chromosome aberrations in
leukemia and lymphoma. Ter Arkh. 55:30–35. 1983.(In Russian).
PubMed/NCBI
|
|
4
|
Palanisamy N: Chromosomal translocations
in AML: Detection and prognostic significance. Cancer Treat Res.
145:41–58. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martelli MP, Sportoletti P, Tiacci E,
Martelli MF and Falini B: Mutational landscape of AML with normal
cytogenetics: Biological and clinical implications. Blood Rev.
27:13–22. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cancer Genome Atlas Research Network, ;
Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A,
Hoadley K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic
landscapes of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ilyas AM, Ahmad S, Faheem M, Naseer MI,
Kumosani TA, Al-Qahtani MH, Gari M and Ahmed F: Next generation
sequencing of acute myeloid leukemia: Influencing prognosis. BMC
Genomics. 16 Suppl 1(Suppl 1):S52015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
O'Donnell MR, Tallman MS, Abboud CN,
Altman JK, Appelbaum FR, Arber DA, Attar E, Borate U, Coutre SE, et
al: Acute myeloid leukemia, version 2.2013. J Natl Compr Canc Netw.
11:1047–1055. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li H and Durbin R: Fast and accurate short
read alignment with Burrows-Wheeler transform. Bioinformatics.
25:1754–1760. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38:e1642010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Roberts A, Pimentel H, Trapnell C and
Pachter L: Identification of novel transcripts in annotated genomes
using RNA-Seq. Bioinformatics. 27:2325–2329. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Trapnell C, Williams BA, Pertea G,
Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ and Pachter
L: Transcript assembly and quantification by RNA-Seq reveals
unannotated transcripts and isoform switching during cell
differentiation. Nat Biotechnol. 28:511–515. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Trapnell C, Hendrickson DG, Sauvageau M,
Goff L, Rinn JL and Pachter L: Differential analysis of gene
regulation at transcript resolution with RNA-seq. Nat Biotechnol.
31:46–53. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Reiner A, Yekutieli D and Benjamini Y:
Identifying differentially expressed genes using false discovery
rate controlling procedures. Bioinformatics. 19:368–375. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tang H, Wang X, Bowers JE, Ming R, Alam M
and Paterson AH: Unraveling ancient hexaploidy through
multiply-aligned angiosperm gene maps. Genome Res. 18:1944–1954.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu J, Peatman E, Tang H, Lewis J and Liu
Z: Profiling of gene duplication patterns of sequenced teleost
genomes: Evidence for rapid lineage-specific genome expansion
mediated by recent tandem duplications. BMC Genomics. 13:2462012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Aickin M and Gensler H: Adjusting for
multiple testing when reporting research results: The Bonferroni
vs. Holm methods. Am J Public Health. 86:726–728. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie C, Mao X, Huang J, Ding Y, Wu J, Dong
S, Kong L, Gao G, Li CY and Wei L: KOBAS 2.0: A web server for
annotation and identification of enriched pathways and diseases.
Nucleic Acids Res. 39((Web Server issue)): W316–W322. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ostrovsky O, Korostishevsky M, Levite I,
Leiba M, Galski H, Vlodavsky I and Nagler A: Association of
heparanase gene (HPSE) single nucleotide polymorphisms with
hematological malignancies. Leukemia. 21:2296–2303. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mitra AK, Crews KR, Pounds S, Cao X,
Feldberg T, Ghodke Y, Gandhi V, Plunkett W, Dolan ME, Hartford C,
et al: Genetic variants in cytosolic 5′-nucleotidase II are
associated with its expression and cytarabine sensitivity in HapMap
cell lines and in patients with acute myeloid leukemia. J Pharmacol
Exp Ther. 339:9–23. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu CY, Hsu YH, Pan PC, Wu MT, Ho CK, Su
L, Xu X, Li Y and Christiani DC; Kaohsiung Leukemia Research Group,
: Maternal and offspring genetic variants of AKR1C3 and the risk of
childhood leukemia. Carcinogenesis. 29:984–990. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
te Poele EM, Siedlinski M, Anne de Pagter
PJ, Bierings MB, Scherpen FJ, Meeuwsen-de Boer TG, Koppelman GH,
Postma DS, Kamps WA, Boezen HM and de Bont ES: MBL2 and fever
during neutropenia in children with acute lymphoblastic leukaemia.
Br J Haematol. 157:132–135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cao S, Yang J, Qian X, Jin G and Ma H: The
functional polymorphisms of ARID5B and IKZF1 are associated with
acute myeloid leukemia risk in a Han Chinese population. Gene.
647:115–120. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Abdel-Wahab O, Mullally A, Hedvat C,
Garcia-Manero G, Patel J, Wadleigh M, Malinge S, Yao J, Kilpivaara
O, Bhat R, et al: Genetic characterization of TET1, TET2, and TET3
alterations in myeloid malignancies. Blood. 114:144–147. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Petiti J, Rosso V, Lo Iacono M, Calabrese
C, Signorino E, Gaidano V, Berger M, Saglio G and Cilloni D:
Prognostic significance of The Wilms' Tumor-1 (WT1) rs16754
polymorphism in acute myeloid leukemia. Leuk Res. 67:6–11. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wu H, Deng J, Zheng J, You Y, Li N, Li W,
Wu D and Zhou Y: Functional polymorphisms in the CD44 gene and
acute myeloid leukemia cancer risk in a Chinese population. Mol
Carcinog. 54:102–110. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim B, Kim S, Lee ST, Min YH and Choi JR:
FLT3 internal tandem duplication in patients with acute myeloid
leukemia is readily detectable in a single next-generation
sequencing assay using the pindel algorithm. Ann Lab Med.
39:327–329. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tien FM, Hou HA, Tsai CH, Tang JL, Chiu
YC, Chen CY, Kuo YY, Tseng MH, Peng YL, Liu MC, et al: GATA2 zinc
finger 1 mutations are associated with distinct clinico-biological
features and outcomes different from GATA2 zinc finger 2 mutations
in adult acute myeloid leukemia. Blood Cancer J. 8:872018.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Celton M, Forest A, Gosse G, Lemieux S,
Hebert J, Sauvageau G and Wilhelm BT: Epigenetic regulation of
GATA2 and its impact on normal karyotype acute myeloid leukemia.
Leukemia. 28:1617–1626. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lee JS, Cheong HS, Koh Y, Ahn KS, Shin HD
and Yoon SS: MCM7 polymorphisms associated with the AML relapse and
overall survival. Ann Hematol. 96:93–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Oshrine BR, Olsen MN, Heneghan M, Wertheim
G, Daber R, Wilmoth DM, Biegel JA, Pawel B, Aplenc R and King RL:
Acquired isochromosome 12p, somatic TP53 and PTEN mutations, and a
germline ATM variant in an adolescent male with concurrent acute
megakaryoblastic leukemia and mediastinal germ cell tumor. Cancer
Genet. 207:153–159. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Foster N, Paulsson K, Sales M, Cunningham
J, Groves M, O'Connor N, Begum S, Stubbs T, McMullan DJ, Griffiths
M, et al: Molecular characterisation of a recurrent, semi-cryptic
RUNX1 translocation t(7;21) in myelodysplastic syndrome and acute
myeloid leukaemia. Br J Haematol. 148:938–943. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Stirewalt DL, Pogosova-Agadjanyan EL,
Tsuchiya K, Joaquin J and Meshinchi S: Copy-neutral loss of
heterozygosity is prevalent and a late event in the pathogenesis of
FLT3/ITD AML. Blood Cancer J. 4:e2082014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Itzykson R, Kosmider O, Renneville A,
Morabito M, Preudhomme C, Berthon C, Adès L, Fenaux P, Platzbecker
U, Gagey O, et al: Clonal architecture of chronic myelomonocytic
leukemias. Blood. 121:2186–2198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cao T, Jiang N, Liao H, Shuai X, Su J and
Zheng Q: The FLT3-ITD mutation and the expression of its downstream
signaling intermediates STAT5 and Pim-1 are positively correlated
with CXCR4 expression in patients with acute myeloid leukemia. Sci
Rep. 9:122092019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Schnittger S, Bacher U, Haferlach C,
Alpermann T, Kern W and Haferlach T: Diversity of the juxtamembrane
and TKD1 mutations (exons 13–15) in the FLT3 gene with regards to
mutant load, sequence, length, localization, and correlation with
biological data. Genes Chromosomes Cancer. 51:910–924. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Patel RK, Weir MC, Shen K, Snyder D,
Cooper VS and Smithgall TE: Expression of myeloid Src-family
kinases is associated with poor prognosis in AML and influences
Flt3-ITD kinase inhibitor acquired resistance. PLoS One.
14:e02258872019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Levis M and Small D: FLT3: ITDoes matter
in leukemia. Leukemia. 17:1738–1752. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun JY, Mu N, Mu J, Li W, Zhang CG and
Wang DM: Expression and significance of PTEN and BCL-2 in acute
myeloid leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 26:121–125.
2018.(In Chinese). PubMed/NCBI
|