Introduction
Anexelekto (Axl), a member of the Tyro3, Axl and
Mertk family of receptor tyrosine kinases (RTKs), can be activated
by phosphorylation from binding of its natural ligand, growth
arrest-specific protein 6 (Gas6), and has been associated with
tumor cell growth, migration, invasion and immune suppression
(1–5). Upregulation or overactivation of Axl
has been reported in various solid tumors, leukemias and other
types of lymphoid neoplasms, particularly in invasive types of
cancer (1–3,6–8).
More importantly, chemotherapy can indirectly induce
the upregulation and phosphorylation of Axl, which may trigger cell
survival signaling and, ultimately, contribute to chemoresistance
in breast, colon, lung cancer, mesothelioma and acute myeloid
leukemia (2,4,9–11). By contrast, inhibition of Axl can
reduce tumor cell proliferation and migration, as well as maintain
the sensitivity of tumor cells to chemotherapeutic agents (12–14).
Therefore, the critical role of Axl in tumor development,
progression and therapeutic resistance makes it an attractive
target for cancer therapy (2,8,15).
Small molecular tyrosine kinase inhibitors and
monoclonal antibodies (mAbs) are the main Axl inhibitors. A total
of 26 small molecular kinase inhibitors against Axl have been
reported to date, and have either been proven or are under clinical
and preclinical development (4,5,15,16).
However, most of these kinase inhibitors target several RTKs
sharing similar kinase domains and, generally, Axl is not the
primary target (2,4,15). In
total, only three kinase inhibitors have Axl as their selective
target (5). R428 (BGB324) is the
first selective kinase inhibitor to target Axl only and is
currently undergoing phase II clinical trials (4,5,16–18).
Compared with smaller molecular kinase inhibitors,
research into Axl-targeted mAb has developed more recently. In
total, six groups of Axl-targeted mAbs (DAXL-88, 20G7-D9, AXL
antibody, D9/E8, YW327.6S2 and 3G9/8B5/12A11/4F8) have been
reported during the last 10 years. These mAbs have shown
bioactivity in blocking the Gas6-Axl interaction and downstream
signaling (1,3,6,19), inhibiting tumor cell proliferation
and migration (3,6,7,12), attenuating tumor xenograft growth
(1,3,6,19) and reducing the invasion of cancer
cells (1,19) in studies examining triple-negative
breast cancer (TNBC), non-small cell lung cancer (NSCLC) and
pancreatic cancer models. In addition, two antibody-drug conjugates
(Enapotamab vedotin and BA3011) have been reported (20,21). The
development of Axl inhibitors was our main focus, and we
investigated a phage-derived human mAb (DAXL-88) and variants of
the Axl external cell domain (Axl−ECD) fused with
IgG1-Fc (Axl−ECD-Fc) (3,22).
In antibody research, the design and preparation of
the antigen are the most important steps for screening functional
mAbs. For the aforementioned six groups of Axl-targeted mAbs, the
Axl−ECD was used as the antigen in the screening
process. The Axl−ECD [26–451 amino acids (aa)] consists
of 426 aa, and the Axl functional domain, that interacts with Gas6,
is only a small component of Axl−ECD. Antibodies
targeting the Axl functional domain may efficiently block Gas6-Axl
binding and attenuate downstream signals and activities. To the
best of our knowledge, no mAb that targets the Axl functional
domain has been previously reported.
In the present study, the major Axl functional
domain, interacting with Gas6, was determined using bioinformatics
and structural biology methods. Subsequently, anti-Axl mAbs were
generated using hybridoma technology and evaluated using a series
of biological experiments.
Materials and methods
Reagents and antibodies
The pGEX-4T-1-Axl−Ig1 recombinant plasmid
was purchased from Beijing SinoGenoMax Research Center Co., Ltd.
The Axl−ECD-Fc was made in our laboratory (Beijing Key
Laboratory of Therapeutic Gene Engineering Antibody, Beijing,
China). The biotin-labeled Axl−ECD-Fc was purchased from
Beijing Jiaxuan Zhirui Biotechnology Co., Ltd. RPMI-1640 medium
(cat. no. A1049101), DMEM (cat. no. C11995500BT), 0.25%
trypsin-EDTA (cat. no. 25200-072) and FBS (cat. no. 1997802C) were
purchased from Gibco (Thermo Fisher Scientific, Inc.). Freund's
complete adjuvant (cat. no. F5881), Freund's incomplete adjuvant
(cat. no. F5506), polyethylene glycol (PEG1450; cat. no. P7306) and
hypoxanthine-aminopterin-thymidine (HAT) media supplement (cat. no.
H0262) were purchased from Sigma-Aldrich (Merck KGaA). Escherichia
coli BL21 (DE3) competent cells (cat. no. C1400), Glutathione
S-transferase (GST) beads (cat. no. P2020), RIPA buffer (cat. no.
R0010) and Giemsa solution (cat. no. G1010) were purchased from
Beijing Solarbio Science and Technology Co., Ltd. HiTrap Protein G
HP (cat. no. 17-0405-01) was purchased from GE Healthcare (Cytiva).
R428 inhibitor (cat. no. CC2997) was purchased from ChemCatch
(http://www.chemcatch.cn/index.php).
The 3,3′,5,5,-tetramethylbenzidine (TMB) solution (cat. no.
00-4201-56) was purchased from Invitrogen (Thermo Fisher
Scientific, Inc.).
Recombinant human Gas6 protein (cat. no. 885-GSB)
and goat anti-Axl polyclonal antibody (cat. no. AF154) were
purchased from R&D Systems, Inc. Anti-phosphorylated (p)-Axl
(cat. no. 5724), anti-mouse IgG-HRP (cat. no. 7076), anti-rabbit
IgG-HRP (cat. no. 7074) and anti-GAPDH-HRP (cat. no. 51332)
antibodies were purchased from Cell Signaling Technology, Inc.
Streptavidin-HRP (cat. no. 554066) was purchased from BD
Biosciences. Anti-mouse IgG-FITC (cat. no. ab6785), anti-goat
IgG-FITC (cat. no. ab6737) and anti-goat IgG-HRP (cat. no. ab6741)
were purchased from Abcam. Goat anti-human IgG (GAH-IgG; cat. no.
01-10-06) was purchased from KPL, Inc., while mouse anti-GST IgG
(cat. no. 66001-2-Ig) was purchased from ProteinTech Group,
Inc.
Animals
A total of 4 female BALB/c mice were purchased from
JOINN Laboratories. The animal experiments were approved by the
Animal Ethics Committee of Suzhou Vocational Health College
(Jiangsu, China; approval no. SWAE201905) and performed in
accordance with the Guidelines of the Care and Use of Laboratory
Animals of the Ministry of Health, China to minimize animal
suffering and distress. The mice were monitored daily by an animal
keeper and no mice died during the experiment. The mice were
anesthetised with 2% isoflurane and blood was extracted from the
tails to verify the immune titer. A total of 4 mice were euthanized
by cervical dislocation and death was verified by respiratory and
cardiac arrest. The duration of the experiment was 8 weeks and 3
days.
Cell culture
The human breast cancer cell line, MDA-MB-231 (cat.
no. HTB-26) and the human NSCLC cell line H1299 (cat. no. CR-5803)
were purchased from the American Type Culture Collection and were
authenticated by Beijing ZhongYuan Ltd., in 2014. The SP2/0 mouse
myeloma cell line (cat. no. CL-0217) was purchased from Procell
Life Science&Technology Co., Ltd. The cells were cultured in
DMEM containing 10% FBS and 100 U/ml penicillin-streptomycin at
37°C in a humidified incubator with 5% CO2.
Bioinformatics
The protein sequence information for Axl (UniProtKB
P30530) was determined using www.ebi.ac.uk. The crystal complex information for
Gas6-Axl (no. 2C5D) was obtained from the Protein Data Bank
(https://www.ncbi.nlm.nih.gov/Structure/pdb).
Interaction analysis of Gas6-Axl was performed using Insight II
2000 software (Insight Software, Inc.).
Interaction analysis between Axl and
Gas6
Theoretical analyses were performed as described in
our previous study (22). Briefly,
based on the crystal complex structure (23), the Gas6-Axl theoretical complex model
was constructed and optimized using the steepest decent and
conjugate gradient method (discovery mode) under the consistent
valence force field. With the optimized complex structure, the
computer graphics technique and the distance geometry method
(standard mode) were used to analyze the Gas6-Axl interaction mode.
The superimposition method (standard mode) was used to identify the
orientation of the main chain carbon atoms. Furthermore, the
interaction binding free energy calculation method (discovery mode)
was used to calculate the binding energy of Gas6-Axl.
Preparation and identification of the
GST-Axl−Ig1 antigen
The DNA sequence encoding the Axl functional domain
(Axl−Ig1) was confirmed by BLAST analysis (https://blast.ncbi.nlm.nih.gov/Blast.cgi, GenBank:
BC032229.1) and cloned into the pGEX-4T-1 vector between
BamHI and XhoI restiction sites, which allowed for
the addition of a GST tag to the N-terminus of the
Axl−Ig1 domain (GST-Axl−Ig1 fusion protein).
The recombinant plasmid pGEX-4T-1-Axl−Ig1 was
transformed into Escherichia coli BL21 (DE3) competent
cells. After optimization of the expression conditions, the
GST-Axl−Ig1 fusion protein expression was induced by the
addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at 20°C
for 6 h, followed by purification using GST beads according to the
manufacturer's instructions. Then, the GST-Axl−Ig1
fusion protein was analyzed using 12% SDS-PAGE and further
validated using western blot analysis and the goat anti-Axl
polyclonal antibody, AF154 and mouse anti-GST IgG, as separate
primary antibodies, followed by anti-goat IgG-HRP and anti-mouse
IgG-HRP as separate secondary antibodies.
Animal immunization and cell
fusion
Animal immunization was performed as described
previously (24). Briefly, four
BALB/c female mice (8-week-old, 18–20 g) were subcutaneously
immunized with 100 µg GST-Axl−Ig1 antigen in Freund's
complete adjuvant on the first week, and in Freund's incomplete
adjuvant on the fourth and sixth week. When the immune titer was
>1:10,000, the mice were intravenously immunized with 100 µg
GST-Axl−Ig1 antigen without adjuvant for the final
boost. Then, 3 days later, the mice were euthanized by cervical
dislocation and the spleen cells from the immunized mice were fused
with SP2/0 mouse myeloma cells at a ratio of 7:1 with PEG1450. The
fused cells were cultured in RPMI-1640 medium with 20% FBS and HAT,
then screened using ELISA and the hybridoma cell supernatant.
Screening of hybridoma cells and
production of anti-Axl mAbs
The hybridoma cell clones that bound to
GST-Axl−Ig1, but not GST were further screened using a
BD FACSCalibur™ flow cytometer (cat. no. 342975; BD Biosciences)
and FlowJo software (version 7.6; BD Biosciences) using H1299 cells
(NSCLC cell line) with a high expression of Axl. The H1299 cell
line was incubated with the hybridoma cell supernatant containing
anti-Axl mAbs and detected using anti-mouse IgG-FITC (1:1,000).
Goat anti-Axl polyclonal antibody AF154 (1:500) served as a
positive control and was detected using anti-goat IgG-FITC
(1:1,000). ELISAs were always performed before flow cytometry
screening. After three rounds of ELISA, flow cytometry and limited
dilution subclonal screening, hybridoma cell secreting anti-Axl
mAbs were obtained. The hybridoma cells were cultured at 37°C with
5% CO2, and 3×106 hybridoma cells
(6×106 cells/ml) were seeded into the abdominal cavity
of each mouse to boost the production of the antibody protein.
Anti-Axl mAbs were purified from the mouse ascites using Protein G
affinity chromatography and an ÄKTAprime® plus system
(cat. no. 11001313; Cytiva) with 20 mM sodium phosphate (pH 7.0) as
binding buffer, and 0.1 M glycine-HCl (pH 2.7) as elution buffer
according to the manufacturer's instructions. Anti-Axl mAbs were
analyzed via reduced and non-reduced 12% SDS-PAGE, filtered under
sterile conditions and stored at 4°C in PBS. Protein concentrations
were determined using a BCA kit (Applygen Technologies, Inc.).
Binding to Axl fusion proteins
(measured using ELISA)
The anti-Axl mAbs were tested for binding with Axl
fusion proteins, Axl−ECD-Fc and GST-Axl−Ig1.
The ELISA plates were coated with 2 µg/ml Axl−ECD-Fc and
GST-Axl−Ig1 in 0.1 M sodium carbonate buffer (pH 9.6)
overnight at 4°C. After blocking with 10% FBS in PBS buffer at 37°C
for 1 h, the plates were incubated with serial dilutions of mouse
anti-Axl mAb (5.56, 1.85, 0.62, 0.21, 0.069, 0.023, 0.008, 0.0027
and 0.0009 µg/ml) at 37°C for 1 h. Subsequently, anti-mouse IgG-HRP
(1:2,500) was added as the secondary antibody and incubated at 37°C
for 30 min. The binding signals were visualized using the TMB
substrate and the absorbance was measured at 450 nm using a SPECTRA
MAX 190 ELISA reader (Molecular Devices, LLC). The ELISA plates
were separately washed with TBS-Tween-20 (TBST; 50 mM Tris-HCl, 150
mM NaCl, 0.2% Tween-20, pH 7.4) three times for 5 min after
coating, blocking and incubating with the primary and secondary
antibodies.
Binding to Axl proteins (measured
using western blot analysis)
The anti-Axl mAbs were tested for binding to the Axl
fusion proteins. Axl−ECD-Fc and GST-Axl−Ig1
proteins were resolved on 12% SDS-PAGE and transferred onto a PVDF
membrane, which was blocked with 5% skimmed milk at room
temperature for 1 h. The membranes were incubated with mouse
anti-Axl mAbs (10 µg/ml) overnight at 4°C, then with anti-mouse
IgG-HRP (1:2,500) at room temperature for 1 h. The anti-Axl
polyclonal antibody AF154 (1:500) was used as the positive control
to recognize the Axl−ECD and Axl−Ig1 domains
and was detected using the anti-goat IgG-HRP (1:2,500). Finally,
the membranes were developed using an ECL detection system
(SuperSignal Westpico Trial kit; Thermo Fisher Scientific, Inc.).
The membrane was separately washed three times with TBST for 10 min
after transferring, blocking and incubating with the primary and
secondary antibodies.
The anti-Axl mAbs were tested for binding to Axl
molecules on the surface of the cells. After trypsin digestion, the
MDA-MB-231 cell lysates from 5×105 cells were obtained
using ice-cold RIPA buffer and were resolved using 12% SDS-PAGE.
The subsequent steps were the same as aforementioned. The anti-Axl
antibody AF154 was also used as the positive control.
Flow cytometry
After trypsin digestion, 3×105 MDA-MB-231
cells were collected and incubated with mouse anti-Axl mAb (10
µg/ml) as the primary antibody at 4°C for 30 min. The cells were
then incubated with anti-mouse IgG-FITC (1:1,000) as the secondary
antibody at 4°C for 30 min. Finally, the cells were detected using
a BD FACSCalibur™ flow cytometer (cat. no. 342975; BD Biosciences).
The cells were separately washed three times with washing buffer
(PBS containing 2% FBS) after digestion and incubation with the
primary and secondary antibodies.
Competitive ELISA
ELISA was performed as aforementioned. Briefly, the
ELISA plates were coated with 0.5 µg/ml Gas6, and incubated with 2
µg/ml biotin-labelled Axl−ECD-Fc, then with serial
dilutions of anti-Axl mAb (500, 125, 32, 8, 2 and 0.5 µg/ml).
Streptavidin-HRP (1:1,000) was added as the secondary antibody and
binding signals were measured at 450 nm. The ELISA plates were
washed three times separately with TBST for 5 min after coating,
blocking and incubating with the primary and secondary
antibodies.
Detection of Axl and p-Axl proteins
using western blot analysis
Western blot analysis was performed as
aforementioned. The MDA-MD-231 cells were seeded into 6-well plates
at 3×105 cells/well and cultured overnight. After
incubation, 100 µg/ml (0.67 µmol/l) anti-Axl mAbs were added to the
cells for 4 h, followed by the addition of 200 ng/ml Gas6 for 15
min. Neither Gas6 nor anti-Axl mAbs were added to the non-treated
group (NT). Subsequently, the cell lysates were obtained using
ice-cold RIPA buffer, and the proteins were resolved on 12%
SDS-PAGE after quantification using a BCA kit (Applygen
Technologies, Inc.). The proteins were transferred onto a PVDF
membrane, followed by blocking with 5% skimmed milk. Goat anti-Axl
antibody AF154 (1:500) and anti-goat IgG-HRP (1:2,500) were used to
detect the expression level of Axl, and rabbit anti-p-Axl antibody
(1:500) and anti-rabbit IgG-HRP (1:2,500) were used for the
detection of p-Axl (Tyr702). Finally, the signal was detected using
ECL detection reagents and semi-quantified using ImageJ software
(version 18.0; National Institutes of Health). The membrane was
washed three times with TBST for 10 min after transferring,
blocking and incubating with the primary and secondary
antibodies.
Transwell migration assay
To assess cell migration, 8-µm pore size BD-Falcon
24 Fluoroblock Transwell inserts were used. After serum-starved
culture overnight, 1×105 MDA-MB-231 cells were seeded
into the upper chamber containing serum-free DMEM. For
Gas6-dependent migration, 200 ng/ml Gas6 was added to the lower
chamber containing 10% FBS+ DMEM in the presence or absence of
anti-Axl mAbs (100 µg/ml or 0.67 µmol/l). R428 (0.67 µmol/l) was
selected as the positive control. Neither Gas6 nor anti-Axl
mAbs/R428 were added to the NT group. After 4 h of migration at
37°C, the non-migrating cells on the top surface of the filter were
removed using cotton swabs. The migrating cells on the bottom
surface of the filter were fixed with 4% paraformaldehyde for 30
min at room temperature and stained with Giemsa solution for 10 min
at room temperature. Images of the migrating cells were captured
under a light microscope, then quantified using ImageJ software
(version 1.52p; National Institutes of Health), based on five
random visual fields in the different groups.
Wound healing assay
After trypsin digestion, 8×105 MDA-MB-231
cells were seeded into 6-well plates in 2 ml DMEM per well. At 90%
confluence, wounds were created by scratching the cell monolayer
with a sterile p200 µl pipette tip. Then, the cells were washed
with PBS and cultured in serum-free DMEM containing anti-Axl mAbs
(100 µg/ml or 0.67 µmol/l) or R428 (0.67 µmol/l) as the positive
control. In total, five images from random visual fields for each
group were captured at 0 and 24 h post-wounding. The wounded area
was measured using ImageJ software (version 1.52p; National
Institutes of Health), and recorded as S0 and
S24 respectively. The cell migration rate (%) was
calculated with the following formula:
(S0-S24)/S0 × 100%.
Statistical analysis
All assays related to bioactivities were repeated
three times. The data were presented as the mean ± SD. Statistical
analysis was performed using GraphPad Prism software v.8.0
(GraphPad Software, Inc.). For the Axl, p-Axl level detection and
cell migration assays, one-way ANOVA was performed and Dunnett's
multiple comparisons test was used to compare all other groups with
the Gas6 group or NT group. P<0.05 was considered to indicate a
statistically significant difference.
Results
Determination of the Axl functional
domain
The natural ligand, Gas6, binds to Axl and actives
downstream signaling pathways. Thus, the current study first
determined the functional domain of Axl that interacts with Gas6,
before preparing an antigen containing the specific domain.
Schematic representation of the Gas6-Axl interaction is showed in
Fig. 1A. The protein sequence
information of Axl (UniProtKB P30530) was obtained by searching
www.ebi.ac.uk. As shown in Fig. 1A, the Axl protein consisted of 894
aa, with a signal peptide (1–25 aa) and a mature chain (26–894 aa).
The mature chain of Axl consisted of an extracellular domain
(26–451 aa), a transmembrane region (452–472 aa) and an
intracellular domain (473–894 aa). There were two Ig-like domains
in the extracellular domain, Axl Ig-like domain
1(Axl−Ig1, 27–128 aa) and Axl Ig-like domain 2
(Axl−Ig2, 139–222 aa).
Based on the crystal complex information (PDB no.
2C5D) (23) and the theoretical
complex structure of Gas6-Axl (Fig.
1C), Axl−Ig1 (27–128 aa) was determined as the major
functional domain interacting with Gas6 using interaction mode
analysis and the standard mode. As presented in Fig. 1B, 23 aa residues from Gas6 laminin
G-like domains 1 (Gas6−LG1) and 22 aa residues from
Axl−Ig1 interacted at the interface, and resulted in the
formation of 13 hydrogen bonds and 120 non-banded contacts between
Gas6 and Axl, which was a relatively stable interaction.
It was identified that R308,
R310, K312, R313 and
L314 of Gas6−LG1 served important roles in
interacting with ≥3 aa residues of Axl−Ig1 via hydrogen
bonds and non-bonded contacts (Fig.
1B), and were considered as the key aa. With respect to
Axl−Ig1, E56, E59, D73,
S74, T75, Q76, T77 and
Q78 interacted with ≥1 of the key aa from
Gas6−LG1 (Fig. 1B), and
were considered as the key aa. Thus, the antibody against this
Axl−Ig1 domain may block the Gas6-Axl binding and
attenuate the downstream signals and bioactivities.
Furthermore, amino acids involved in Gas6-Axl
interactions were analyzed and compared between a previous EMBO
study (23) and the present study.
As presented in Table I, according
to Sasaki et al (23), 12 aa
residues of Gas6−LG1 and 9 aa residues of
Axl−Ig1 were involved in Gas6-Axl interactions,
including 6 hydrogen bonds (no data available for the other
contacts). Furthermore, 10 aa residues of Gas6 and 8 aa residues of
Axl (italicized in Table I) were
shared in the study by Sasaki et al and the present study.
Consistently, R308, R310 and K312
of Gas6 and E56, E59 and T77 of
the Axl−Ig1 domain were considered as the key residues
in Gas6-Axl binding by both studies (underlined in Table I). Furthermore, the key role of
E56, E59 and T77 of the
Axl−Ig1 domain in binding to Gas6 was confirmed by the
study by Sasaki et al (23)
and our previous study using mutants assays (22).
 | Table I.Amino acids involved in Gas6-Axl
interactions. |
Table I.
Amino acids involved in Gas6-Axl
interactions.
|
|
|
|
Gas6−LG1 |
Axl−Ig1 |
|
|---|
|
|
|
|
|
|
|
|---|
| Study | Interaction | No. of
contacts | Involved aa | No. of aa |
| No. of aa | (Ref.) |
|---|
| Sasaki et
al | Hydrogen bonds | 6 |
R299,
S302, R308,
R310,
K312 | 5 | E56,
E59,
D73, P80 | 4 | (23) |
|
| Other contacts | – |
I307,
L309, F311,
T457, M468, I458,
T461 | 7 |
T75, T77,
V79, V92, I90 | 5 |
|
| Present study | Hydrogen bonds | 13 |
R299,
S302, R308,
R310,
K312,
R313, L314,
E460 | 8 | E56,
D73,
T75,
P80, R48, S74, Q76, Q78,
E83, | 9 | – |
|
| Other contacts | 120 |
I307,
L309, F311,
T457, M468, M300,
T304, P305, V306, R414,
L416, D455, T456, N465,
S663 | 15 | E59,
T77,
V79, V92, E70,
L71, A72, L81, G82,
E85, W89, Q94, R96 | 13 |
|
Generation of the
GST-Axl−Ig1 protein
GST-Axl−Ig1 protein expression was
induced by 1 mM IPTG at 20°C for 6 h. As expected, an obvious
expression band at 34 kDa was visible after induction following
SDS-PAGE analysis, and most of the GST-Axl−Ig1 protein
was soluble in the ultrasonic supernatant (Fig. 2A). GST-Axl−Ig1 protein
with high purity was obtained after affinity purification using GST
beads. Further western blot analysis revealed that the
GST-Axl−Ig1 protein could specifically bind with the
anti-Axl antibody AF154 and anti-GST IgG (Fig. 2B), indicating that the
GST-Axl−Ig1 protein was correctly expressed and could be
used as an antigen for immunizing animals.
 | Figure 2.Generation of the
GST-Axl−Ig1 protein. Western blot analysis of the (A)
GST-Axl−Ig1 protein and the (B) purified
GST-Axl−Ig1 protein. The GST-Axl−Ig1 protein
was validated by incubating with anti-Axl antibody AF154 and
anti-GST IgG. M, protein marker; 1, total lysate without IPTG
induction; 2 and 3, total lysate after 1 mM IPTG induction; 4,
soluble supernatant after induction; 5, insoluble fraction after
induction; 6, purified protein. GST, glutathione S-transferase;
Axl−Ig1, Axl Ig-like domains 1; Axl, anexelekto; IPTG,
isopropyl-β-D-thiogalactopyranoside. |
Generation of anti-Axl mAb
In total, seven hybridoma cell clones that bound to
GST-Axl−Ig1, but not GST, were selected from six plates
of 96-well cells from two rounds of ELISA screening (Fig. 3A) and were further screened using
flow cytometry by binding to the H1299 cells with high Axl
expression. After three rounds of ELISA and flow cytometry
screening in turn, two clones were obtained with 39.42 and 38.80%
binding ratio to the H1299 cells, separately (Fig. 3C). Finally, the hybridoma cell clones
that specifically bound to the Axl−Ig1 functional domain
were obtained, named as Am1 and Am2. The two mAbs had the same
isotype, IgG3 heavy chain and κ light chain.
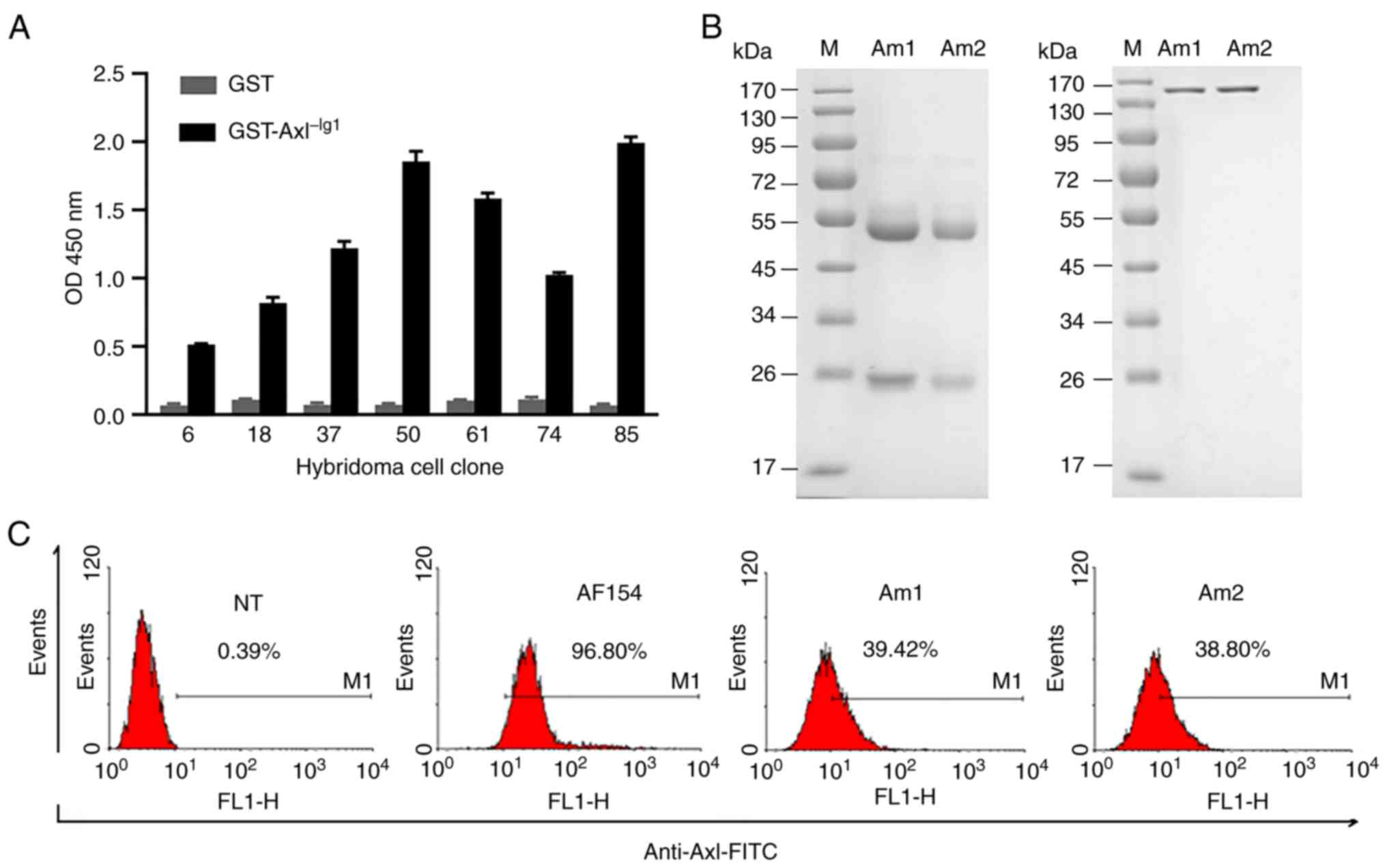 | Figure 3.Generation of the anti-Axl mAb. (A)
In total, seven positive cell clones, that bound to
GST-Axl−Ig1 but not GST, were selected by two rounds of
ELISA screening. (B) Results of reduced and non-reduced SDS-PAGE
analysis of Am1 and Am2 mAbs after protein G purification. (C) A
total of seven positive cell clones were further screened using
flow cytometry by binding to the H1299 cell line with high
expression of Axl. The H1299 cells were incubated with the mouse
anti-Axl mAb supernatant and were detected using anti-mouse
IgG-FITC. The goat anti-Axl polyclonal antibody, AF154 served as a
screening control and was detected using anti-goat IgG-FITC. After
three rounds of ELISA and flow cytometry screening, two hybridoma
cell clones, that bound specifically to human Axl were obtained,
named as Am1 and Am2. NT, non-treated group; mAb, monoclonal
antibody; GST, glutathione S-transferase; Axl−Ig1, Axl
Ig-like domains 1; Axl, anexelekto. |
After protein G purification, the specific antibody
protein bands were observed at ~50 kDa and 25 kDa in reduced
SDS-PAGE analysis, and at ~150 kDa in non-reduced SDS-PAGE
analysis, and as expected with a high purity (Fig. 3B).
Binding to Axl fusion proteins
As demonstrated by ELISA and western blot analysis
(Fig. 4A and B), Am1 or Am2 could
bind with the Axl fusion proteins, Axl−ECD-Fc and
GST-Axl−Ig1, which indicated that Am1 or Am2 could
recognize the Axl−ECD domain and Axl−Ig1
functional domain. In the present study, goat anti-human IgG
(GAH-IgG) was used as the control for recognizing the human IgG-Fc
domain and anti-GST IgG was used as the control for recognizing the
GST tag.
Blocking Gas6-Axl interaction
Using competitive ELISA, anti-Axl mAbs were detected
for their ability to inhibit Gas6 binding with
Axl−ECD-Fc. Biotin-labeled Axl−ECD-Fc and Am1
or Am2 were incubated with the ELISA plates coated with Gas6, and
the captured biotin-labeled Axl−ECD-Fc was detected
using streptavidin-HRP. As shown in Fig.
4C, the decreasing signal indicated that Am1 and Am2 blocked
Axl−ECD-Fc binding to Gas6. Thus, the inhibitory effect
of Am1 and Am2 on the Gas6-Axl interaction made it possible to
neutralize the activity of Axl. Given that 125 µg/ml Am1 and Am2
had a notable inhibitory effect on the Gas6-Axl interaction, 100
µg/ml Am1 and Am2 was used in the subsequent assays.
Binding to Axl and reducing the levels
of Axl and p-Axl
Axl was notably overexpressed in highly invasive
cells (1–3,6–8). The anti-Axl mAbs were detected for
their binding to the natural Axl proteins on the surface of highly
invasive MDA-MB-231 cell using western blot analysis and flow
cytometry. As presented in Fig. 5A,
Am1 or Am2 could bind to Axl proteins in the cell lysate or on the
cell surface.
 | Figure 5.Am1 and Am2 inhibit Axl signaling and
cell migration. (A) Am1 or Am2 could bind to the natural Axl
protein in the cell lysate or on the cell surface, as determined by
western blot and flow cytometry assays. MDA-MB-231 cell lysate or
cells were incubated with Am1 or Am2 antibody, then with anti-mouse
IgG-HRP or anti-mouse IgG-FITC, separately. (B) Am1 and Am2 reduced
the level of Axl and inhibited Axl phosphorylation. MDA-MD-231
cells were incubated with Am1 or Am2 (0.67 µmol/l), followed by 200
ng/ml Gas6 (the NT group was left untreated). GAPDH was used as the
loading control. Semi-quantification using ImageJ software of each
protein band was normalized to GAPDH and then represented as
fold-change relative to the NT group; the ratio of p-Axl/Axl was
also shown. (C) Am1 and Am2 attenuated MDA-MD-231 cell migration,
as detected in the Transwell assay, with Gas6 (200 ng/ml and either
Am1 or Am2 (0.67 µmol/l), or R428 (0.67 µmol/l) as a positive
control (the NT group was left untreated). Migrating cells were
stained with Giemsa solution, imaged and quantified using ImageJ.
(D) Am1 and Am2 attenuated MDA-MD-231 cell migration in the
wound-healing assay and the cells were cultured with Am1 or Am2
(0.67 µmol/l), or R428 (0.67 µmol/l) as a positive control, and the
results were statistically analyzed. *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001. NT, non-treated group; Gas6, growth
arrest-specific protein 6; Axl, anexelekto. |
Gas6 binds to Axl and induces its phosphorylation,
which leads to activation of the downstream signaling pathways and
the proliferation and migration of tumor cells (2). Axl and p-Axl proteins were detected
using western blot analysis to determine the effect of anti-Axl mAb
binding with Axl in the MDA-MB-231 cells. For the NT group with no
Gas6 stimulation, Axl protein expression was observed at a high
level in the MDA-MB-231 cells, but p-Axl was almost undetectable
(Fig. 5B). Furthermore, Gas6
stimulation for 15 min had no effect on Axl expression but
increased p-Axl expression up to 165-fold compared with that in the
NT group. It was also identified that Am1 or Am2 significantly
decreased Axl expression from 0.930 to 0.483 or 0.480 folds
compared with that in the NT group respectively (P<0.001), and
p-Axl expression from 165.35 to 9.99 or 21.91 folds compared with
that in the NT group, respectively (P<0.0001). The ratio of
p-Axl/Axl is also shown in Fig. 5B.
Therefore, it was suggested that Am1 and Am2 could reduce the
expression level of Axl and significantly inhibit Axl
phosphorylation, which may be the main mechanism preventing
tumorigenesis.
Inhibiting cell migration
Anti-Axl mAbs were evaluated for their ability to
inhibit cell migration using a Transwell assay. The MDA-MD-231
cells, plated in the upper chamber of Transwell inserts, were
stimulated to migrate into the lower chamber containing Gas6 and
Am1 or Am2. Compared with that in the Gas6 group, the migration of
the MDA-MD-231 cells was significantly attenuated by Am1 or Am2
(P<0.001) to a similar extent as that observed with the Axl
inhibitor R428 (P=0.865 and P=0.735, respectively) (Fig. 5C). In addition, cell migration in the
Gas6+Am1 or Gas6+Am2 groups was weaker compared with that in the NT
group without Gas6 stimulation, indicating that Am1 and Am2 almost
completely blocked the stimulation of Gas6.
In the wound healing assay, Am1 or Am2 were added to
scratched MDA-MD-231 cells to verify their inhibitory effect on
cell migration during the wound healing progress. Compared with
that in the NT group, the wound healing speed at 24 h post-wounding
was significantly impeded by Am1 (P<0.01) or Am2 (P<0.05),
although the inhibitory effect of Am1 or Am2 on cell migration was
a little weaker to that observed in the group treated with the Axl
inhibitor R428 (Fig. 5D).
Collectively, both the Transwell and wound healing assays indicated
that Am1 and Am2 could effectively inhibit MDA-MD-231 cell
migration.
Discussion
Axl-targeted mAbs have been attracting increasing
interest due to the important role of Axl in tumor development and
chemotherapy resistance (1,2,4,6,7).
Furthermore, accurate determination of the Axl functional domain
may make the screening of mAb more effective and feasible. In the
present study, Axl−Ig1, consisting of 102 aa, was
predicted as the major Axl functional domain interacting with Gas6,
based on theoretical analysis of the Gas6-Axl crystal structure and
complex model using bioinformatics and structural biology methods.
This finding was consistent with a previous EMBO study that
reported major contact between Axl−Ig1-Gas6 and minor
contact between Axl−Ig2-Gas6 (23).
In the present study, the mAbs targeting the
Axl−Ig1 functional domain were generated and evaluated
in tumor cells with high Axl expression. Since anti-Axl mAb against
the Axl−Ig1 domain was not commercially available,
anti-Axl polyclonal antibody AF154 was used as the positive control
to recognize the Axl−Ig1 domain for
GST-Axl−Ig1 protein validation, clone screening and
western blot analysis, as the Axl−Ig1 domain may be
recognized by the polyclonal antibody, AF154 immunized by
recombinant human Axl domain (33–440 aa; accession no. AAA61243),
that overlaps with the Axl−Ig1 domain (27–128 aa).
Furthermore, the first selective kinase inhibitor R428 (undergoing
clinical phase II trials) (16–18) was
selected as a positive control in the cell migration assays, since
no neutralizing anti-Axl mAb is commercially available.
MDA-MB-231 is a highly invasive TNBC cell line with
upregulated Axl expression (1,15).
Consistent with previous reports (1,25,26), Axl
was significantly upregulated in the MDA-MB-231 cell line without
Gas6 stimulation and the phosphorylation of Axl was dependent on
Gas6. Am1 or Am2 significantly reduced the protein expression level
of Axl and almost completely blocked the stimulation of Gas6 in Axl
phosphorylation and the Transwell migration assays, as well as
producing similar activities to the positive control drug, R428, in
the Transwell and wound healing migration assays. Thus, it was
suggested that Am1 or Am2 attenuated MDA-MB-231 cell migration by
blocking Axl signaling.
TNBC, accounting for 15–20% of breast cancer cases,
is characterized by the absence of estrogen receptors, progesterone
receptors and human epidermal growth factor receptor 2 (27,28). Due
to the lack of therapeutic targets, no targeted therapy is
currently available for TNBC. Fortunately, TNBC is sensitive to
chemotherapy, which remains the most practicable therapeutic option
for patients with TNBC (1,27). However, these patients remain
susceptible to chemotherapy resistance (27,29,30).
An increasing number of studies have reported that
the activation of Axl was associated with chemotherapy resistance
in breast, colon, lung and other types of cancer (2,31–34).
Therefore, Axl inhibitors can directly reduce tumor cell migration,
as well as the development of chemotherapy resistance by blocking
Axl activity. Previous studies have revealed that the Axl
inhibitor, R428 enhanced the efficacy of chemotherapy by increasing
the sensitivity of tumor cells to chemotherapeutic reagents in
mesothelioma, uterine serous cancer and pancreatic cancer (9,13,14).
Furthermore, the combination of chemotherapy and Axl inhibitors
could improve clinical outcomes for patients, particularly those
with TNBC, for which no targeted agents have been currently
approved (1,26,35).
In addition, Axl has been associated with
resistance not only to chemotherapy, but also to immunotherapy,
targeted therapy and radiation therapy (2,11,36,37).
The application of Axl inhibition combined with chemotherapy or
targeted therapy has provided a promising treatment strategy for
patients with breast and lung cancer (12,19,36,38).
Compared with the previous Axl-targeted mAbs, that
are against Axl−ECD, anti-Axl mAbs in the present study
directly target Axl−Ig1, the major Axl functional domain
interacting with Gas6. To the best of our knowledge, no mAb that
targets the Axl functional domain has been previously reported,
which is the main advantage of the antibodies reported in the
present study.
In conclusion, the present study investigated two
anti-Axl mAbs targeting Axl−Ig1, the major Axl
functional domain interacting with Gas6. Anti-Axl mAbs almost
completely neutralized the stimulation of Gas6 in Axl
phosphorylation and cell migration assays, and displayed an almost
similar activity to the positive control drug, R428 in the cell
migration assays. Future studies are required to further evaluate
the two anti-Axl mAbs for their bioactivity in vitro and
in vivo, particularly in reducing the development of
chemotherapy resistance. The anti-Axl mAbs targeting the
Axl−Ig1 domain might serve as an option for the combined
therapy strategy.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 31771010).
Availability of data and materials
The datasets used and/or analyzed in the current
study are available from the first author upon reasonable
request.
Authors' contributions
ML and JF designed the study. JF performed the
computer-guided analysis. RA, XYL and XLL performed the
experiments. HC and RA analyzed the data, drafted and revised the
article. JF, ML and HC approved the version to be published. HC and
ML confirm the authenticity of all the raw data. All authors have
read and approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Animal
Ethics Committee of Suzhou Vocational Health College (Jiangsu,
China; approval no, SWAE201905).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Gas6
|
growth arrest-specific protein 6
|
|
mAb
|
monoclonal antibody
|
|
Axl−ECD
|
Axl external cell domain
|
|
Axl−ECD-Fc
|
Axl−ECD fused with
IgG1-Fc
|
|
Axl−Ig1
|
Axl Ig-like domains 1
|
|
Gas6−LG1
|
Gas6 laminin G-like domain 1
|
|
TNBC
|
triple-negative breast cancer
|
|
NSCLC
|
non-small cell lung cancer
|
|
IPTG
|
isopropyl-β-D-thiogalactopyranoside
|
|
HAT
|
hypoxanthine/aminopterin/thymidine
|
References
|
1
|
Leconet W, Chentouf M, du Manoir S,
Chevalier C, Sirvent A, Ait-Arsa I, Busson M, Jarlier M,
Radosevic-Robin N, Theillet C, et al: Therapeutic activity of
anti-AXL antibody against triple-negative breast cancer
patient-derived xenografts and metastasis. Clin Cancer Res.
23:2806–2816. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gay CM, Balaji K and Byers LA: Giving AXL
the axe: Targeting AXL in human malignancy. Br J Cancer.
116:415–423. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Duan Y, Luo L, Qiao C, Li X, Wang J, Liu
H, Zhou T, Shen B, Lv M and Feng J: A novel human anti-AXL
monoclonal antibody attenuates tumour cell migration. Scand J
Immunol. 90:e127772019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Myers SH, Brunton VG and Unciti-Broceta A:
AXL inhibitors in cancer: A Medicinal Chemistry Perspective. J Med
Chem. 59:3593–3608. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Colavito SA: AXL as a target in breast
cancer therapy. J Oncol. 2020:52919522020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leconet W, Larbouret C, Chardes T, Thomas
G, Neiveyans M, Busson M, Jarlier M, Radosevic-Robin N, Pugniere M,
Bernex F, et al: Preclinical validation of AXL receptor as a target
for antibody-based pancreatic cancer immunotherapy. Oncogene.
33:5405–5414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iida S, Miki Y, Suzuki T, Mori K, Saito M,
Niikawa H, Kondo T, Yamada-Okabe H and Sasano H: Activation of AXL
and antitumor effects of a monoclonal antibody to AXL in lung
adenocarcinoma. Anticancer Res. 34:1821–1827. 2014.PubMed/NCBI
|
|
8
|
Wei J, Sun H, Zhang A, Wu X, Li Y, Liu J,
Duan Y, Xiao F, Wang H, Lv M, et al: A novel AXL chimeric antigen
receptor endows T cells with anti-tumor effects against triple
negative breast cancers. Cell Immunol. 331:49–58. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Oien DB, Garay T, Eckstein S and Chien J:
Cisplatin and pemetrexed activate AXL and AXL inhibitor BGB324
enhances mesothelioma cell death from chemotherapy. Front
Pharmacol. 8:9702018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hong J, Peng D, Chen Z, Sehdev V and
Belkhiri A: ABL regulation by AXL promotes cisplatin resistance in
esophageal cancer. Cancer Res. 73:331–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hong CC, Lay JD, Huang JS, Cheng AL, Tang
JL, Lin MT, Lai GM and Chuang SE: Receptor tyrosine kinase AXL is
induced by chemotherapy drugs and overexpression of AXL confers
drug resistance in acute myeloid leukemia. Cancer Lett.
268:314–324. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li Y, Ye X, Tan C, Hongo JA, Zha J, Liu J,
Kallop D, Ludlam MJ and Pei L: Axl as a potential therapeutic
target in cancer: Role of Axl in tumor growth, metastasis and
angiogenesis. Oncogene. 28:3442–3455. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Palisoul ML, Quinn JM, Schepers E,
Hagemann IS, Guo L, Reger K, Hagemann AR, McCourt CK, Thaker PH,
Powell MA, et al: Inhibition of the receptor tyrosine kinase AXL
restores paclitaxel chemosensitivity in uterine serous cancer. Mol
Cancer Ther. 16:2881–2891. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ludwig KF, Du W, Sorrelle NB,
Wnuk-Lipinska K, Topalovski M, Toombs JE, Cruz VH, Yabuuchi S,
Rajeshkumar NV, Maitra A, et al: Small-molecule inhibition of axl
targets tumor immune suppression and enhances chemotherapy in
pancreatic cancer. Cancer Res. 78:246–255. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shen Y, Chen X, He J, Liao D and Zu X: Axl
inhibitors as novel cancer therapeutic agents. Life Sci.
198:99–111. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Falcone I, Conciatori F, Bazzichetto C,
Bria E, Carbognin L, Malaguti P, Ferretti G, Cognetti F, Milella M
and Ciuffreda L: AXL receptor in breast cancer: Molecular
involvement and therapeutic limitations. Int J Mol Sci.
21:84192020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sheridan C: First Axl inhibitor enters
clinical trials. Nat Biotechnol. 31:775–776. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen F, Song Q and Yu Q: Axl inhibitor
R428 induces apoptosis of cancer cells by blocking lysosomal
acidification and recycling independent of Axl inhibition. Am J
Cancer Res. 8:1466–1482. 2018.PubMed/NCBI
|
|
19
|
Ye X, Li Y, Stawicki S, Couto S,
Eastham-Anderson J, Kallop D, Weimer R, Wu Y and Pei L: An anti-Axl
monoclonal antibody attenuates xenograft tumor growth and enhances
the effect of multiple anticancer therapies. Oncogene.
29:5254–5264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koopman LA, Terp MG, Zom GG, Janmaat ML,
Jacobsen K, Gresnigt-van den Heuvel E, Brandhorst M, Forssmann U,
de Bree F, Pencheva N, et al: Enapotamab vedotin, an AXL-specific
antibody-drug conjugate, shows preclinical antitumor activity in
non-small cell lung cancer. JCI Insight. 4:e1281992019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ahnert JR, Taylor MH, O'Reilly E, Zhang J,
Doebele R, Ben Y, Sharp L, Boyle WJ, Chang CY, Frey G, et al: A
phase 1/2 dose-escalation and expansion study of a conditionally
active anti-AXL humanized monoclonal antibody (BA3011) in patients
with advanced solid tumors. J Clin Oncol. 36 (Suppl
15):TPS121262018. View Article : Google Scholar
|
|
22
|
Duan Y, Hu B, Qiao C, Luo L, Li X, Wang J,
Liu H, Zhou T, Shen B, Lv M and Feng J: Engineered
AXL−ECD-Fc variants that abolish the AXL/Gas6
interaction suppress tumor cell migration. Oncol Lett.
17:5784–5792. 2019.PubMed/NCBI
|
|
23
|
Sasaki T, Knyazev PG, Clout NJ, Cheburkin
Y, Gohring W, Ullrich A, Timpl R and Hohenester E: Structural basis
for Gas6-Axl signalling. EMBO J. 25:80–87. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suzuki K, Nakamura K, Kato K, Hamada H and
Tsukamoto T: Exploration of target molecules for prostate cancer
gene therapy. Prostate. 67:1163–1173. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wilson C, Ye X, Pham T, Lin E, Chan S,
McNamara E, Neve RM, Belmont L, Koeppen H, Yauch RL, et al: AXL
inhibition sensitizes mesenchymal cancer cells to antimitotic
drugs. Cancer Res. 74:5878–5890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
D'Alfonso TM, Hannah J, Chen Z, Liu Y,
Zhou P and Shin SJ: Axl receptor tyrosine kinase expression in
breast cancer. J Clin Pathol. 67:690–696. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Nakhjavani M, Hardingham JE, Palethorpe
HM, Price TJ and Townsend AR: Druggable molecular targets for the
treatment of triple negative breast cancer. J Breast Cancer.
22:341–361. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tovey H and Cheang MCU: Identifying
biomarkers to pair with targeting treatments within triple negative
breast cancer for improved patient stratification. Cancers (Basel).
11:18642019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Carey L, Winer E, Viale G, Cameron D and
Gianni L: Triple-negative breast cancer: Disease entity or title of
convenience? Nat Rev Clin Oncol. 7:683–692. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ding L, Gu H, Xiong X, Ao H, Cao J, Lin W,
Yu M, Lin J and Cui Q: MicroRNAs involved in carcinogenesis,
prognosis, therapeutic resistance and applications in human
triple-negative breast cancer. Cells. 8:14922019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kim KC, Baek SH and Lee C:
Curcumin-induced downregulation of Axl receptor tyrosine kinase
inhibits cell proliferation and circumvents chemoresistance in
non-small lung cancer cells. Int J Oncol. 47:2296–2303. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Asiedu MK, Beauchamp-Perez FD, Ingle JN,
Behrens MD, Radisky DC and Knutson KL: AXL induces
epithelial-to-mesenchymal transition and regulates the function of
breast cancer stem cells. Oncogene. 33:1316–1324. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heckmann D, Maier P, Laufs S, Li L,
Sleeman JP, Trunk MJ, Leupold JH, Wenz F, Zeller WJ, Fruehauf S and
Allgayer H: The disparate twins: A comparative study of CXCR4 and
CXCR7 in SDF-1α-induced gene expression, invasion and
chemosensitivity of colon cancer. Clin Cancer Res. 20:604–616.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sun ZG, Liu JH, Zhang JM and Qian Y:
Research progress of Axl inhibitors. Curr Top Med Chem.
19:1338–1349. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yang PW, Liu YC, Chang YH, Lin CC, Huang
PM, Hua KT, Lee JM and Hsieh MS: Cabozantinib (XL184) and R428
(BGB324) inhibit the growth of esophageal squamous cell carcinoma
(ESCC). Front Oncol. 9:11382019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang ZF, Lee JC, Lin L, Olivas V, Au V,
LaFramboise T, Abdel-Rahman M, Wang X, Levine AD, Rho JK, et al:
Activation of the AXL kinase causes resistance to EGFR-targeted
therapy in lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Schoumacher M and Burbridge M: Key Roles
of AXL and MER receptor tyrosine kinases in resistance to multiple
anticancer therapies. Curr Oncol Rep. 19:192017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu L, Greger J, Shi H, Liu Y, Greshock J,
Annan R, Halsey W, Sathe GM, Martin AM and Gilmer TM: Novel
mechanism of lapatinib resistance in HER2-positive breast tumor
cells: Activation of AXL. Cancer Res. 69:6871–6878. 2009.
View Article : Google Scholar : PubMed/NCBI
|
















