Introduction
The proteins p16INK4a and
p21WAFI/Cip1 inhibit cyclin/cyclin-dependent kinase
(CDK) complexes, in which CDKs depend on cyclins. These proteins
affect cell cycle progression in the G1/S phase by
directly interfering with CDK activation and inhibiting DNA
replication. Thus, p16INK4a and
p21WAFI/Cip1 potentially act as tumor suppressor
genes (1–5). p16INK4a belongs to the
INK4a family and regulates the cell cycle by specifically attaching
to and inhibiting the expression of CDK4 and CDK6 (6). p21WAFI/Cip1 regulates the
cell cycle by inhibiting multiple CDKs, including CDK1, CDK2 and
CDK4 (3,4,7–9).
Aberrant regulation of these proteins, which have a low molecular
mass, is characteristic of cervical carcinoma that expresses human
papillomavirus (HPV) E6 and E7 oncogenes and their precursor E2
(4,10–12).
Although the p16INK4a tumor
suppressor gene is inactivated by mutations or epigenetic changes
that lead to excessive cellular proliferation in most tumors, it is
expressed at high levels in cervical cancer cells infected with HPV
in which the oncoprotein E7 is expressed (4,5,13–19).
Consequently, in HPV-transformed cervical cancer,
p16INK4a has oncogenic activity through the
CDK6-HuR-IL 1A axis and represents a diagnostic marker of cervical
neoplasia. Although the p16INK4a gene is highly
expressed in this case, it does not exert a negative physiological
effect on the cell cycle (10,12).
Transcriptional silencing of
p16INK4a results from DNA hypermethylation of the
gene promoter in various tumors (20). However, in cervical cancer induced
by HPV infection, in which the p16INK4a protein is
highly expressed, complete DNA methylation has been reported in the
p16INK4a promoter without any influence on its
expression, thus indicating no association between this epigenetic
marker and reduced expression of p16INK4a
(10,21). The increased expression of
p16INK4a may also be regulated by histone
modifications, such as H3K4me3 (22), which is a histone mark that has been
associated with gene activation (23). A reduction in H3K27me3, a
transcriptionally repressive epigenetic mark (24), has been reported to occur in the
promoter of p16INK4a and involves the
participation of the histone demethylase KDM6B (18,25,26).
Downregulation of p21WAFI/Cip1 has
been directly associated with cervical cancer compared with normal
epithelium; specifically, in HeLa cells,
p21WAFI/Cip1 is weakly expressed and is
associated with the progression of malignant transformation
(4,27). Epigenetic alterations in the
p21WAFI/Cip1 promoter, including DNA
hypermethylation and histone H3 hypoacetylation, are key events in
the inactivation of this gene (28,29).
Histone acetylation is generally associated with chromatin opening
and activated gene expression, although inactivation of inducible
promoters enriched for H3K14 acetylation has been reported
(30).
Histone deacetylase (HDAC) inhibitors (HDACis), such
as sodium valproate (VPA), have been reported to induce increased
expression of p21WAF1/Cip1 in cervical and breast
cancer cell lines and chronic lymphocytic leukemia (31–35).
It has been suggested that HDACis stimulate
p21WAF1/Cip1 expression through a selective
increase in the degree of histone H3 acetylation (H3Ac) and a
decrease in DNA methylation at the gene promoter in rat
hippocampus, colon and bladder cancer cell lines, and human lung
carcinoma cells (11,36–40).
Increased expression of p21WAF1/Cip1 has also
been reported to be associated with enhanced methylation levels of
H3K4me2/me3, and decreased levels of H3K9me2/me3 in rat kidney
cells, suggesting a role of methylated H3K in TGF-β1-mediated
p21 gene expression and its protective potential in managing
chronic renal diseases (41,42).
VPA is an anticonvulsive drug that has been reported
to exhibit antitumor effects, either alone or in combination with
other drugs, against several cancer types (31,43–51).
This drug acts through various mechanisms that involve inhibition
of the neurotransmitter γ-aminobutyric acid, and blockage of T-type
calcium and voltage-gated sodium channels, and that affect several
epigenetic markers and chromatin supraorganization (49,52–54).
VPA may also directly interact with isolated DNA and histones H1
and H3 in vitro, and affect chromatin at the nucleosome
level (55–58).
VPA acts on epigenetic marks by inhibiting class I
and II HDACs, often favoring the acetylation of histones H3 and H4
(31,44,59).
Moreover, in HeLa cells, a widely used model of cervical cancer,
VPA can promote DNA demethylation with the participation of TET and
DNMT1 enzymes, and can change the methylation status of different
lysine residues in histone H3, in addition to histone acetylation
(60–64). Consequently, VPA alters the
epigenetic landscape of HeLa cells by modulating the expression of
their genes (60).
Considering that VPA promotes cell cycle arrest at
the G1 phase and induces changes in the methylation
levels of histones in HeLa cells (63,64),
it would be relevant to detect whether this drug induces changes in
the expression of genes such as p21WAF1/Cip1,
which participates negatively in the cell cycle, and
p16INK4a, which is a biomarker of cervical
neoplasia (65). In the present
study, the effects of VPA on changes in p16INK4a
and p21WAF1/Cip1 genes were investigated in HeLa
cells to demonstrate whether VPA modulates the expression of a
cervical carcinoma biomarker and a tumor suppressor gene. The
enzymatic activity of HDAC and the acetylation of histone H3 were
also evaluated in this context. This investigation intended to
improve understanding of the antitumorigenic effects of VPA, in
addition to the alterations in DNA and histone methylation status,
and chromatin supraorganization previously reported for these cells
(60–64).
Materials and methods
Cell culture and VPA treatments
HeLa cells were acquired at passage 10 from the
Emerging Virus Studies Laboratory, University of Campinas
(Campinas, Brazil) and were validated at the Technical Division of
Support for Teaching, Research, and Innovation, Faculty of Medicine
Foundation, University of São Paulo (São Paulo, Brazil). The cells
were used at passages 11–45 and were cultured in high-glucose
Dulbecco's modified Eagle's medium (Sigma-Aldrich; Merck KGaA)
supplemented with 10% bovine fetal calf serum (FCS; Vitrocell
Embriolife), penicillin/streptomycin (100 IU and 100 µg/ml,
respectively; Sigma-Aldrich; Merck KGaA) and 1% sodium pyruvate
(Sigma-Aldrich; Merck KGaA) at 37°C in 5% CO2. For cell
treatment, the cells were cultured for 24 h in medium containing 1%
FCS and 0.5 or 2.0 mM VPA (Santa Cruz Biotechnology, Inc.),
preceded by cell culture for 24 h in the absence of the drug. When
HeLa cells were cultured for 24 h in the presence of 1 and 2 mM
VPA, cell viability reached values of 94 and 89%, respectively, as
detected using the MTT assay (66).
Based on previously reported analyses, under 0.5 and 2.0 mM VPA
treatment conditions for 24 h, HeLa cells exhibited G1
phase cell cycle arrest and no induction of apoptotic cell death
(67,68). When quantifying DNA fragmentation
using the TUNEL assay or calculating cell death ratios in
preparations subjected to the Feulgen reaction, the exposure of
HeLa cells to 1 mM VPA for 24 h did not result in an increase of
apoptosis (61). In the present
study, control cells were cultured in the absence of VPA. For
immunofluorescence assays, the cells were seeded onto round
coverslips in 24-well plates at a concentration of 5×104
cells/ml and 100 µl/well. For western blotting (WB) and HDAC
activity assays, the cells were cultured in 6-well plates at a
concentration of 1.0×105 cells/ml and 4 ml/plate. For
reverse transcription-quantitative PCR (RT-qPCR), the cells were
cultured in 25-cm2 culture flasks at a concentration of
6×104 cells/ml and 5 ml/flask.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde in phosphate
buffer (pH 7.4) for 10 min at 25°C, rinsed in PBS, permeabilized
with 0.2% Triton X-100 (MilliporeSigma) for 10 min at 25°C and
blocked with 5% bovine serum albumin (BSA; Sigma-Aldrich; Merck
KGaA) for 30 min at 25°C. The cells were then incubated overnight
with mouse anti-p16INK4a (1:100 dilution; cat. no.
sc-56330), mouse anti-p21WAFI/Cip1 (1:100 dilution; cat.
no. sc-6246) (both from Santa Cruz Biotechnology, Inc.) and rabbit
anti-H3Ac (1:1,000 dilution; cat. no. 06-599; Sigma-Aldrich; Merck
KGaA) primary antibodies in 1% BSA blocking solution at 4°C in the
dark, followed by extensive PBS washes. To detect
p16INK4a and p21WAFI/Cip1, the cells were
incubated with a FITC-conjugated goat anti-mouse antibody (1:50
dilution; cat. no. F0257; Sigma-Aldrich; Merck KGaA) for 1 h at 4°C
in the dark, followed by nuclear counterstaining with TO-PRO-3
(1:1,000 dilution; Thermo Fisher Scientific, Inc.) for 1 h at 4°C.
To detect H3Ac, an Alexa-Fluor 488-conjugated goat anti-rabbit
secondary antibody (1:1,000; cat. no. A-11008; Thermo Fisher
Scientific, Inc.) was used to incubate the cells for 1 h at 4°C in
the dark, followed by counterstaining with DAPI for 5–10 min at
25°C. The preparations were then rinsed in PBS and mounted using
VECTASHIELD (Vector Laboratories, Inc.). The images were captured
using a Leica TCS SP5 II confocal microscope (Leica Microsystems
GmbH) at the Central Laboratory of High-Performance Technology in
Life Science (University of Campinas).
WB
The p16INK4a, p21WAFI/Cip1 and
H3Ac proteins were examined after total proteins were extracted
from HeLa cells using RIPA buffer [50 mM Tris-HCl (pH 8.0), 150 mM
NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM
EDTA, 0.5 mM EGTA, and 1 mM PMSF] for ≥30 min on ice. The Bradford
assay (Sigma-Aldrich; Merck KGaA) was used to detect protein
concentrations, using BSA as a standard. Absorbance values were
quantified after all samples were incubated for 1 h at room
temperature at 595 nm using a Multiskan™ FC Microplate
Photometer (Thermo Fisher Scientific, Inc.). Protein samples (60
µg) were then incubated in heated sample buffer [0.06 M Tris-HCl
(pH 6.8), 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.025%
Bromophenol Blue] for 5 min and were separated by SDS-PAGE on 15%
polyacrylamide gels. The proteins were transferred to
nitrocellulose membranes (Thermo Fisher Scientific, Inc.), which
were blocked in 4% BSA for 2 h, at 25°C and separately incubated
with mouse anti-p16INK4a, (1:150; cat. no. MA5-17054;
Thermo Fisher Scientific, Inc.) mouse anti-p21WAFI/Cip1
(1:100; cat. no. 1026-MSM11-P1; Thermo Fisher Scientific, Inc.) and
rabbit anti-H3Ac (1:4,000; cat. no. PA5-114693; Thermo Fisher
Scientific, Inc.) primary antibodies overnight in 1X Tris-buffered
saline −0.1% Tween 20 (TBST; cat. no. 91414; Sigma-Aldrich; Merck
KGaA) blocking solution at 4°C. After extensive washing with TBST,
the membranes were incubated with horseradish peroxidase-conjugated
goat anti-mouse (1:2,000; cat. no. 1706516; Bio-Rad Laboratories,
Inc.) and anti-rabbit (cat. no. 31460; Invitrogen; Thermo Fisher
Scientific, Inc.) secondary antibodies to detect
p16INK4a, or p21WAFI/Cip1 and H3Ac,
respectively; for detection of p21WAFI/Cip1 a dilution
of 1:2,000 was used, whereas for H3Ac a dilution of 1:4,000 was
used. In all cases, incubation was performed in 1% BSA blocking
solution for 2 h at 25°C. Protein blots were imaged using an ECL
Western Blotting Detection System (Amersham; Cytiva) and were
visualized by chemiluminescence using a ChemiDoc Imaging System
(Bio-Rad Laboratories, Inc.) at the Laboratory of Tissue Biology of
the University of Campinas. As a control for differences in protein
loading, the membranes were incubated overnight at 4°C with rabbit
anti-β-actin primary antibody (1:1,000 dilution; cat. no. 4970;
Cell Signaling Technology, Inc.), followed by incubation with a
horseradish peroxidase-conjugated goat anti-rabbit secondary
antibody (1:4,000 dilution; cat. no. 31460; Invitrogen Thermo
Fisher Scientific, Inc.) for 1 h at 4°C. ImageJ version IJ 1.46r
software (National Institutes of Health) was used to estimate
p16INK4a/β-actin, p21WAFI/Cip1/β-actin and
H3Ac/β-actin ratios. The assays were repeated five times.
HDAC assay
The enzymatic activity of HDAC in VPA-treated HeLa
cells, expressed relative to untreated controls, was detected using
an HDAC assay kit (cat. no. CS1010; Sigma-Aldrich; Merck KGaA)
according to the manufacturer's instructions. Cells were lysed in
RIPA buffer and incubated in 96-well plates with the reaction
substrate (peptide with acetylated lysine and a fluorescent group
attached) for 30 min at 30°C. The revealing reaction solution was
then added, promoting the breakage of the deacetylated substrate by
the HDACs present in the samples and the liberation of the
fluorescent group. Subsequently, the solution was incubated for 10
min at room temperature. Fluorescence was measured at 360 nm (test
wavelength) and 460 nm (reference wavelength) using a Multiskan FC
Microplate Photometer (Thermo Fisher Scientific, Inc.).
RT-qPCR
Total RNA was isolated using the PureLink RNA Mini
Kit (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions. RNA integrity number was evaluated
using a Nano-Vue spectrophotometer (Cytiva). RNA was reverse
transcribed using a High-Capacity cDNA Reverse Transcription Kit
(Thermo Fisher Scientific, Inc.), according to the manufacturer's
protocol. The PCR primers were obtained from data reported in the
literature (Table I) (32,69,70).
Subsequently, 1 µl cDNA (4 ng/µl) was amplified using the Real Q
Plus 2X Master Mix Green, High ROX kit (cat. no. A323402; Ampliqon
A/S) and 400 nM of each primer in a final volume of 20 µl. The
cycling conditions were as follows: 10 min at 95°C, followed by 40
cycles of denaturation at 95°C for 15 sec, and annealing and
extension at 60°C for 1 min. Expression levels were detected using
Bio-Rad CFX Maestro (Bio-Rad Laboratories, Inc.). The dissociation
curve was evaluated to confirm specific amplification. The data
were normalized using the Q-Gene program version 4.3 (71,72).
Cycle threshold values were calculated from experiments performed
in triplicate and normalized with respect to the housekeeping gene
GAPDH. Relative quantification was achieved using the
comparative 2−ΔΔCq method (73).
 | Table I.Primers used for reverse
transcription-quantitative PCR. |
Table I.
Primers used for reverse
transcription-quantitative PCR.
| Genes | Sequences | (Refs.) |
|---|
| p16 | F:
CAACGCACCGAATAGTTACGG | (69) |
|
| R:
GCGCAGTTGGGCTCCG | (69) |
| p21 | F:
TGATGCGCTAATGGCGGGCT | (32) |
|
| R:
TGCTGGTCTGCCGCCGTTTT | (32) |
| GADPH | F:
GAATGGGCAGCCGTTAGGAA | (70) |
|
| R:
ATCACCCGGAGGAGAAATCG | (70) |
Statistical analysis
GraphPad Prism version 9.5.0 (525) (Dotmatics) was
used for statistical analysis. For comparisons between more than
two groups, one-way ANOVA followed by Dunnett's test was used for
WB data, and Kruskal-Wallis followed by Dunn's test was used for
fluorescence intensity (FI) and RT-qPCR data. Mann-Whitney U test
was applied to compare H3Ac FI data between two groups. To compare
HDAC activity between more than two groups, one-way ANOVA and
Dunnett's post hoc test were used. P<0.05 was considered to
indicate a statistically significant difference.
Results
VPA affects p16INK4a
protein and gene expression in HeLa cells
HeLa cells cultured in the presence of 2 mM VPA
exhibited a significant average decrease of ~11% in the nuclear
protein abundance of p16INK4a, and an average decrease
of ~45% in the mRNA expression levels of p16INK4a
in comparison to untreated controls, based on the
immunofluorescence data and RT-qPCR results (Fig. 1A-D). Although the results obtained
by WB did not indicate a statistically significant difference in
the protein expression levels of p16INK4a, there was a
tendency for p16INK4a expression to decrease in response
to VPA treatment (Fig. 1E).
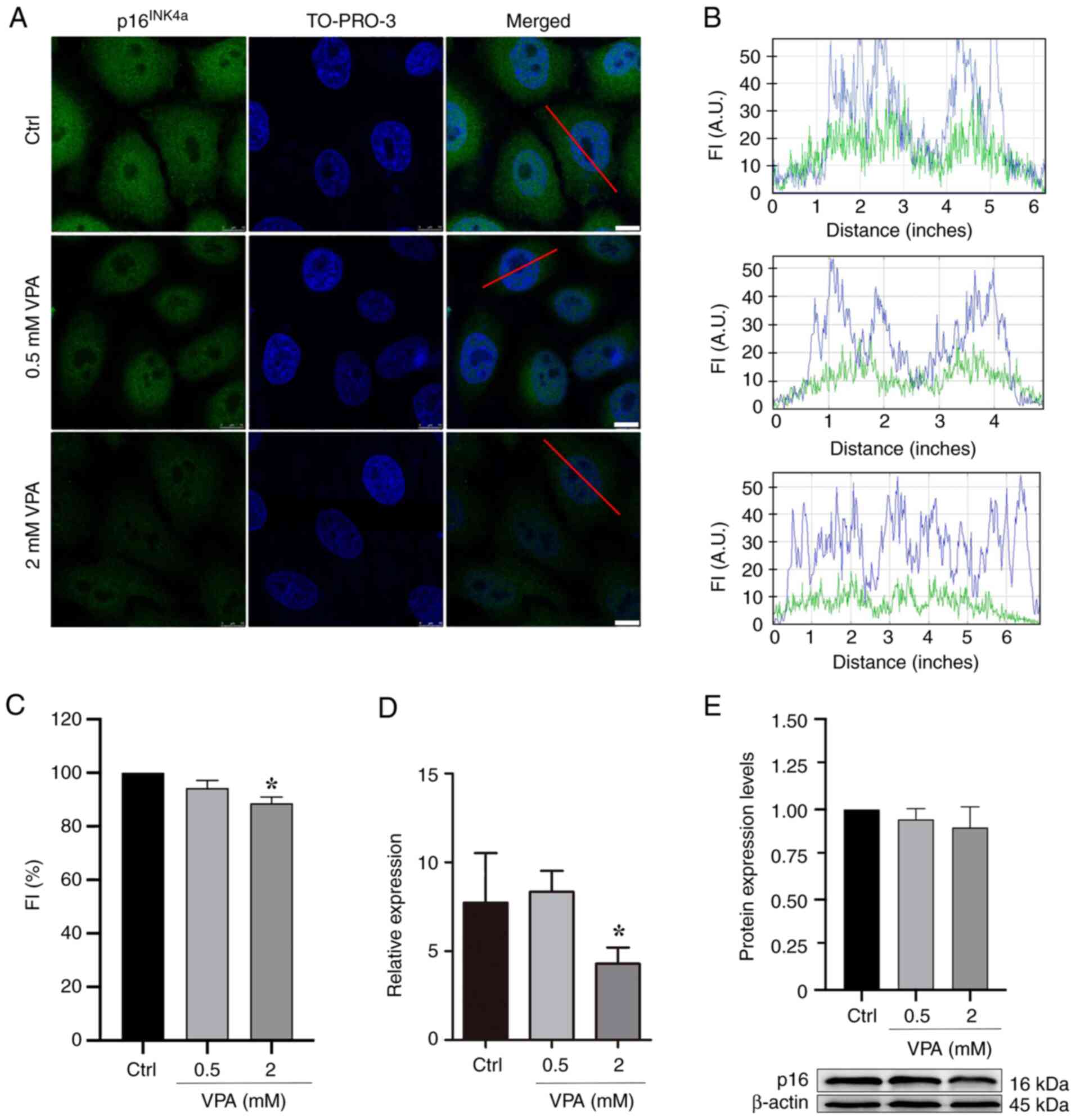 | Figure 1.p16INK4a protein abundance
and gene expression levels in VPA-treated HeLa cells as assessed
using confocal microscopy, RT-qPCR and WB. (A) Confocal microscopy
images. Images are representative of three independent experiments,
comprising the analysis of 60 nuclei. Scale bars, 10 µm. (B) Graphs
representing FI profiles along the red line drawn in the merged
image of selected nuclear images to identify the immunofluorescence
signals for p16INK4a (green) and the TO-PRO-3-stained
DNA (blue). (C) Fluorescence intensity of p16INK4a
signals decreased in response to 2 mM VPA treatment relative to
untreated control. (D) mRNA expression levels of the
p16INK4a gene analyzed using RT-qPCR and
normalized to endogenous GADPH control decreased significantly
after cell treatment with 2 mM VPA. (E) WB and respective
densitometry of five independent experiments indicated no
statistically significant changes in p16INK4a protein
abundance following VPA treatment, although a trend toward
decreased values was observed. β-actin was used as a loading
control. Data are presented as the mean ± standard error of the
mean. *P<0.05. A.U., arbitrary units; Ctrl, control; FI,
fluorescence intensity; RT-qPCR, reverse transcription-quantitative
PCR; VPA, valproate; WB, western blotting. |
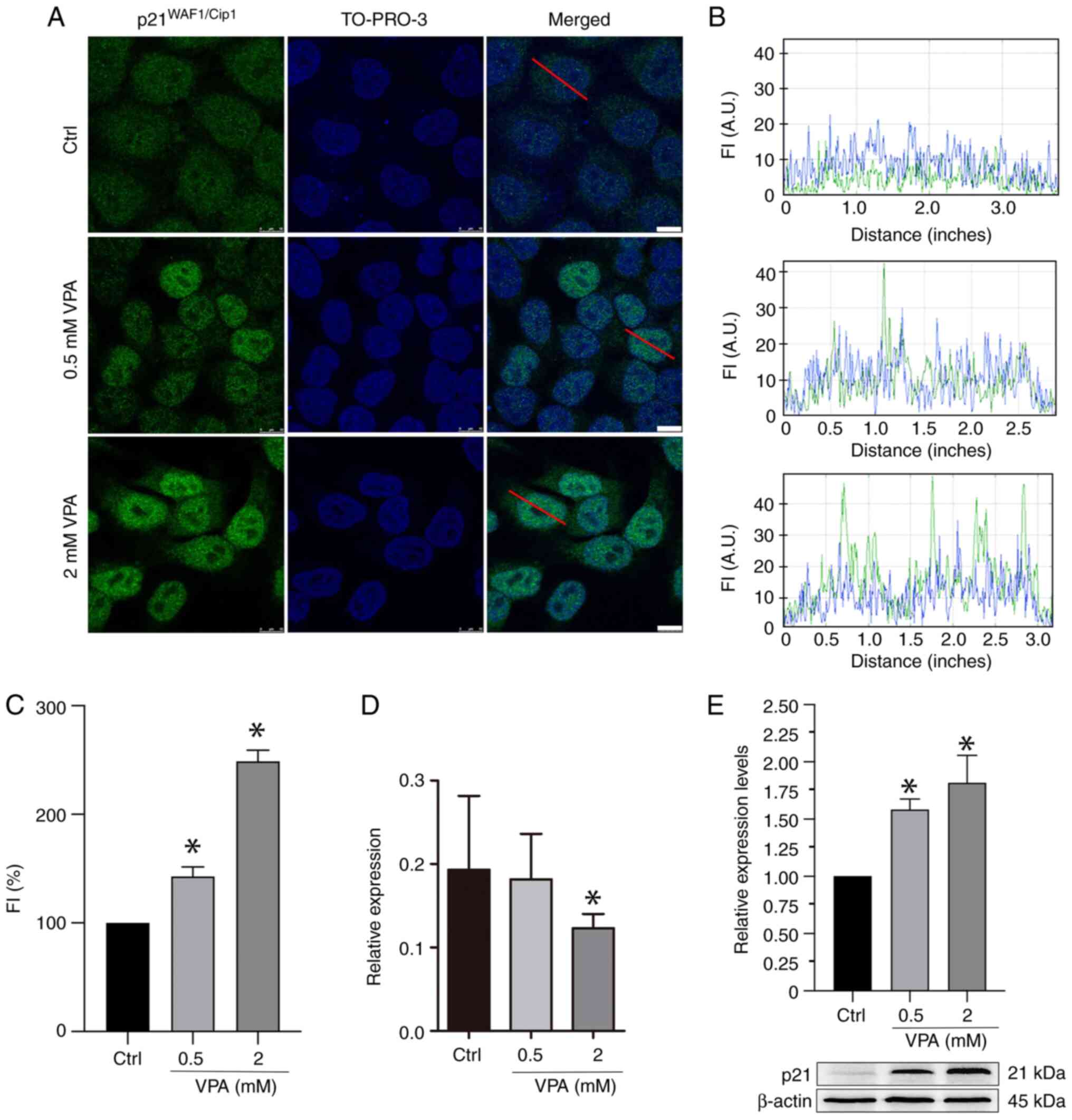
VPA affects p21WAFI/Cip1
protein and gene expression in HeLa cells
Immunofluorescence and WB analyses revealed that VPA
treatment increased p21WAFI/Cip1 protein abundance in a
dose-dependent manner (Fig. 2A-C and
E). When considering the immunofluorescence data, average
increases of ~42 and 148% were detected after treatment with 0.5
and 2 mM VPA, respectively. When considering the WB data, average
increases of ~62 and 88% were detected after treatment with 0.5 and
2 mM VPA, respectively. However, the results of RT-qPCR indicated
that the mRNA expression levels of p21WAFI/Cip1
were reduced by an average of 37% when cells were treated with 2 mM
VPA (Fig. 2D).
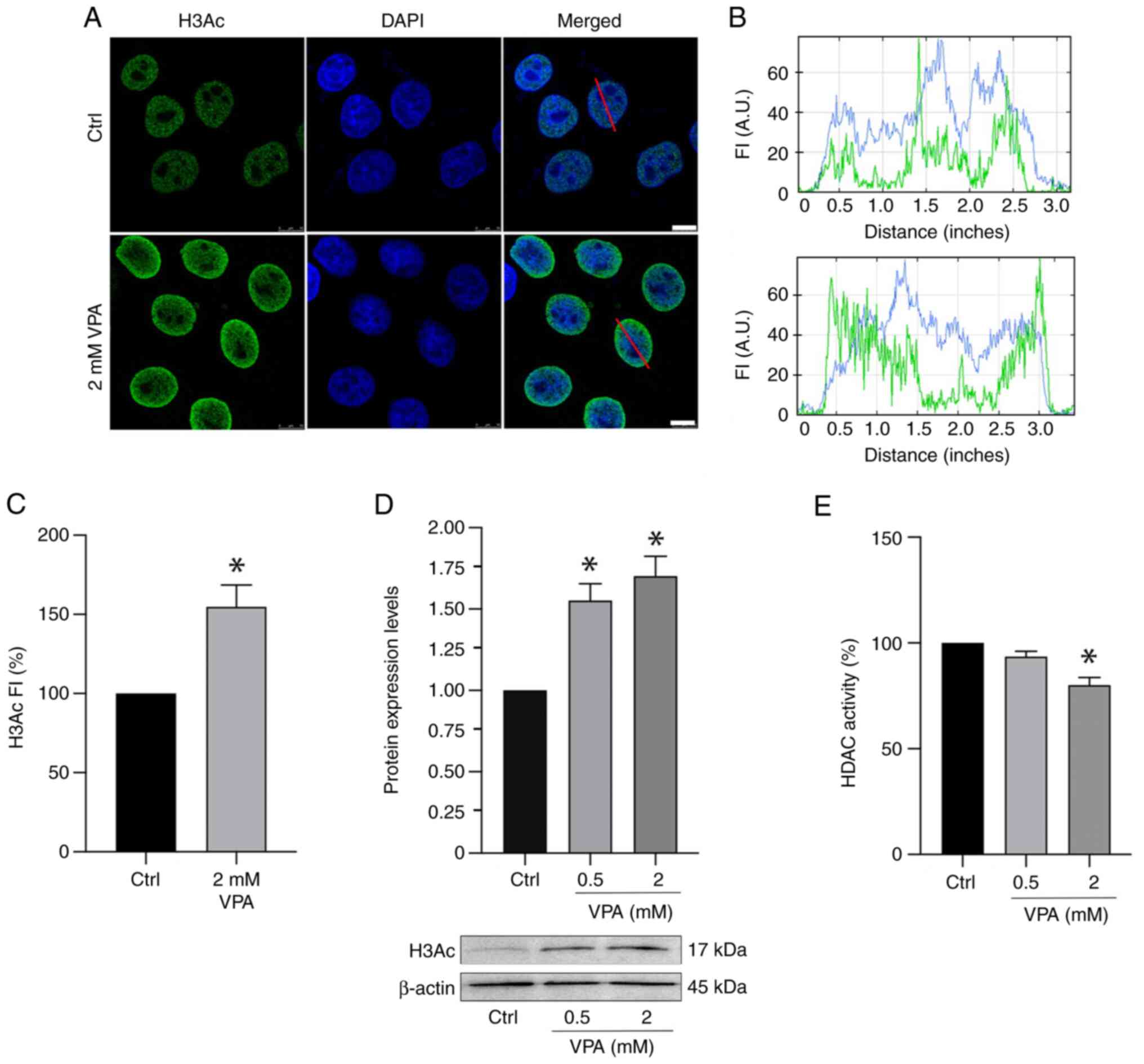
Reduced HDAC activity concomitant with
increased H3Ac is induced in VPA-treated HeLa cells
Immunofluorescence signals for H3Ac intensified ~58%
when HeLa cells were cultured in the presence of 2 mM VPA (Fig. 3A-C). Notably, no effect on H3Ac
nuclear signals was detected in HeLa cells cultured in the presence
of 0.5 mM VPA (data not shown). WB results demonstrated an increase
in H3Ac abundance in a dose-dependent manner in response to VPA
(Fig. 3D), whereas HDAC activity
was significantly inhibited, with an average decrease of 20%
following 2 mM VPA treatment (Fig.
3E).
Discussion
The present results indicated that, in addition to
VPA affecting epigenetic markers by inducing histone acetylation,
DNA demethylation, and histone methylation or demethylation in HeLa
cells (60–64), it may induce suppression of a gene
that acts on oncogenic activity (p16INK4a) and
increase the protein abundance of a tumor suppressor gene
(p21WAFI/Cip1) in these cells, thus contributing
to evidence of the pharmacological potential of VPA.
The present results detected decreased expression
levels of p16INK4a in response to VPA treatment.
Although p16INK4a is often considered a tumor
suppressor gene (4,6), it has been reported to participate in
the oncogenic activity of cervical cancer (5,12,13,65).
It has been demonstrated that silencing p16INK4a
with small interfering RNA can inhibit the proliferation of
cervical tumor cells, causing apoptosis and cell cycle arrest at
the G1 phase (12,18).
In human fibroblasts, p16INK4a levels have been
reported to diminish following exposure to relatively high
concentrations of HDACis, such as Trichostatin A and sodium
butyrate (74).
Further experiments are required to confirm the
effects of VPA on p16INK4a protein expression levels,
since, in the present study, they were shown to decrease in
response to VPA; however, this finding was not statistically
significant. If the present results are not verifiable, and
significantly decreased protein expression levels of
p16INK4a are not demonstrated under the same conditions
as those reported in the present study, or in response to >24 h
treatments or >2 mM VPA concentrations, this may be due to
ineffective p16INK4a protein degradation. Such an event
could result, for instance, from proteasome ineffectiveness, thus
impairing protein degradation. Although proteasomes are abundant in
HeLa cells (75),
ubiquitin/proteasome pathway impairments are currently under focus
in the literature in other cell types, such as U2OS human bone
osteosarcoma cells and 293 cells, and in bacteria (Mycobacterium
tuberculosis) (76,77). A recent study demonstrated that
valproate treatment (5 mM) for 36 h may mediate proteasome
dysfunction, resulting in the accumulation of abnormal
ubiquitinated proteins in Cos-7 and A549 cell lines (78).
Although VPA is known to affect DNA methylation in
HeLa cells (61), previous studies
have identified no association between p16INK4a
DNA methylation and expression of p16INK4a protein
(10,21). The decreased expression of
p16INK4a in response to VPA treatment appears to
be more concerned with changes in the methylation levels of H3K4
and H3K27 (25,26). If H3Ac, which was found to be
increased in VPA-treated HeLa cells, is also involved in this
epigenetic modulation, further experiments involving chromatin
immunoprecipitation (ChIP) assays are required for a better
understanding of such an event. Histone acetylation is generally
associated with chromatin opening and activated gene expression,
although an exception relating inactivation of inducible promoters
enriched for H3K14 acetylation has been reported (30).
The low p21WAFI/Cip1 protein levels in
untreated HeLa cells, as detected by immunofluorescence, were
supported by a previous report on the same cell line (27). These levels were increased following
treatment with VPA for 24 h, as revealed using immunofluorescence
and WB; this finding is consistent with published results obtained
in several tumor cell lines, including HeLa cells, cultured under
different experimental conditions (treatment with 1.2, 4 and 5 mM
VPA for 72 h) (31–34). However, the discordant results
between p21WAFI/Cip1 gene and protein expression
detected in triplicate assays were unexpected, and the mechanism
underlying this difference remains unclear. It may be the case that
treatment for >24 h with >2 mM VPA is required for the
attainment of the expected increase in
p21WAFI/Cip1 gene expression associated with the
increased protein abundance. Upregulation of p21 has been
detected in HeLa cells treated with >2 mM VPA for 48 and 72 h
(68). It may be hypothesized that,
if p21WAFI/Cip1 expression decreases were
sustained in further experiments under the same experimental
conditions as those described in the present study, the drug
treatment initially triggered a decrease in
p21WAFI/Cip1 mRNA expression and that, due to
post-transcriptional regulation, reduced protein degradation or
enhanced protein stabilization influenced by proteasome dysfunction
may have resulted in the accumulation of the
p21WAFI/Cip1 protein, thus causing the discrepancy
between RT-qPCR, and WB and immunofluorescence data (78). Further experiments to provide a
deeper understanding of the present results are thus required.
The p21WAFI/Cip1 gene, which is
responsible for translation of a CDK inhibitor (CKI) that inhibits
cyclin-CDK complexes, is crucial for the control of cell
proliferation mediated by HDACs, which are enzymes that attach to
the gene promoter and negatively regulate its expression (79). When HDAC abundance diminishes in
human hepatocellular carcinoma, the expression of the CKI p21 can
induce cell cycle blockage at G1 phase (80). VPA-inhibited HDAC activity in HeLa
cells is well known (60,61). HDACis, such as VPA, are a class of
promising antitumor agents that, through epigenetic modulation, can
regulate the expression of tumor suppressor genes and genes that
participate in the oncogenic process (12,38,81–84).
Decreased HDAC activity concomitant with increased
H3Ac was observed in the present study. However, because
upregulation of p21WAFI/Cip1 gene expression
could not be detected under the present experimental conditions,
although p21WAFI/Cip1 protein abundance was shown to be
increased in response to VPA treatment, and previous reports have
indicated a G1 phase arrest and no acceleration of
apoptosis under treatment with this drug (61,67,68),
participation of VPA-induced HDAC inhibition in the decreased
expression of the p21WAFI/Cip1 gene could not be
considered. Therefore, based only on the results detected in the
present study, it could not be concluded that VPA-induced global
acetylation of histone H3 exerted a direct effect on the expression
of the p21WAFI/Cip1 gene under the present
experimental conditions.
In conclusion, the present study makes a
significant contribution to the indication that VPA can act as a
multitarget drug. In addition to the well-known effects of VPA
inducing decreased HDAC activity, increased histone acetylation,
and changes in DNA and histone methylation status (60–64),
the present study indicated that this drug may suppress the
p16INK4a gene, which acts on oncogenic activity,
and increase the abundance of the p21WAFI/Cip1 protein,
which is a product of a tumor-suppressing gene in HeLa cells. Given
that these findings provide novel data on the activity of VPA, and
that HDACis have emerged as promising agents in cervical cancer
therapy (84), the present study is
relevant, and may contribute to the fields of cell, molecular and
cervical cancer biology. Since the expression of
p16INK4a and p21WAFI/Cip1 may
be regulated in HeLa cells by HDACis, which are known to affect
epigenetic marks, including histones and non-histone proteins, an
investigation into the effects of VPA directly on the promoters of
these genes would be relevant. Studies involving H3K4me2/me3,
H3K9me2/me3, H3K27me3 and H3Ac levels at the
p16INK4a and p21WAFI/Cip1
promoters, as determined using ChIP assays, may contribute
additional important information to complement the present
results.
Acknowledgements
The authors would like to thank Dr Aline M. dos
Santos for helpful discussions, Mrs. Camila B.M. de Oliveira for
their assistance with cell culture, and Mr. Eli H.M. dos Anjos for
formatting Fig. 1, Fig. 2, Fig.
3 (all Department of Structural and Functional Biology,
Institute of Biology, University of Campinas, São Paulo, Brazil).
The present study has been presented at the 67th Brazilian Congress
of Genetics and was part of the PhD thesis of Marina A. Rocha.
Funding
This work was supported by the Fundação de Amparo à Pesquisa do
Estado de São Paulo (FAPESP, Brazil; grants no. 2015/10356-2 and
2015/16661-1) and Conselho Nacional de Pesquisa e Desenvolvimento
(CNPq, Brazil; grant no. 421299/2018-5). MAR received a PhD
fellowship from Coordenação de Aperfeiçoamento de Pessoal de Nível
Superior (CAPES, Brazil; Finance code 001). ALC received a
postdoctoral fellowship from FAPESP (grant no. 2017/07484-4) and
MLSM received a fellowship from CNPq (grant no. 304797/2019-7). The
funders had no role in study design, data collection and analysis,
decision to publish or preparation of the manuscript.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
MAR and ALC conceived, designed, and performed the
experiments, and confirm the authenticity of all the raw data. MAR,
ALC, CM and MLSM analyzed the data. MLSM and CM contributed the
reagents/materials/analysis tools. MLSM and MAR wrote the original
draft of the manuscript. MLSM revised the manuscript. All authors
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Chen YQ, Cipriano SC, Arenkiel JM and
Miller FR: Tumor suppression by p21WAF1. Cancer Res. 55:4536–4539.
1995.PubMed/NCBI
|
|
2
|
Yang ZY, Perkins ND, Ohno T, Nabel EG and
Nabel GJ: The p21 cyclin-dependent kinase inhibitor suppresses
tumorigenicity in vivo. Nat Med. 1:1052–1056. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim YT, Cho NH, Park SW and Kim JW:
Underexpression of cyclin-dependent kinase (CDK) inhibitors in
cervical carcinoma. Gynecol Oncol. 71:38–45. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim YT and Zhao M: Aberrant cell cycle
regulation in cervical carcinoma. Yonsei Med J. 46:597–613. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huo W, Zhai S, Wang Y, Qiang X, Na R, Gui
H, Wu N, Cao Y and Bai H: Relevance research between the expression
of p16INK4a, Notch1, and hTERC genes: The development of
HPV16-positive cervical cancer. J Clin Lab Anal. 34:e232072020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Medema RH, Herrera RE, Lam F and Weinberg
RA: Growth suppression by p16ink4 requires functional
retinoblastoma protein. Proc Natl Acad Sci USA. 92:6289–6293. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Israels ED and Israels LG: The cell cycle.
Stem Cells. 19:88–91. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pei XH and Xiong Y: Biochemical and
cellular mechanisms of mammalian CDK inhibitors: A few unresolved
issues. Oncogene. 24:2787–2795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nehls K, Vinokurova S, Schmidt D, Kommoss
F, Reuschenbach M, Kisseljov F, Einenkel J, von Knebel Doeberitz M
and Wentzeusen N: p16 methylation does not affect protein
expression in cervical carcinogenesis. Eur J Cancer. 44:2496–2505.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin CK, Liu ST, Chang CC and Huang SM:
Regulatory mechanisms of fluvastatin and lovastatin for the p21
induction in human cervical cancer HeLa cells. PLoS One.
14:e02144082019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li M, Yang J, Liu K, Yang J, Zhan X, Wang
L, Shen X, Chen J and Mao Z: p16 promotes proliferation in cervical
carcinoma cells through CDK6-HuR-IL1A axis. J Cancer. 11:1457–1467.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klaes R, Friedrich T, Spitkovsky D, Ridder
R, Rudy W, Petry U, Dallenbach-Hellweg G, Schmidt D and von Knebel
Doeberitz M: Overexpression of p16(INK4A) as a specific marker for
dysplastic and neoplastic epithelial cells of the cervix uteri. Int
J Cancer. 92:276–284. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van de Putte G, Holm R, Lie AK, Tropé CG
and Kristensen GB: Expression of p27, p21, and p16 protein in early
squamous cervical cancer and its relation to prognosis. Gynecol
Oncol. 89:140–147. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Volgareva G, Zavalishina L, Andreeva Y,
Frank G, Krutikova E, Golovina D, Bliev A, Spitkovsky D, Ermilova V
and Kisseljov F: Protein p16 as a marker of dysplastic and
neoplastic alterations in cervical epithelial cells. BMC Cancer.
4:582004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bahnassy AA, Zekri AR, Alam El-Din HM,
Aboubakr AA, Kamel K, El-Sabah MT and Mokhtar NM: The role of
cyclins and cyclins inhibitors in the multistep process of
HPV-associated cervical carcinoma. J Egypt Natl Cancer Inst.
18:292–302. 2006.PubMed/NCBI
|
|
17
|
Yoruker EE, Mert U, Bugra D, Yamaner S and
Dalay N: Promoter and histone methylation and p16(INK4A) gene
expression in colon cancer. Exp Ther Med. 4:865–870. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang CY, Bao W and Wang LH:
Downregulation of p16(ink4a) inhibits cell proliferation and
induces G1 cell cycle arrest in cervical cancer cells. Int J Mol
Med. 33:1577–1585. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu H, Zhang J and Shi H: Expression of
cancer stem markers could be influenced by silencing of p16 gene in
HeLa cervical carcinoma cells. Eur J Gynaecol Oncol. 37:221–225.
2016.PubMed/NCBI
|
|
20
|
Merlo A, Herman JG, Mao L, Lee DJ,
Gabrielson E, Burger PC, Baylin SB and Sidransky D: 5′ CpG island
methylation is associated with transcriptional silencing of the
tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med.
1:686–692. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lin Z, Gao M, Zhang X, Kim YS, Lee ES, Kim
HK and Kim I: The hypermethylation and protein expression of p16
INK4A and DNA repair gene O6-methylguanine-DNA
methyltransferase in various uterine cervical lesions. J Cancer Res
Clin Oncol. 131:364–370. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beyer S, Zhu J, Mayr D, Kuhn C, Schulze S,
Hofmann S, Dannecker C, Jeschke U and Kost BP: Histone H3 acetyl K9
and histone H3 tri methyl K4 as prognostic markers for patients
with cervical cancer. Int J Mol Sci. 18:4772017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santos-Rosa H, Schneider R, Bannister AJ,
Sherriff J, Bernstein BE, Tolga Emre NC, Schreiber SL, Mellor J and
Kouzarides T: Active genes are tri-methylated at K4 of histone H3.
Nature. 419:407–411. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cai Y, Zhang Y, Loh YP, Tng JQ, Lim MC,
Cao Z, Raju A, Aiden EL, Li S, Manikandan L, et al: H3K27me3-rich
genomic regions can function as silencers to repress gene
expression via chromatin interactions. Nature Commun. 12:7192021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
McLaughlin-Drubin ME, Crum CP and Münger
K: Human papillomavirus E7 oncoprotein induces KDM6A and KDM6B
histone demethylase expression and causes epigenetic reprogramming.
Proc Natl Acad Sci USA. 108:2130–2135. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McLaughlin-Drubin ME, Park D and Munger K:
Tumor suppressor p16INK4A is necessary for survival of cervical
carcinoma cell lines. Proc Natl Acad Sci USA. 110:16175–16180.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yokoyama Y, Takahashi Y, Morishita S,
Hashimoto M and Tamaya T: Introduction of p21(Waf1/Cip1) gene into
a carcinoma cell line of the uterine cervix with inactivated p53.
Cancer Lett. 116:233–239. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fang JY and Lu YY: Effects of histone
acetylation and DNA methylation on p21(WAF1) regulation. World J
Gastroenterol. 8:400–405. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen YX, Fang JY, Lu R and Qiu DK:
Expression of p21(WAF1) is related to acetylation of histone H3 in
total chromatin in human colorectal cancer. World J Gastroenterol.
13:2209–2213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Karmodiya K, Krebs AR, Oulad-Abdelghani M,
Kimura H and Tora L: H3K9 and H3K14 acetylation co-occur at many
gene regulatory elements, while H3K14ac marks a subset of inactive
inducible promoters in mouse embryonic stem cells. BMC Genomics.
13:4242012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sami S, Höti N, Xu HM, Shen Z and Huang X:
Valproic acid inhibits the growth of cervical cancer both in vitro
and in vivo. J Biochem. 144:357–362. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tsai C, Leslie JS, Franko-Tobin LG,
Prasnal MC, Yang T, Vienna Mackey L, Fuselier JA, Coy DH, Liu M, Yu
C and Sun L: Valproic acid suppresses cervical cancer tumor
progression possibly via activating Notch1 signaling and enhances
receptor-targeted cancer chemotherapeutic via activating
somatostatin receptor type II. Arch Gynecol Obstetr. 288:393–400.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mawatari T, Ninomiya I, Inokuchi M, Harada
S, Hayashi H, Oyama K, Makino I, Nakagawara H, Miyashita T, Tajima
H, et al: Valproic acid inhibits proliferation of HER2-expressing
breast cancer cells by inducing cell cycle arrest and apoptosis
through Hsp70 acetylation. Int J Oncol. 47:2073–2081. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lipska K, Filip A and Gumieniczek A: The
impact of chlorambucil and valproic acid on cell viability,
apoptosis, and expression of p21, HDM2, BCL2 and MCL1
genes in chronic lymphocytic leukemia. Cells. 10:10882021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luna-Palencia GR, Correa-Basurto J,
Trujillo-Ferrara J, Meraz-Ríos MA and Vásquez-Moctezuma I:
Epigenetic evaluation of N-(2-hydroxyphenyl)-2-propylpentanamide, a
valproic acid aryl derivative with activity against HeLa cells.
Curr Mol Pharmacol. 14:570–578. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Richon VM, Sandhoff TW, Rifkind RA and
Marks PA: Histone deacetylase inhibitor selectively induces p21WAF1
expression and gene-associated histone acetylation. Proc Natl Acad
Sci USA. 97:10014–10019. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Minucci S and Pelicci PG: Histone
deacetylase inhibitors and the promise of epigenetic (and more)
treatments for cancer. Nat Rev Cancer. 6:38–51. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lin YC, Lin JH, Chou CW, Chang YF, Yeh SH
and Chen CC: Statins increase p21 through inhibition of histone
deacetylase activity and release of promoter-associated HDAC1/2.
Cancer Res. 68:2375–2383. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee S, Park JR, Seo MS, Roh KH, Park SB,
Hwang JW, Sun B, Seo K, Lee YS, Kang SK, et al: Histone deacetylase
inhibitors decrease proliferation potential and multilineage
differentiation capability of human mesenchymal stem cells. Cell
Prolif. 42:711–720. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Aizawa S and Yamamuro Y: Valproate
administration to mice increases hippocampal p21 expression by
altering genomic DNA methylation. Neuroreport. 26:915–920. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Guo Q, Li X, Han H, Li C, Liu S, Gao W and
Sun G: Histone lysine methylation in TGF-β1 mediated p21 gene
expression in rat mesangial cells. Biomed Res Int.
2016:69272342016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li X, Li C, Li X, Cui P, Li Q, Guo Q, Han
H, Liu S and Sun G: Involvement of histone lysine methylation in
p21 gene expression in rat kidney in vivo and rat mesangial cells
in vitro under diabetic conditions. J Diabetes Res.
2016:38532422016. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Göttlicher M, Minucci S, Zhu P, Krämer OH,
Schimpf A, Giavara S, Sleeman JP, Lo Coco F, Nervi C, Pelicci PG
and Heinzel T: Valproic acid defines a novel class of HDAC
inhibitors inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Phiel CJ, Zhang F, Huang EY, Guenther MG,
Lazar MA and Klein PS: Histone deacetylase is a direct target of
valproic acid, a potent anticonvulsant, mood stabilizer, and
teratogen. J Biol Chem. 276:36734–36741. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Peterson GM and Naunton M: Valproate: A
simple chemical with so much to offer. J Clin Pharm Therap.
30:417–421. 2005. View Article : Google Scholar
|
|
46
|
Terbach N and Williams RSB:
Structure-function studies for the panacea, valproic acid. Biochem
Soc Trans. 37:1126–1132. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tomson T, Battino D and Perucca E:
Valproic acid after five decades of use in epilepsy: Time to
reconsider the indications of a time-honoured drug. Lancet Neurol.
15:210–218. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Makarević J, Rutz J, Juengel E, Maxeiner
S, Tsaur I, Chun FKH, Bereiter-Hahn J and Blaheta RA: Influence of
the HDAC inhibitor valproic acid on the growth and proliferation of
temsirolimus-resistant prostate cancer cells in vitro. Cancers
(Basel). 11:5662019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Romoli M, Mazzocchetti P, D'Alonzo R,
Siliquini S, Rinaldi VE, Verrotti A, Calabresi P and Costa C:
Valproic acid and epilepsy: From molecular mechanisms to clinical
evidences. Curr Neuropharmacol. 17:926–946. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang Y, Zhang Y, Li M, Meng F, Yu Z, Chen
Y and Cui G: Combination of SB431542, CHIR99021 and PD0325901 has a
synergic effect on abrogating valproic acid-induced
epithelial-mesenchymal transition and stemness in HeLa, 5637 and
SCC-15 cells. Oncol Rep. 41:3545–3554. 2019.PubMed/NCBI
|
|
51
|
Han W, Yu F, Wang R, Guan W and Zhi F:
Valproic acid sensitizes glioma cells to luteolin through induction
of apoptosis and autophagy via Akt signaling. Cell Mol Neurobiol.
41:1625–1634. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Johannessen CU and Johannessen SI:
Valproate: Past, present, and future. CNS Drug Rev. 9:199–216.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Chateauvieux S, Morceau F, Dicato M and
Diederich M: Molecular and therapeutic potential and toxicity of
valproic acid. J Biomed Biotechnol. 2010:4793642010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mello MLS: Sodium valproate-induced
chromatin remodeling. Front Cell Dev Biol. 9:6455182021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sargolzaei J, Rabbani-Chadegani A, Mollaei
H and Deezagi A: Spectroscopic analysis of the interaction of
valproic acid with histone H1 in solution and in chromatin
structure. Int J Biol Macromol. 99:427–432. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
de Campos Vidal B and Mello MLS: Sodium
valproate (VPA) interactions with DNA and histones. Int J Biol
Macromol. 163:219–231. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Baumann C, Zhang X, Zhu L, Fan Y and De La
Fuente R: Changes in chromatin accessibility landscape and histone
H3 core acetylation during valproic acid-induced differentiation of
embryonic stem cells. Epigenetics Chromatin. 14:582021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Vidal BC and Mello MLS: Data on FTIR
spectra of mixtures of sodium valproate (VPA) and histones H1 and
H3. Latin Amer Data Sci. 1:102–109. 2022. View Article : Google Scholar
|
|
59
|
Gurvich N, Tsygankova OM, Meinkoth JL and
Klein PS: Histone deacetylase is a target of valproic acid-mediated
cellular differentiation. Cancer Res. 64:1079–1086. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dejligbjerg M, Grauslund M, Litman T,
Collins L, Qian X, Jeffers M, Lichenstein H, Jensen PB and Sehested
M: Differential effects of class I isoform histone deacetylase
depletion and enzymatic inhibition by belinostat or valproic acid
in HeLa cells. Mol Cancer. 7:702008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Felisbino MB, Tamashiro WMSC and Mello
MLS: Chromatin remodeling, cell proliferation and cell death in
valproic acid-treated HeLa cells. PLoS One. 6:e291442011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Veronezi GMB, Felisbino MB, Gatti MSV,
Mello MLS and Vidal BC: DNA methylation changes in valproic
acid-treated HeLa cells as assessed by image analysis,
immunofluorescence and vibrational microspectroscopy. PLoS One.
12:e01707402017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Rocha MA, Veronezi GMB, Felisbino MB,
Gatti MSV, Tamashiro WMSC and Mello MLS: Sodium valproate and
5-aza-2′-deoxycytidine differentially modulate DNA demethylation in
G1 phase-arrested and proliferative HeLa cells. Sci Rep.
9:182362019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Rocha MA, Vidal BC and Mello MLS: Sodium
valproate modulates the methylation status of lysine residues 4, 9
and 27 in histone H3 of HeLa cells. Curr Mol Pharmacol. 16:197–210.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Tringler B, Gup CJ, Singh M, Groshong S,
Shroyer AL, Heinz DE and Shroyer KR: Evaluation of p16INK4a and pRb
expression in cervical squamous and glandular neoplasia. Hum
Pathol. 35:689–696. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Rocha MA, Oliveira CBM and Mello MLS:
Sodium valproate cytotoxicity effects as assessed by the MTT assay.
Repositório de Dados de Pesquisa da Unicamp; version 2, . 2021
|
|
67
|
Han BR, You BR and Park WH: Valproic acid
inhibits the growth of HeLa cervical cancer cells via
caspase-dependent apoptosis. Oncol Rep. 30:2999–3005. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hashemi N, Zoshk MY, Rahidian A, Laripour
R, Fasihi H, Hami Z and Chamanara M: Anti-proliferative and
apoptotic effects of valproic acid on HeLa cells. Int J Cancer
Manag. 15:e1202242022. View Article : Google Scholar
|
|
69
|
Kondo Y, Shen L and Issa JPJ: Critical
role of histone methylation in tumor suppressor gene silencing in
colorectal cancer. Mol Cell Biol. 23:206–215. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sanmukh SG, Dos Santos NJ, Barquilha CN,
Cucielo MS, de Carvalho M, Dos Reis PP, Delella FK, Carvalho HF and
Felisbino SL: Bacteriophages M13 and T4 increase the expression of
anchorage-dependent survival pathway genes and down regulate
androgen receptor expression in LNCaP prostate cell line. Viruses.
13:17542021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Muller PY, Janovjak H, Miserez AR and
Dobbie Z: Processing of gene expression data generated by
quantitative real-time RT-PCR. Biotechniques. 32:1372–1374.
13761378–1379. 2002.PubMed/NCBI
|
|
72
|
Simon P: Q-Gene: Processing quantitative
real-time RT-PCR data. Bioinformatics. 19:1439–1440. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Matheu A, Klatt P and Serrano M:
Regulation of the INK4a/ARF locus by histone deacetylase
inhibitors. J Biol Chem. 280:42433–42441. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yewdell JW: Not such a dismal science: The
economics of protein synthesis, folding, degradation and antigen
processing. Trends Cell Biol. 11:294–297. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Sun Y, Chen J, Huang SYN, Su YP, Wang W,
Agama K, Saha S, Jenkins LM, Pascal JM and Pommier Y: PARylation
prevents the proteasomal degradation of topoisomerase I DNA-protein
crosslinks and induces their deubiquitylation. Nat Commun.
12:50102021. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Block MF, Delley CL, Keller LML,
Stuehlinger TT and Weber-Ban E: Electrostatic interactions guide
substrate recognition of the prokaryotic ubiquitin-like protein
ligase PafA. Nat Commun. 14:52662023. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kinger S, Jagtap YA, Dubey AR, Kumar P,
Choudhary A, Karmakar S, Lal G, Prajapti VK, Jha HC, Gutti RK and
Mishra A: Valproate mediated proteasome dysfunctions induce
apoptosis. Adv Therap. 23004212024. View Article : Google Scholar
|
|
79
|
Zupkovitz G, Grausenburger R, Brunmeir R,
Senese S, Tischler J, Jurkin J, Rembold M, Meunier D, Egger G,
Lagger S, et al: The cyclin-dependent kinase inhibitor p21 is a
crucial target for histone deacetylase 1 as a regulator of cellular
proliferation. Mol Cell Biol. 30:1171–1181. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Fan J, Lou B, Chen W, Zhang J, Lin S, Lv
FF and Chen Y: Down-regulation of HDAC5 inhibits growth of human
hepatocellular carcinoma by induction of apoptosis and cell cycle
arrest. Tumor Biol. 35:11523–11532. 2014. View Article : Google Scholar
|
|
81
|
Chun SM, Lee JY, Choi J, Lee JH, Hwang JJ,
Kim CS, Suh YA and Jang SJ: Epigenetic modulation with HDAC
inhibitor CG200745 induces anti-proliferation in non-small cell
lung cancer cells. PLoS One. 10:e01193792015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Han JW, Ahn SH, Park SH, Wang SY, Bae GU,
Seo DW, Kwon HK, Hong S, Lee HY, Lee YW and Lee HW: Apicidin, a
histone deacetylase inhibitor, inhibits proliferation of tumor
cells via induction of p21WAF1/Cip1 and gelsolin. Cancer Res.
60:6068–6074. 2000.PubMed/NCBI
|
|
83
|
Kim YB, Ki SW, Yoshida M and Horinouchi S:
Mechanism of cell cycle arrest caused by histone deacetylase
inhibitors in human carcinoma cells. J Antibiot (Tokyo).
53:1191–1200. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Psilopatis I, Garmpis N, Garmpi A, Vrettou
K, Sarantis P, Koustas E, Antoniou EA, Dimitroulis D, Kourakis G,
Karamouzis MV, et al: The emerging role of histone deacetylases
inhibitors in cervical cancer therapy. Cancers (Basel).
15:22222023. View Article : Google Scholar : PubMed/NCBI
|