Introduction
Colorectal cancer (CRC), which accounts for ~10% of
all cancer cases, ranks second as a global cause of cancer-related
fatalities (1). In China, CRC ranks
third among malignant tumors and its occurrence is steadily rising,
particularly among younger individuals (2). It is estimated that by 2030, the
number of CRC cases under 50 years of age will account for 11% of
the total number of colon cancer and 23% of the total number of
rectal cancer cases (3). The
advanced stage of most diagnosed cases of CRC can be attributed to
the absence of early screening (4).
Starting from the 1950s, significant changes in lifestyle factors,
such as antibiotics consumption, decreased physical activity and
increased obesity, have had an impact on the gut microbiome,
potentially playing a significant role in the occurrence of CRC at
a younger age (5,6). A molecular biology perspective
highlights the significant involvement of colon epithelial
proto-oncogene mutation, tumor suppressor gene inactivation and
genome epigenetic modification in the initiation and progression of
CRC (7).
In the development of CRC, the inactivation of the
adenomatous polyposis coli (APC) gene, a crucial tumor suppressor
gene, is regarded as an initial and significant step (8). The APC gene spans 8,535 nucleotides
and is situated on chromosome 5q21-q22. It comprises 21 exons
(9). The protein encoded by the APC
gene is a large 310-kDa molecule, made up of 2,843 amino acids. A
noticeable portion of the genetic code, ~75%, is found in exon 15,
which is also the most frequently affected area for mutations in
the APC gene. When there are mutations in the germ cells of the APC
gene, it leads to familial adenomatous polyposis (FAP), a
remarkable genetic predisposition to the development of CRC
(10). More than 80% of sporadic
CRC cases were discovered to have somatic APC gene mutations. APC
is a versatile protein with multiple functions facilitated by
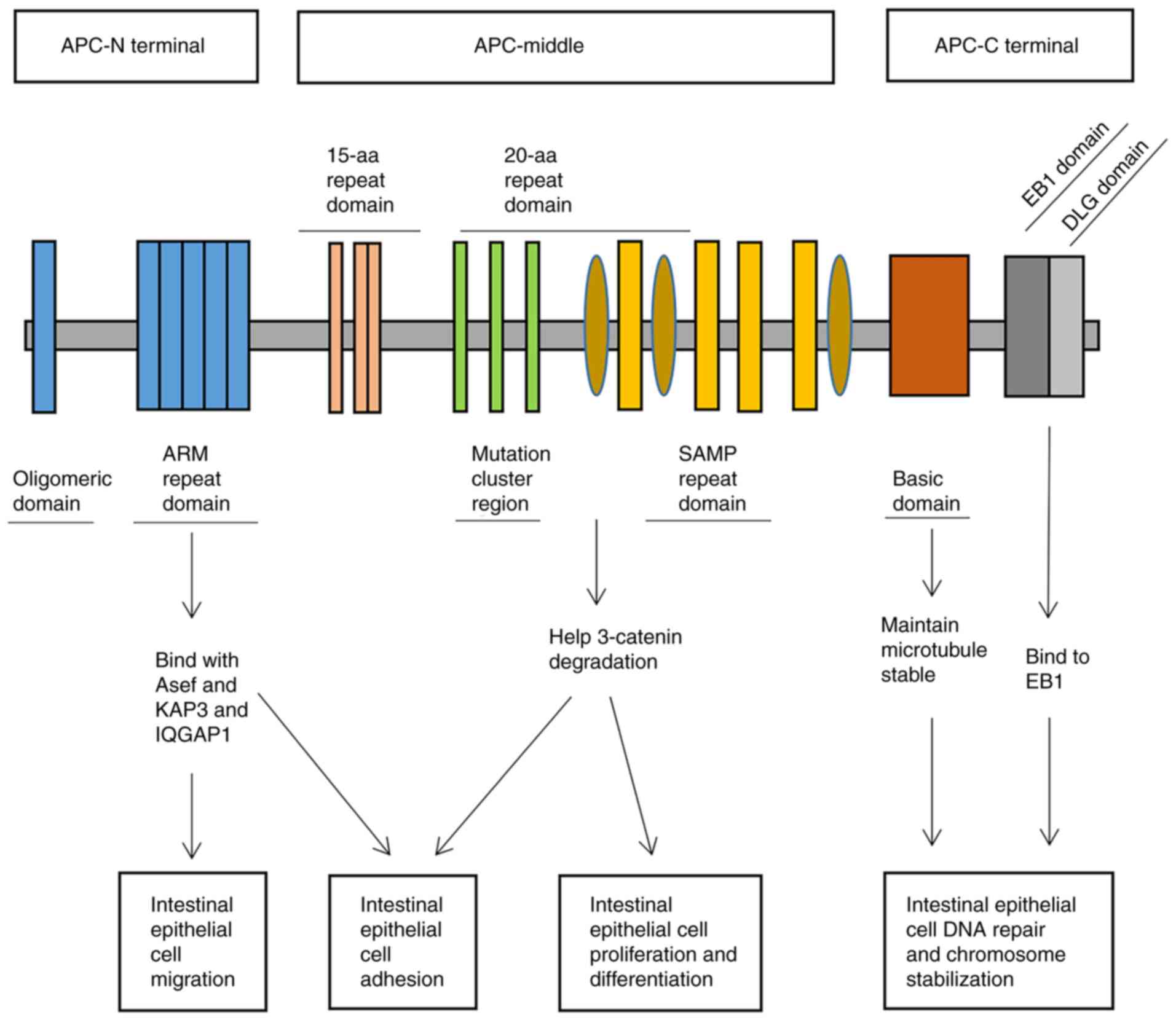
various binding partners. The APC structure extends from its
N-terminus to its C-terminus and includes an oligomerization
region, an armadillo (ARM) repeat region, a domain with repeats of
either 15 or 20 residues, a Ser-Ala-Met-Pro (SAMP) repeat domain, a
basic domain and a domain that interacts with end-binding protein 1
(EB1) and Discs large homolog (DLG) (Fig. 1) (11,12).
Experimental evidence has illustrated that the binding site for APC
mutants is the oligomerization domain. APC mutant proteins that
retain at least the initial 171 amino acids can interact with this
region, potentially leading to a dominant negative effect (13). The most highly conserved domain of
APC proteins, known as the ARM repeat domain, has been proven to
have the ability to bind with various proteins, including IQ motif
containing GTPase activating protein 1, protein phosphatase type 2A
and asef and kinesin-associated protein 3 (KAP3), which all contain
IQ motifs and are associated with GTP enzyme activation.
These interactions noticeably affect the stimulation
of cell migration and adhesion (14,15).
The 15- or 20-residue repeat domain and the SAMP repeat sequence
have a crucial function in negatively regulating the canonical Wnt
signaling pathway by promoting the proteasome degradation of
β-catenin (16,17). By interacting with EB1, the basic
and C-terminal domains have the ability to directly or indirectly
bind to microtubules, thereby preserving microtubule stability,
ensuring proper kinetochore function and facilitating chromosome
segregation (18). APC is involved
in a wide range of cellular activities, such as cell growth,
programmed cell death, movement, cell attachment, DNA fixing and
separation of chromosomes. It achieves these functions by
interacting with different proteins. Individuals with CRC have
shorter versions of the APC protein that are missing specific
regions needed for attaching to microtubules, EB1 and β-catenin.
This deficiency causes an unstable genetic structure, leading to
increased cell growth and decreased differentiation (19,20). A
notable finding indicated that a remarkable percentage (73%) of
metastatic CRC cases had an accumulation of APC mutations, the
majority of which were truncated mutations. It was revealed that
patients with N-terminal APC mutations had a notably lower tumor
mutational burden than those with C-terminal mutations. In
addition, patients with N-terminal APC mutations exhibited
prolonged overall survival in comparison to those with C-terminal
mutations. Further analysis of tumor gene pathways demonstrated
that patients with C-terminal mutations had markedly higher
frequency of gene mutations in the RTK/RAS, Wnt and TGF-β signaling
pathways than patients with N-terminal mutations. In addition, the
presence of KRAS proto-oncogene, GTPase, APC membrane recruitment
protein 1, transforming growth factor (TGF)-β receptor type 2 and
AT-rich interactive domain-containing protein 1A-driven mutations
were found to be higher among patients with C-terminal APC
mutations. Consequently, the truncation of the APC gene's
C-terminal region, leading to the loss of its tumor suppressor
function, may have a crucial role in CRC development (21). However, there have been preclinical
animal studies demonstrating that the C-terminus of APC does not
influence the formation or advancement of intestinal adenomas.
Scholars developed two sets of mouse models by genetically altering
them. In the first set, they deleted the SAMP repeat sequence while
keeping the C-terminus intact. In the second set, they removed the
entire C-terminus while maintaining the other domains.
Surprisingly, both sets of mice developed numerous intestinal
adenomas, which were similar in terms of quantity, placement and
abnormal cell growth. Strikingly, no signs of cancer were detected
in either set of mice. Although the tumors in mice showed a
comparable disruption of the Wnt signaling pathway, there was no
indication that the C-terminus had any functional differences. This
includes aspects such as cell migration, chromosomal instability
(CIN) or the localization of APC and EB1 (22). The Wnt/β-catenin signaling pathway,
as detailed by Nusse and Clevers (23) and Klaus and Birchmeier (24), plays a crucial role in regulating
cell fate, proliferation, migration and polarity. This pathway is
activated when Wnt proteins bind to Frizzled receptors and
low-density lipoprotein-related receptors 5 and 6 co-receptors,
leading to the inhibition of the β-catenin destruction complex
[comprising APC, Axin, glycogen synthase kinase 3β (GSK-3β) and
casein kinase 1 (CK1)]. Consequently, β-catenin accumulates in the
cytoplasm and translocates to the nucleus, where it interacts with
transcription factor (TCF)/lymphoid enhancer-binding factor (LEF)
transcription factors to regulate target gene expression. This
signaling is essential for embryonic development, tissue
homeostasis and stem cell renewal. However, aberrant activation of
the Wnt/β-catenin pathway, often due to mutations in APC or
β-catenin, is a key driver in the development of various cancers,
including CRC. While the Hippo, Notch and Hedgehog pathways play
significant roles in CRC progression, their relationship with APC
truncations is less direct compared to the well-characterized
impact of APC mutations on Wnt/β-catenin signaling (19). APC truncations primarily dysregulate
Wnt signaling, driving tumorigenesis. Although dysregulation of the
Hippo, Notch and Hedgehog pathways can contribute to CRC through
various mechanisms, current evidence does not directly link APC
truncations to the modulation of these pathways (20). Instead, their cooperative
interaction with Wnt signaling, which is altered by APC
truncations, may exacerbate cancer progression, with each pathway
contributing to cellular proliferation, differentiation and
metastasis in distinct manners (21).
Despite extensive research on APC's involvement in
CRC, recent studies continued to reveal new dimensions of its role,
emphasizing its ongoing relevance in the field of oncology.
Emerging evidence highlights the intricate interactions of APC with
various signaling pathways, its impact on CIN and its influence on
the tumor microenvironment, all of which are crucial for developing
novel therapeutic strategies. Given the extensive coverage of the
canonical Wnt/β-catenin signaling pathway in the prior literature,
the present review did not concentrate on this aspect. This review
emphasized recent preclinical findings, particularly new insights
from animal models that challenge previous assumptions about the
C-terminal region of APC gene and its role in CRC formation and
progression, as well as emerging evidence on the therapeutic
implications of specific APC mutations. This review concentrated on
the roles and implications of APC mutations beyond the
well-established Wnt/β-catenin signaling pathway. While the
canonical role of APC in the Wnt/β-catenin signaling pathway has
been extensively studied, this review uniquely explored the
multifaceted impact of APC truncation across various cellular
processes, including cell adhesion, migration, apoptosis, DNA
repair and CIN. By integrating these diverse pathways, this review
aimed to provide a holistic view of APC's contribution to CRC
progression and identify novel therapeutic targets. Specifically,
it highlighted the differential impact of N-terminal and C-terminal
truncations of the APC gene on tumor mutational burden, overall
survival and associated genetic mutations in patients with CRC. In
addition, it explored the non-canonical roles of APC, including its
interactions with various signaling pathways, such as RTK/RAS and
TGF-β, its role in maintaining chromosomal stability and
microtubule dynamics and its involvement in cell adhesion.
APC truncation and inactivation of the
canonical Wnt signaling pathway
One of the most prevalent pathways mutated in cancer
is the Wnt/β-catenin signaling pathway, which has a pivotal role in
coordinating crucial processes during early embryonic development,
tissue stability and regeneration, as well as governing the
maintenance of stem cells, determination of cell fate and
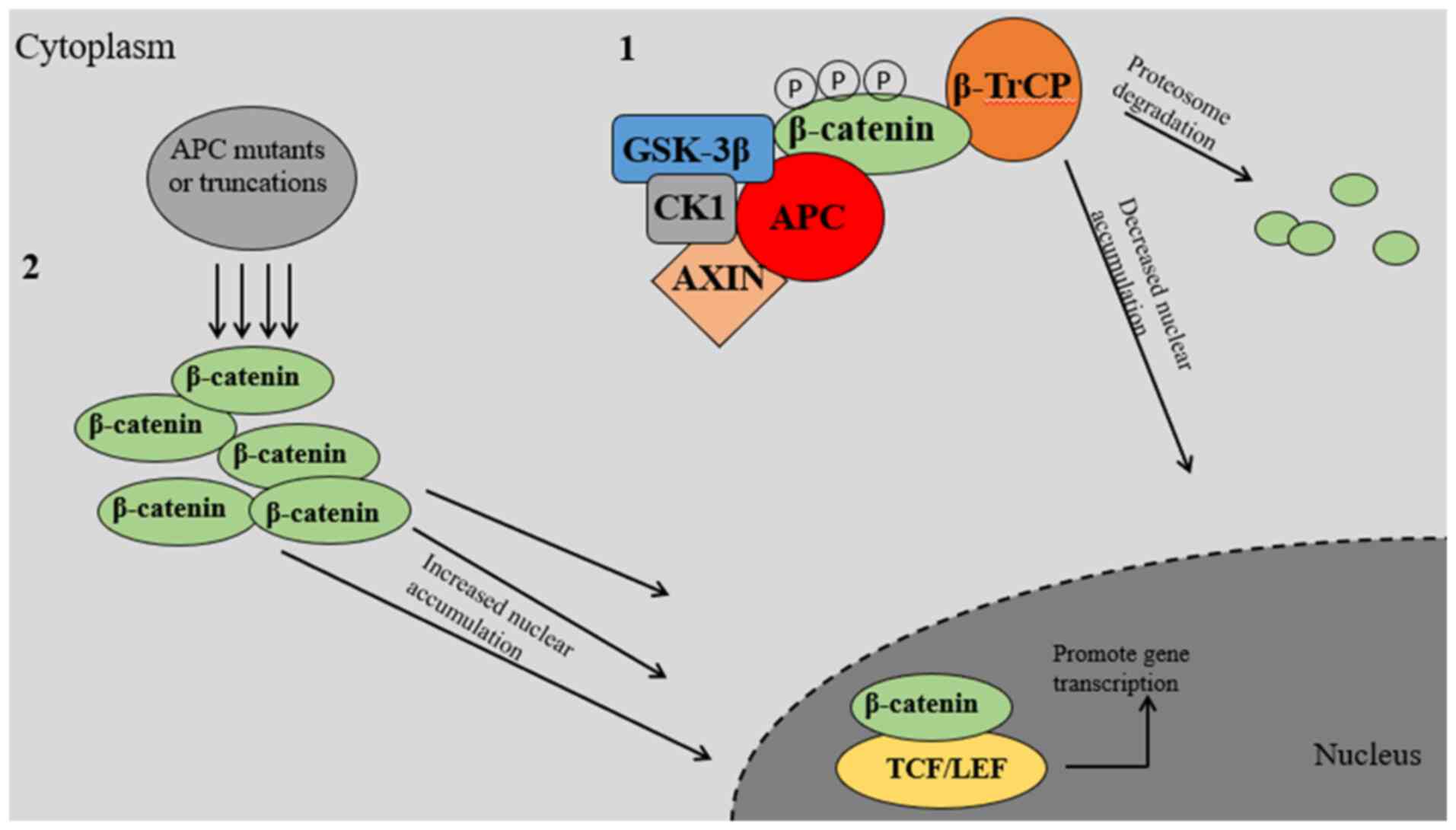
regulation of cell proliferation (23). The APC protein plays a crucial role
in controlling cell proliferation and differentiation in the
gastrointestinal tract by acting as a significant inhibitor of the
canonical Wnt signaling pathway (Fig.
2) (25,26). To achieve this, cytoplasmic
complexes involving β-catenin, APC and GSK-3β are necessary to
facilitate serine phosphorylation and consequent degradation of
β-catenin (27). GSK-3β, a kinase,
is responsible for phosphorylating β-catenin and APC. The
cooperation between the kinase and the substrate heavily relies on
the presence of the APC protein. The formation of the complex
β-catenin-GSK-3β-APC involves another protein called Axin (28). Under normal circumstances, the Wnt
signaling pathway promotes the degradation of β-catenin by
inhibiting GSK-3β activity. However, under pathological conditions,
mutations in APC hinder β-catenin degradation (29,30).
APC promotes AXIN1 multimerization and stabilizes the AXIN1
complex, thus increasing the efficiency of this disruption
mechanism (31). In the canonical
Wnt/β-catenin signaling pathway, the cytoplasmic protein β-catenin
serves as a crucial switch. Its stability and level of activation
are regulated by both the β-catenin disruption complex and the Wnt
growth factor receptor. Within this intricate process, AXIN1, a
protein known for suppressing tumor growth, functions as a scaffold
by interacting with various molecules, such as β-catenin, APC and
two serine-threonine kinases (CK1α/δ and GSK-3α/β). This
interaction leads to the labeling of β-catenin for degradation by
the proteasome through a process called phosphorylation-dependent
ubiquitination (31,32). Furthermore, the involvement of the
Wnt growth factor receptor is crucial in the transformation of the
disruption complex into the receptor-associated Wnt complex. In
this scenario, the phosphorylation and ubiquitination of β-catenin
are significantly diminished, leading to elevated levels of
β-catenin and its accumulation in the nucleus. This, in turn,
enhances the activation of β-catenin/TCF/LEF target genes. Within
the disruption complex, the scaffold protein and tumor suppressor
AXIN1 collaborate with APC to enlist casein kinase 1 (CK1) and
GSK-3β, which ultimately affect the substrate β-catenin (33–35).
AXIN1, a pivotal scaffold for the disruption complex, possesses the
remarkable ability to directly associate and assemble all the
constituent elements of the core disruptive complex. Furthermore,
it markedly enhances the phosphorylation of β-catenin by impeding
the phosphorylation of other molecules that via with β-catenin to a
certain degree (36). APC, the
second scaffold of the disruption complex, has the ability to bind
to up to 10 β-catenin sites, some of which are controlled by kinase
phosphorylation of the disruption complex. In addition, APC
contains three docking motifs for AXIN1. Within a sequence of amino
acid repeats binding β-catenin, the APC protein also includes three
motifs for binding AXIN1 (37). The
function of the β-catenin disruption complex heavily relies on the
essential role of APC. When APC truncation mutations occur in
intestinal stem cells, these mutations cause the loss of different
motifs to varying degrees. As a result, the levels of β-catenin
increase significantly, which has been linked to the occurrence and
development of up to 80% of CRC cases.
The APC gene in colonic adenomas and CRCs is
shortened, leading to the impairment of the β-catenin disruption
complex. This results in the accumulation of active β-catenin in
the cytoplasm and nucleus, where it can form complexes with TCF/LEF
family DNA-binding proteins. As a result, β-catenin acts as a
co-activator for TCF, assisting in transcriptional processes.
However, in APC-mutant tumor cells, the regulatory proteins lose
their ability to inhibit the Wnt signaling pathway, as they are
either upstream or at the level of the APC protein in the pathway.
The APC mutation causes an increase in the activation of
β-catenin/TCF transcriptional activity by enhancing the levels of
nuclear β-catenin and reducing the inhibitory effects of
C-terminal-binding protein on the repressive complex. This
activation results in the elevation of cyclin D1 and Myc, which
play crucial roles in promoting cell proliferation, apoptosis and
cell cycle progression, thus driving the development of tumors
(38). The strong selectivity of
the retention of the initial 20 amino acid repeat sequences of APC,
which can bind to β-catenin and control its transcriptional
activity, is strikingly favored. The selection of the APC genotype
was specifically aimed at achieving a particular level of
β-catenin, which is highly favorable for the development of tumors.
However, in the absence of any binding sites between APC and
β-catenin, the continuous activation of the β-catenin pathway may
result in extensive alterations in gene regulation, consequently
raising the likelihood of cell demise. By contrast, maintaining
certain β-catenin binding sites could result in a partial decrease
in activity, thereby enabling the Wnt signaling pathway to still
confer a growth advantage to CRC tumor cells without triggering
cell death (39). This led to the
suggestion of the ‘triple hit’ hypothesis, which suggests that the
optimal level of Wnt in CRC tumors may vary as the tumor
progresses, either due to alterations in the surrounding
environment or the acquisition of new genetic mutations (40). Certain types of CRC can regulate the
Wnt signaling pathway by modifying the copy numbers or experiencing
other types of genetic alterations known as ‘third hits’ in APC
genes. In addition, a different hypothesis called ‘independent
nuclear export activities’ has been suggested, proposing that the
truncation of APC reduces the ability to export proteins from the
nucleus (41). This reduction is
caused by the loss of the central nuclear export signal located
next to the mutation cluster region, leading to a significant
impairment of its tumor suppressor function (42).
APC mutations predominantly consist of missense
mutations that introduce premature stop signals, causing a
shortened APC protein to be produced. The majority of these
mutations are found within a specific region known as the
mutation-cluster region, which houses crucial binding sites for
β-catenin, Axin and other important proteins involved in the Wnt
signaling pathway. The truncation of APC due to these mutations
disrupts essential interactions with Wnt signaling proteins, actin
and the microtubule cytoskeleton that occur at the protein's
C-terminus. Recent findings have solidified the notion that the
truncation of APC produces irregular control over β-catenin,
resulting in heightened transcription of Wnt target genes and
consequently contributing to the onset and progression of CRC
(43).
APC truncation and intestinal epithelial
cell proliferation
The C-terminal binding protein (CtBP) plays a
crucial role in the functional mechanism of APC in its ability to
counteract β-catenin. In vivo, APC is present and interacts
with CtBP through a conserved sequence comprising 15 amino acids.
When APC is truncated, it loses its ability to bind to CtBP.
Consequently, an increase in CtBP levels leads to the formation of
a higher amount of β-catenin/TCF complexes and an elevation in
TCF-mediated transcription. It is worth highlighting that in
vivo, there is no association between CtBP and TCF, and
mutating the CtBP binding motif in TCF-4 does not affect its
transcriptional activity. This casts doubt on the notion that CtBP
directly enhances TCF function. Evidence suggests that APC
facilitates the interaction between β-catenin and CtBP, and CtBP
hinders the binding of free nuclear β-catenin to TCF by forming a
complex with APC/β-catenin (44).
CtBP1 and CtBP2 are transcriptional co-regulators that have been
conserved throughout evolution. They interact with DNA-binding
transcription factors and chromatin remodeling factors, such as
histone methyltransferases and histone deacetylases. This
interaction allows them to either activate or suppress gene
expression (45). Overexpression of
CtBP1 and CtBP2 is frequently observed in various solid tumors,
indicating that CtBP is a crucial gene involved in promoting cancer
growth in solid tumors. One significant aspect of tumor development
is the sensitivity of truncated APC to CtBP oligomerization
(46). In colon cancer cases where
truncated APC is expressed, the interaction between APC and CtBP is
disrupted, leading to the promotion of CRC occurrence and
progression (47). CtBP's capacity
to induce tumor growth arises from its ability to alter the gene
expression patterns of cells, leading to the suppression of genes
involved in apoptotic processes, such as Bcl-2 interacting killer,
p53 upregulated modulator of apoptosis, NADPH oxidase activator and
p53 apoptosis effector related to peripheral myelin protein 22. In
addition, it inhibits the activity of tumor suppressors, such as
phosphatase and tensin homolog, p16Ink4a,
p15Ink4b and p21waf1/cip1. CtBP also
activates the metastasis-associated gene TIAM Rac1 associated GEF
1, facilitating cell migration and invasion (48). Mutations of the APC gene in FAP
cases lead to the production of shortened APC proteins that cannot
effectively control the activity of the β-catenin transcription
factor. Notably, even though CRC cells in FAP cases continue to
have the shortened APC protein, it was found that CtBP could
facilitate the assembly of these shortened APC proteins by binding
to a specific sequence of 15 amino acids that was repeated in the
APC protein. Surprisingly, CtBP has the ability to attach itself to
the initial, third, or fourth sequence of the 15-amino acid
repeats, but it is unable to bind to the second sequence. The
formation of CtBP oligomers necessitates the removal of the
dimerization region in APC, in addition to CtBP's own dimerization
process. A comprehensive examination of APC sequence mutations in
individuals with FAP revealed that the truncated APC products
consistently favored the initial 15 amino acid repeat sequences
(49). Scholars attempted to
develop a short hairpin (sh)RNA targeting a particular subtype of
APC that is truncated, while preserving the original wild-type APC.
They discovered that when they reduced the level of this truncated
APC, the transcriptional activity of β-catenin increased in 5 out
of 6 CRC cell lines. This demonstrates that the truncated APC is
still capable of regulating Wnt signaling by controlling β-catenin
levels. Consequently, the truncated APC facilitates the
proliferation of CRC cells by moderating β-catenin to a ‘suitable’
extent (50). Furthermore,
scientists have recently identified a selective inhibitor called
TASIN-1, targeting APC-truncated cells, while leaving normal human
colon epithelial cells and certain cancer cells unaffected, which
is quite remarkable. Animal experiments conducted in vivo
demonstrated that treatment with TASIN-1 effectively hindered the
growth of CRC cells with truncated APC, without showing significant
toxicity towards CRC cells that had wild-type APC (51). Mizutani et al (52) found that a newly developed inhibitor
called RK-287107 effectively caused AXIN2 accumulation, suppressed
β-catenin expression, reduced TCF/LEF activity, attenuated Myc
expression and restrained the growth of CRC cells with APC
mutations.
In addition, Novellasdemunt et al (53) utilized clustered regularly
interspaced short palindromic repeats (CRISPR)/CRISPR-associated
protein 9 (Cas9) technology to create different APC truncated
isogenic lines and observed that the inhibitory domain of β-catenin
(CID) in APC is responsible for determining the pathological levels
of Wnt activation and the transformation of tumors. Through a
specific mechanism, the depletion of CID in APC truncation leads to
the deubiquitination of β-catenin by facilitating the reverse
binding of β-transducin repeat-containing protein and ubiquitin
specific peptidase 7 (USP7) to the disruption complex. In CRC with
APC mutations, the deletion of USP7 effectively suppresses Wnt
activation, induces differentiation and hinders xenograft tumor
growth by promoting the ubiquitination of β-catenin. Of note, the
role of USP7 in Wnt activation is specific to APC mutations, making
it a potential therapeutic target specific to CRC.
APC truncation and intestinal epithelial
cell apoptosis
Contrary to full-length APC, APC mutants exhibit
antiapoptotic capabilities through mechanisms that do not involve
transcription. TASIN-1, on the other hand, has been found to
trigger apoptosis in truncated APC human CRC cells by inducing
endoplasmic reticulum stress-related JNK activation, accompanied by
the generation of reactive oxygen species. Furthermore, TASIN-1
hampers AKT activity in a cholesterol-dependent fashion. When
examining human CRC xenografts in immunodeficient mice, it becomes
evident that the molecular mechanism underlying TASIN-1-induced
tumor cell death is consistent with what has been observed in
vitro (54). Multiple studies
have demonstrated that APC truncation hinders the process of
apoptosis-related caspase cleavage, independent of
β-catenin-mediated transcription (55). The introduction of truncated APC, as
opposed to full-length APC, provided protection against
Sulindac-induced apoptosis in SW480 cells. Conversely, temporarily
reducing the levels of APC truncation in SW480 cells led to a
decrease in Bcl-2 expression in the mitochondria and an increase in
apoptosis (56).
APC truncation and intestinal epithelial
cell migration
APC exerts control over cell migration through
various mechanisms, such as regulating the actin cytoskeleton
(57), governing the microtubule
network (58) and interacting with
APC-stimulated guanine nucleotide-exchange factor (Asef) (59), a specific guanine
nucleotide-exchange factor for Rac. A notable observation has been
made that the introduction of truncated APC, as opposed to
full-length APC, triggers Asef-mediated cell migration in
Madin-Darby canine kidney cells. Furthermore, when shRNA was
utilized to silence truncated APCs, it noticeably reduced cell
migration of SW480 and WiDr cells. However, when shRNA specifically
targeted truncated APC, there was no impact on the migration of
HCT116 and LS180 cells that had wild-type APC (60). These findings demonstrate that
truncated APC, not full-length one, is strong activator of Asef,
potentially causing abnormal cell migration in CRC cells. Recent
evidence also showed that N-terminal truncation of APC plays a
significant role in promoting directed cell migration in various
model systems (61). In addition,
it was noted that APC, when acting as a kinesin carrier, can be
found at the ends of microtubules. This was found in A6 Xenopus
epithelial cells, where both full-length APC and APC mutants
without the C-terminus could move towards the plus-end of
microtubules in a manner dependent on ATP. This movement aligned
with the plus-end-directed motility activity was typically
associated with kinesin (62). The
translocation of APC C-terminal deletion constructs may rely on
either heterotrimeric [kinesin family member 3A (KIF3A)/3B/KAP3] or
homodimeric (KIF17) kinesin-2, both of which have the capability to
interact with the APC-N terminus and facilitate the accumulation of
APC at the outer edges of the cell (63,64).
The interaction between APC and KIF5 occurs through the C-terminal
region and plays a role in stabilizing APC at the end of
microtubules, impacting cell migration (65). The presence of the APC C-terminus
alone, with or without the Dlg1 binding motif, is capable of
correcting the disrupted epithelial cell extrusion direction caused
by the expression of APC with a truncated C-terminus. This
indicates that the stabilization of microtubules by the APC
C-terminus alone is sufficient to restore cell polarity (66).
APC truncation and intestinal epithelial
cell adhesion
Collective cell remodeling and motility rely on the
actin cytoskeleton, exerting a crucial role in the dynamic
reorganization of cell contacts. The initiation and growth of actin
filaments (F-actin) are facilitated by APC, leading to the enhanced
stability and movement of cell junctions. Consequently, APC may
contribute significantly to the processes of cellular remodeling
and motility. The presence of the APC-dependent actin pool has a
crucial role in maintaining appropriate levels of F-actin,
E-cadherin and occludin proteins at cell junctions. These proteins
are responsible for preserving the length and angle of cell
junctions and ensuring the motility and integrity of cellular tips.
When the APC protein is truncated, the actin pool is lost,
resulting in slower and more random movement of larger cells. This
highlights the significance of APC-driven cytoskeletal function in
understanding the process of intestinal morphogenesis (57). Epithelial cells possess an exclusive
intracellular reservoir of β-catenin, known as Drosophila
armadillo, serving as a structural element in junctions of adhesion
molecules. It is noteworthy that APC proteins potentially
participate in the assembly of these adhesion molecules and
emerging evidence suggests that APC has a role in facilitating cell
adhesion (67). Therefore, the
potential function of APC-β-catenin-Armadillo in both Wnt signaling
and cell adhesion suggests its possible involvement as a tumor
suppressor. It has been suggested that APC mutation truncation
could contribute to tumorigenesis by interfering with cell-cell
adhesion. Additionally, APC interacts with Drosophila
armadillo, a specific subset of β-catenin located within cells, to
establish a connection between E-cadherin, α-catenin and the actin
cytoskeleton (68,69). In animal models with mutant Apc
copies, scientists observed a decrease in E-cadherin levels in both
intestinal cells and tumor cell membranes. Additionally, the
connection between β-catenin and E-cadherin was found to be
weakened (70,71). The presence of complete APCs in CRC
cells, as opposed to truncated APCs, led to a notable increase in
E-cadherin levels on the cell membrane. This resulted in the
relocation of β-catenin from the nucleus and cytoplasm to the outer
edge of the cell, ultimately enhancing cell adhesion (72,73).
Thus, APC may regulate the distribution of β-catenin and E-cadherin
between the cytoplasm and the cell membrane in a notable manner,
ultimately affecting cell adhesion. A mutant APC that lacks the
β-catenin binding domain leads to reduced cell adhesion.
APC truncation, DNA repair and CIN
APCs are mainly present within the cytoplasm, while
they have the ability to translocate to the nucleus in order to
regulate nuclear activities (74).
The direct binding between complete APC molecules and polymerase β
(Pol-β), flap structure-specific endonuclease 1 (FEN1) and APE1
endonuclease can lead to the prevention of the assembly of base
excision repair (BER) proteins on damaged DNA and the hindrance of
long-patch BER (75). The region of
APC that inhibits DNA repair, which binds to Pol-β and FEN1, can be
found in the N-terminal section and remains present in mutated
forms of APC (76). Studies have
demonstrated that in cancer cells (e.g., LoVo) expressing truncated
APC protein, the assembly of BER proteins is sped up, and APE1,
FEN1 and Pol-β are more efficient. However, when full-length APC is
reintroduced, FEN1 expression decreases and makes this cell line
more responsive to 5-fluorouracil (77). An imbalance in the BER pathway and
the potential for CIN and cancer progression may occur as a
consequence of heightened APE1 activity (78). Furthermore, APC has the ability to
interact with replication protein A32 in order to regulate the
response to replication stress (79). In addition, APC plays a direct role
in DNA double-strand break repair by being a component of the
nuclear complex that contains DNA-dependent protein kinase
(80). To sum up, mutations in APC
genes can weaken the functions of BER and DSB repair, resulting in
the accumulation of genetic changes in CRC cells. As a result, CRC
cells with mutant APCs may be more vulnerable to the effects of
DNA-damaging chemotherapy drugs (e.g., oxaliplatin and
5-fluorouracil). In addition, APCs have the ability to directly
attach to and stabilize microtubules, or indirectly by binding to
EB1, a protein found abundantly at the tips of microtubules
(81). Of note, scholars also
discovered that in cells going through mitosis, APC is localized to
kinetochores and forms complexes with BUB1 mitotic checkpoint
serine/threonine kinase (Bub1) and Bub3 (82). When APC is depleted in cancer cells,
such as U2OS and HCT116, the association between checkpoint
proteins Bub1 and BubR1 with kinetochores is reduced, resulting in
alterations in the progression of mitosis and an increase in
mitotic sliding (83).
Consequently, cells carrying the shortened APC gene exhibit a
defect in the proper segregation of chromosomes. In addition, when
APC and/or EB1 are targeted using small interfering RNA, alignment
issues in the mitotic spindle and chromosomes occur. Abnormal
spindle structure and weakened attachment of kinetochore
microtubules were observed in CRC cells that bear the truncated APC
gene (84). Furthermore, the
absence of APC led to an elevation in the chromosomal count in
mouse hepatocytes (85). To
summarize, the presence of APCs with a truncated C-terminus can
promote the advancement of CRC by impairing spindle formation and
progression during cell division, attributed to the absence of a
microtubule-binding domain.
Role of APC in CRC
APC plays a pivotal role in CRC by regulating the
Wnt/β-catenin signaling pathway, which is critical for maintaining
cellular homeostasis and controlling cell proliferation (11,32).
Mutations in the APC gene, particularly truncating mutations,
result in the loss of functional domains in the APC protein. This
typically occurs when truncations remove the C-terminal regions
responsible for binding β-catenin, axin and other regulatory
proteins, leading to the stabilization and accumulation of
β-catenin in the cytoplasm. The accumulated β-catenin translocates
to the nucleus, where it activates the transcription of Wnt target
genes that promote tumorigenesis (51). For instance, truncations at codon
1,309, one of the most common mutations in FAP and sporadic CRC,
result in a premature stop codon. This truncation removes the
majority of the C-terminal domains responsible for β-catenin
regulation and other cellular processes. Similarly, truncations at
codon 1,450 lead to a loss of microtubule-binding domains, which
affects APC's role in cytoskeletal dynamics, impairing cell
migration and adhesion (38). This
structural disruption not only affects β-catenin regulation but
also destabilizes APC's interactions with the cytoskeleton,
promoting increased metastatic potential. Furthermore, APC
mutations are associated with CIN, which is partly driven by the
loss of its ability to interact with microtubules, contributing to
aneuploidy and tumor heterogeneity (26). Truncations affecting the armadillo
repeat and basic domains of APC, crucial for its role in
microtubule attachment and spindle formation, contribute to the
chromosomal missegregation observed in CRC. The loss of functional
domains due to truncating mutations also has implications for the
tumor microenvironment, altering the way tumor cells interact with
the stroma and immune cells. This disruption can create a
microenvironment that supports tumor growth and metastasis
(59). Understanding these
structure-function relationships in APC truncations is critical for
developing more targeted therapeutic approaches in CRC. In this
way, novel therapeutic targets may be discovered and strategies may
be developed to enhance the efficacy of CRC treatments.
The four most common truncating mutations in APC and
their implications in CRC are as follows: The codon 1,309 mutation
is found in ~20–30% of CRC cases and causes truncation in the
mutation cluster region (MCR), which includes the β-catenin binding
and downregulation domains (67).
This leads to the stabilization and nuclear accumulation of
β-catenin, promoting the transcription of oncogenic target genes,
such as c-Myc and cyclin D1. The codon 1,450 mutation, present in
~10-20% of CRC cases, also truncates in the MCR, affecting
β-catenin binding sites and Axin interaction domains, resulting in
increased β-catenin activity and enhanced tumorigenesis through
upregulation of Wnt target genes (69). The codon 1,556 mutation is observed
in 5–10% of CRC patients and involves truncation beyond the MCR,
affecting additional regions involved in cytoskeletal interactions.
This disrupts the role of APC in microtubule stabilization and
chromosomal segregation, contributing to CIN and tumor progression
(72). Lastly, the codon 1,061
mutation, detected in 5–15% of CRC cases, results in truncation
within the armadillo repeats and the MCR, impacting β-catenin and
other interaction sites, thereby enhancing Wnt signaling due to
defective β-catenin degradation, promoting cell proliferation and
survival (81). These mutations are
primarily responsible for the disruption of APC's tumor suppressor
functions, including its roles in Wnt signaling, cell adhesion,
migration and genomic stability, ultimately driving the development
and progression of CRC.
Summary
The role of APC as a tumor suppressor has long been
a significant focus in cancer research. Recently, there has been
growing interest in exploring the cancer-causing potential of APC
mutants due to emerging evidence. An increasing array of evidence
suggested that APC mutants have a noticeable function in the
development and advancement of CRC tumors by hindering the tumor
suppressor function of APC. As cancer cells gradually become
reliant on the cancer-causing characteristics of truncated APC
protein to survive and sustain their malignancy, they experience
changes in their signaling network patterns and continuous
activation of oncogenes. The utilization of proteomics and the
ongoing progress in genome-wide high-throughput screening methods
can potentially assist in the identification of novel interaction
partners of truncated APCs, contributing to a thorough
understanding of the signaling network in CRC cells. While the
fundamental role of APC in CRC pathogenesis is well-established,
recent research has unveiled new aspects of its function, including
its interactions with emerging signaling pathways such as the
Hippo, Notch and Hedgehog pathways, and its broader impact on
genomic stability. The evolving understanding of APC's role in CRC
underscores its remarkable importance in cancer research,
highlighting the potential for novel therapeutic approaches and
personalized medicine based on APC-related mechanisms. This review
consolidated these recent findings, demonstrating that APC plays
notable roles in improving CRC diagnosis, treatment and prevention.
Acquiring knowledge about how these mutated forms of APC cooperate
with downstream effector proteins will be vital in elucidating the
molecular mechanisms underlying CRC tumorigenesis, ultimately
revealing new targets for drug development and improving targeted
therapies for CRC. The evolving understanding of APC's role in CRC
underscores its remarkable importance in cancer research,
highlighting the potential for novel therapeutic approaches and
personalized medicine based on APC-related mechanisms. This review
consolidated these recent findings, demonstrating that APC plays
notable roles in improving CRC diagnosis, treatment and prevention.
The novelties of this review lie in its detailed exploration of APC
truncation-specific mechanisms and their unique contributions to
CRC progression. In contrast to previous reviews that broadly
concentrated on APC mutations, this review highlighted how
truncation mutations affect key protein interactions, such as those
with AXIN1, β-catenin and the Wnt signaling pathway, leading to
profound effects on cellular processes, involving proliferation,
migration and adhesion. The review presented novel insights into
the ‘triple hit’ hypothesis, emphasizing the critical role of
subsequent genetic alterations following APC truncation in
promoting tumor progression. Additionally, the review highlighted
emerging findings on the role of CtBP oligomerization in APC
truncation, a less explored yet crucial pathway in CRC.
Furthermore, the review provided notable perspectives on
therapeutic strategies targeting truncated APC, including the
development of TASIN-1, which selectively targets APC-mutant cells.
It also covered underexplored areas, such as the impact of APC
truncation on cytoskeletal interactions, CIN and DNA repair
mechanisms, while incorporating recent advancements in experimental
techniques (e.g., CRISPR/Cas9). Consequently, these aspects present
a comprehensive and updated view of how APC truncation drives CRC,
providing new directions for understanding tumorigenesis and
potential therapeutic interventions.
Acknowledgements
Not applicable.
Funding
This work was supported by the Scientific Research Project of
Colleges and Universities in Inner Mongolia Autonomous Region
(grant no. NJZZ21062) and Scientific Research Project of Hetao
College (grant no. HYZZ201929).
Availability of data and materials
Not applicable.
Authors' contributions
TW conceived and designed the study. CF, JF and YH
wrote and edited the manuscript. Data authentication is not
applicable. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ciardiello F, Ciardiello D, Martini G,
Napolitano S, Tabernero J and Cervantes A: Clinical management of
metastatic colorectal cancer in the era of precision medicine. CA
Cancer J Clin. 72:372–401. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qu R, Ma Y, Zhang Z and Fu W: Increasing
burden of colorectal cancer in China. Lancet Gastroenterol Hepatol.
7:7002022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Spaander MCW, Zauber AG, Syngal S, Blaser
MJ, Sung JJ, You YN and Kuipers EJ: Young-onset colorectal cancer.
Nat Rev Dis Primers. 9:212023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shaukat A and Levin TR: Current and future
colorectal cancer screening strategies. Nat Rev Gastroenterol
Hepatol. 19:521–531. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wong CC and Yu J: Gut microbiota in
colorectal cancer development and therapy. Nat Rev Clin Oncol.
20:429–452. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Z, Dan W, Zhang N, Fang J and Yang Y:
Colorectal cancer and gut microbiota studies in China. Gut
Microbes. 15:22363642023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Ginkel J, Tomlinson I and Soriano I:
The evolutionary landscape of colorectal tumorigenesis: Recent
paradigms, models, and hypotheses. Gastroenterology. 164:841–846.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weiss JM, Gupta S, Burke CA, Axell L, Chen
LM, Chung DC, Clayback KM, Dallas S, Felder S, Gbolahan O, et al:
NCCN Guidelines® Insights: Genetic/Familial High-Risk
Assessment: Colorectal, Version 1.2021. J Natl Compr Canc Netw.
19:1122–1132. 2021.PubMed/NCBI
|
|
9
|
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ,
Ellis A, Gorman P, Lucibello FC, Murday VA, Rider SH, Scambler P,
et al: Localization of the gene for familial adenomatous polyposis
on chromosome 5. Nature. 328:614–616. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Karstensen JG, Burisch J, Pommergaard HC,
Aalling L, Hojen H, Jespersen N, Schmidt PN and Bülow S: Colorectal
cancer in individuals with familial adenomatous polyposis, based on
analysis of the danish polyposis registry. Clin Gastroenterol
Hepatol. 17:2294–2300.e1. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rowan AJ, Lamlum H, Ilyas M, Wheeler J,
Straub J, Papadopoulou A, Bicknell D, Bodmer WF and Tomlinson IP:
APC mutations in sporadic colorectal tumors: A mutational ‘hotspot’
and interdependence of the ‘two hits’. Proc Natl Acad Sci USA.
97:3352–3357. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aoki K and Taketo MM: Adenomatous
polyposis coli (APC): A Multi-functional tumor suppressor gene. J
Cell Sci. 120:3327–3335. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bressler SG, Mitrany A, Wenger A, Nathke I
and Friedler A: The oligomerization domains of the APC protein
mediate Liquid-liquid phase separation that is phosphorylation
controlled. Int J Mol Sci. 24:64782023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Z, Lin K, Gao L, Chen L, Shi X and
Wu G: Crystal structure of the armadillo repeat domain of
adenomatous polyposis coli which reveals its inherent flexibility.
Biochem Biophys Res Commun. 412:732–736. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kawasaki Y, Senda T, Ishidate T, Koyama R,
Morishita T, Iwayama Y, Higuchi O and Akiyama T: Asef, a link
between the tumor suppressor APC and G-protein signaling. Science.
289:1194–1197. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rowling PJE, Murton BL, Du Z and Itzhaki
LS: Multivalent interaction of Beta-Catenin with its intrinsically
disordered binding partner adenomatous polyposis Coli. Front Mol
Biosci. 9:8964932022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kunttas-Tatli E, Von Kleeck RA, Greaves
BD, Vinson D, Roberts DM and McCartney BM: The two SAMP repeats and
their phosphorylation state in Drosophila Adenomatous polyposis
coli-2 play mechanistically distinct roles in negatively regulating
Wnt signaling. Mol Biol Cell. 26:4503–4518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Juanes MA, Fees CP, Hoeprich GJ, Jaiswal R
and Goode BL: EB1 directly regulates APC-Mediated actin nucleation.
Curr Biol. 30:4763–4772.e8. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Honnappa S, John CM, Kostrewa D, Winkler
FK and Steinmetz MO: Structural insights into the EB1-APC
interaction. EMBO J. 24:261–269. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lui C, Ashton C, Sharma M, Brocardo MG and
Henderson BR: APC functions at the centrosome to stimulate
microtubule growth. Int J Biochem Cell Biol. 70:39–47. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peng H, Ying J, Zang J, Lu H, Zhao X, Yang
P, Wang X, Li J, Gong Z, Zhang D and Wang Z: Specific mutations in
APC, with prognostic implications in metastatic colorectal cancer.
Cancer Res Treat. 55:1270–1280. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lewis A, Davis H, Deheragoda M, Pollard P,
Nye E, Jeffery R, Segditsas S, East P, Poulsom R, Stamp G, et al:
The C-terminus of Apc does not influence intestinal adenoma
development or progression. J Pathol. 226:73–83. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nusse R and Clevers H: Wnt/β-Catenin
signaling, disease, and emerging therapeutic modalities. Cell.
169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Klaus A and Birchmeier W: Wnt signalling
and its impact on development and cancer. Nat Rev Cancer.
8:387–398. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kennell J and Cadigan KM: APC and
beta-catenin degradation. Adv Exp Med Biol. 656:1–12. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mishra L: STRAP: A bridge between mutant
APC and Wnt/ß-Catenin signaling in intestinal cancer.
Gastroenterology. 162:44–46. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Hou Y, Ming M, Yu L, Seba A and Qian
Z: Apc regulates the function of hematopoietic stem cells largely
through beta-catenin-dependent mechanisms. Blood. 121:4063–4072.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Odenwald MA, Prosperi JR and Goss KH:
APC/β-catenin-rich complexes at membrane protrusions regulate
mammary tumor cell migration and mesenchymal morphology. BMC
Cancer. 13:122013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bakker ER, Hoekstra E, Franken PF,
Helvensteijn W, van Deurzen CH, van Veelen W, Kuipers EJ and Smits
R: β-Catenin signaling dosage dictates tissue-specific tumor
predisposition in Apc-driven cancer. Oncogene. 32:4579–4585. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cole JM, Simmons K and Prosperi JR: Effect
of adenomatous polyposis coli loss on tumorigenic potential in
pancreatic ductal adenocarcinoma. Cells. 8:10842019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ranes M, Zaleska M, Sakalas S, Knight R
and Guettler S: Reconstitution of the destruction complex defines
roles of AXIN polymers and APC in β-catenin capture,
phosphorylation, and ubiquitylation. Mol Cell. 81:3246–3261.e11.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Roberts DM, Pronobis MI, Poulton JS,
Waldmann JD, Stephenson EM, Hanna S and Peifer M: Deconstructing
the sscatenin destruction complex: Mechanistic roles for the tumor
suppressor APC in regulating Wnt signaling. Mol Biol Cell.
22:1845–1863. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Montagne J, Preza M, Castillo E, Brehm K
and Koziol U: Divergent Axin and GSK-3 paralogs in the beta-catenin
destruction complexes of tapeworms. Dev Genes Evol. 229:89–102.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Faux MC, Coates JL, Catimel B, Cody S,
Clayton AH, Layton MJ and Burgess AW: Recruitment of adenomatous
polyposis coli and beta-catenin to axin-puncta. Oncogene.
27:5808–5820. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nong J, Kang K, Shi Q, Zhu X, Tao Q and
Chen YG: Phase separation of Axin organizes the beta-catenin
destruction complex. J Cell Biol. 220:e2020121122021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li VS, Ng SS, Boersema PJ, Low TY,
Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi
T and Clevers H: Wnt signaling through inhibition of beta-catenin
degradation in an intact Axin1 complex. Cell. 149:1245–1256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ain QU, Seemab U, Rashid S, Nawaz MS and
Kamal MA: Prediction of structure of human WNT-CRD (FZD) complex
for computational drug repurposing. PLoS One. 8:e546302013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Aghabozorgi AS, Bahreyni A, Soleimani A,
Bahrami A, Khazaei M, Ferns GA, Avan A and Hassanian SM: Role of
adenomatous polyposis coli (APC) gene mutations in the pathogenesis
of colorectal cancer; current status and perspectives. Biochimie;
157. pp. 64–71. 2019, PubMed/NCBI
|
|
39
|
Albuquerque C, Breukel C, van der Luijt R,
Fidalgo P, Lage P, Slors FJ, Leitao CN, Fodde R and Smits R: The
‘just-right’ signaling model: APC somatic mutations are selected
based on a specific level of activation of the beta-catenin
signaling cascade. Hum Mol Genet. 11:1549–1560. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Segditsas S, Rowan AJ, Howarth K, Jones A,
Leedham S, Wright NA, Gorman P, Chambers W, Domingo E, Roylance RR,
et al: APC and the Three-hit hypothesis. Oncogene. 28:146–155.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Henderson BR and Fagotto F: The ins and
outs of APC and beta-catenin nuclear transport. EMBO Rep.
3:834–839. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rosin-Arbesfeld R, Cliffe A, Brabletz T
and Bienz M: Nuclear export of the APC tumour suppressor controls
beta-catenin function in transcription. EMBO J. 22:1101–1113. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Elliott KL, Catimel B, Church NL, Coates
JL, Burgess AW, Layton MJ and Faux MC: Immunopurification of
adenomatous polyposis coli (APC) proteins. BMC Res Notes.
6:4292013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hamada F and Bienz M: The APC tumor
suppressor binds to C-terminal binding protein to divert nuclear
beta-catenin from TCF. Dev Cell. 7:677–685. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sumner ET, Chawla AT, Cororaton AD,
Koblinski JE, Kovi RC, Love IM, Szomju BB, Korwar S, Ellis KC and
Grossman SR: Transforming activity and therapeutic targeting of
C-terminal-binding protein 2 in Apc-mutated neoplasia. Oncogene.
36:4810–4816. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nadauld LD, Phelps R, Moore BC, Eisinger
A, Sandoval IT, Chidester S, Peterson PW, Manos EJ, Sklow B, Burt
RW and Jones DA: Adenomatous polyposis coli control of C-terminal
binding protein-1 stability regulates expression of intestinal
retinol dehydrogenases. J Biol Chem. 281:37828–37835. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Serre L, Stoppin-Mellet V and Arnal I:
Adenomatous polyposis coli as a scaffold for microtubule
End-binding proteins. J Mol Biol. 431:1993–2005. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chinnadurai G: The transcriptional
corepressor CtBP: A foe of multiple tumor suppressors. Cancer Res.
69:731–734. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Schneikert J, Brauburger K and Behrens J:
APC mutations in colorectal tumours from FAP patients are selected
for CtBP-mediated oligomerization of truncated APC. Hum Mol Genet.
20:3554–3564. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chandra SH, Wacker I, Appelt UK, Behrens J
and Schneikert J: A common role for various human truncated
adenomatous polyposis coli isoforms in the control of beta-catenin
activity and cell proliferation. PLoS One. 7:e344792012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang L, Theodoropoulos PC, Eskiocak U,
Wang W, Moon YA, Posner B, Williams NS, Wright WE, Kim SB, Nijhawan
D, et al: Selective targeting of mutant adenomatous polyposis coli
(APC) in colorectal cancer. Sci Transl Med. 8:361ra1402016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Mizutani A, Yashiroda Y, Muramatsu Y,
Yoshida H, Chikada T, Tsumura T, Okue M, Shirai F, Fukami T,
Yoshida M and Seimiya H: RK-287107, a potent and specific tankyrase
inhibitor, blocks colorectal cancer cell growth in a preclinical
model. Cancer Sci. 109:4003–4014. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Novellasdemunt L, Foglizzo V, Cuadrado L,
Antas P, Kucharska A, Encheva V, Snijders AP and Li VSW: USP7 is a
Tumor-specific WNT activator for APC-mutated colorectal cancer by
mediating β-Catenin deubiquitination. Cell Rep. 21:612–627. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang L, Kim SB, Luitel K and Shay JW:
Cholesterol depletion by TASIN-1 induces apoptotic cell death
through the ER Stress/ROS/JNK signaling in colon cancer cells. Mol
Cancer Ther. 17:943–951. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qian J, Steigerwald K, Combs KA, Barton MC
and Groden J: Caspase cleavage of the APC tumor suppressor and
release of an Amino-terminal domain is required for the
Transcription-independent function of APC in apoptosis. Oncogene.
26:4872–4876. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Brocardo M, Lei Y, Tighe A, Taylor SS, Mok
MT and Henderson BR: Mitochondrial targeting of adenomatous
polyposis coli protein is stimulated by truncating cancer
mutations: Regulation of Bcl-2 and implications for cell survival.
J Biol Chem. 283:5950–5959. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Baro L, Islam A, Brown HM, Bell ZA and
Juanes MA: APC-driven actin nucleation powers collective cell
dynamics in colorectal cancer cells. iScience. 26:1065832023.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Juzans M, Cuche C, Rose T, Mastrogiovanni
M, Bochet P, Di Bartolo V and Alcover A: Adenomatous polyposis coli
modulates actin and microtubule cytoskeleton at the immunological
synapse to tune CTL functions. Immunohorizons. 4:363–381. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang X, Zhong J, Zhang Q, Feng L, Zheng Z,
Zhang J and Lu S: Advances and insights of APC-Asef inhibitors for
metastatic colorectal cancer therapy. Front Mol Biosci.
8:6625792021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kawasaki Y, Furukawa S, Sato R and Akiyama
T: Differences in the localization of the adenomatous polyposis
coli-Asef/Asef2 complex between adenomatous polyposis coli
wild-type and mutant cells. Cancer Sci. 104:1135–1138. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nelson SA, Li Z, Newton IP, Fraser D,
Milne RE, Martin DM, Schiffmann D, Yang X, Dormann D, Weijer CJ, et
al: Tumorigenic fragments of APC cause dominant defects in
directional cell migration in multiple model systems. Dis Model
Mech. 5:940–947. 2012.PubMed/NCBI
|
|
62
|
Mimori-Kiyosue Y, Shiina N and Tsukita S:
Adenomatous polyposis coli (APC) protein moves along microtubules
and concentrates at their growing ends in epithelial cells. J Cell
Biol. 148:505–518. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Jaulin F and Kreitzer G: KIF17 stabilizes
microtubules and contributes to epithelial morphogenesis by acting
at MT plus ends with EB1 and APC. J Cell Biol. 190:443–460. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Jimbo T, Kawasaki Y, Koyama R, Sato R,
Takada S, Haraguchi K and Akiyama T: Identification of a link
between the tumour suppressor APC and the kinesin superfamily. Nat
Cell Biol. 4:323–327. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ruane PT, Gumy LF, Bola B, Anderson B,
Wozniak MJ, Hoogenraad CC and Allan VJ: Tumour suppressor
adenomatous polyposis coli (APC) localisation is regulated by both
Kinesin-1 and Kinesin-2. Sci Rep. 6:274562016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Marshall TW, Lloyd IE, Delalande JM,
Nathke I and Rosenblatt J: The tumor suppressor adenomatous
polyposis coli controls the direction in which a cell extrudes from
an epithelium. Mol Biol Cell. 22:3962–3970. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bienz M and Hamada F: Adenomatous
polyposis coli proteins and cell adhesion. Curr Opin Cell Biol.
16:528–535. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wang C, Zhao R, Huang P, Yang F, Quan Z,
Xu N and Xi R: APC Loss-induced intestinal tumorigenesis in
Drosophila: Roles of Ras in Wnt signaling activation and tumor
progression. Dev Biol. 378:122–140. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
De Graeve FM, Van de Bor V, Ghiglione C,
Cerezo D, Jouandin P, Ueda R, Shashidhara LS and Noselli S:
Drosophila apc regulates delamination of invasive epithelial
clusters. Dev Biol. 368:76–85. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Faux MC, Coates JL, Kershaw NJ, Layton MJ
and Burgess AW: Independent interactions of phosphorylated
β-catenin with E-cadherin at Cell-cell contacts and APC at cell
protrusions. PLoS One. 5:e141272010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Restucci B, Martano M, G DEV, Lo Muzio L
and Maiolino P: Expression of E-cadherin, beta-catenin and APC
protein in canine colorectal tumours. Anticancer Res. 29:2919–2925.
2009.PubMed/NCBI
|
|
72
|
Lim JW, Mathias RA, Kapp EA, Layton MJ,
Faux MC, Burgess AW, Ji H and Simpson RJ: Restoration of
full-length APC protein in SW480 colon cancer cells induces
exosome-mediated secretion of DKK-4. Electrophoresis. 33:1873–1880.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Faux MC, Ross JL, Meeker C, Johns T, Ji H,
Simpson RJ, Layton MJ and Burgess AW: Restoration of Full-length
adenomatous polyposis coli (APC) protein in a colon cancer cell
line enhances cell adhesion. J Cell Sci. 117:427–439. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Neufeld KL: Nuclear APC. Adv Exp Med Biol.
656:13–29. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
de Boer HR, Guerrero Llobet S and van Vugt
MA: Controlling the response to DNA damage by the APC/C-Cdh1. Cell
Mol Life Sci. 73:949–960. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Yamada M, Watanabe K, Mistrik M, Vesela E,
Protivankova I, Mailand N, Lee M, Masai H, Lukas J and Bartek J:
ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4)
kinase is required for DNA lesion bypass under replication stress.
Genes Dev. 27:2459–2472. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Das D, Preet R, Mohapatra P, Satapathy SR,
Siddharth S, Tamir T, Jain V, Bharatam PV, Wyatt MD and Kundu CN:
5-Fluorouracil mediated Anti-cancer activity in colon cancer cells
is through the induction of adenomatous polyposis coli: Implication
of the Long-patch base excision repair pathway. DNA Repair (Amst).
24:15–25. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tudek B and Speina E: Oxidatively damaged
DNA and its repair in colon carcinogenesis. Mutat Res. 736:82–92.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Brocardo MG, Borowiec JA and Henderson BR:
Adenomatous polyposis coli protein regulates the cellular response
to DNA replication stress. Int J Biochem Cell Biol. 43:1354–1364.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Stefanski CD, Keffler K, McClintock S,
Milac L and Prosperi JR: APC loss affects DNA damage repair causing
doxorubicin resistance in breast cancer cells. Neoplasia.
21:1143–1150. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Baumann SJ, Grawenhoff J, Rodrigues EC,
Speroni S, Gili M, Komissarov A and Maurer SP: APC couples neuronal
mRNAs to multiple kinesins, EB1, and shrinking microtubule ends for
bidirectional mRNA motility. Proc Natl Acad Sci USA.
119:e22115361192022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Overlack K, Bange T, Weissmann F, Faesen
AC, Maffini S, Primorac I, Muller F, Peters JM and Musacchio A:
BubR1 promotes Bub3-Dependent APC/C inhibition during spindle
assembly checkpoint signaling. Curr Biol. 27:2915–2927.e7. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Dikovskaya D, Schiffmann D, Newton IP,
Oakley A, Kroboth K, Sansom O, Jamieson TJ, Meniel V, Clarke A and
Näthke IS: Loss of APC induces polyploidy as a result of a
combination of defects in mitosis and apoptosis. J Cell Biol.
176:183–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cheng L and Mao Y: mDia3-EB1-APC: A
connection between kinetochores and microtubule plus ends. Commun
Integr Biol. 4:480–482. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Meniel V, Megges M, Young MA, Cole A,
Sansom OJ and Clarke AR: Apc and p53 interaction in DNA damage and
genomic instability in hepatocytes. Oncogene. 34:4118–4129. 2015.
View Article : Google Scholar : PubMed/NCBI
|