Introduction
Glioblastoma (GBM) is one of the most aggressive
types of malignant primary brain tumors, accounting for ~48% of all
primary malignant central nervous system tumors and ~57% of all
gliomas (1). Despite the extensive
use of therapeutic approaches, including surgery, radiation therapy
and chemotherapy, the long-term prognosis of GBM, referring to
overall survival outcomes and quality of life, remains poor, with a
median survival time of ~15 months from diagnosis, primarily due to
tumor recurrence and resistance to therapy (2).
One of the major challenges in treating GBM is the
blood-brain barrier (BBB), a highly selective semipermeable border
consisting of endothelial cells that prevent solutes in the
circulating blood from non-selectively crossing into the
extracellular fluid of the brain. The BBB is composed of brain
microvascular endothelial cells (BMECs), astrocytes and pericytes,
which together form a physical and biochemical barrier that
restricts the entry of most therapeutic agents into the brain
(3). Whilst the BBB allows the
passage of certain small molecules through passive diffusion and
the selective transport of essential nutrients and ions, it also
effectively blocks larger molecules, including many
chemotherapeutic drugs (4). This
characteristic of the BBB poses a significant challenge for the
treatment of GBM, necessitating the development of novel strategies
for safe and effective drug delivery across the BBB.
Phosphatase and tensin homolog (PTEN) is a critical
tumor suppressor gene that encodes a phosphatase enzyme involved in
the dephosphorylation of phosphatidylinositol-3,4,5-trisphosphate,
thereby negatively regulating the phosphatidylinositol 3-kinase
(PI3K)/AKT/mTOR signaling pathway (5). Activation of this pathway in cancer
cells promotes cell proliferation, survival, migration,
angiogenesis and metastasis, whilst inhibiting apoptosis (6). Mutations that affected PTEN protein
destabilization were reported to result in stronger AKT activation
than mutations that affected phosphatase activity. Another patient
study with PTEN gene alteration of GBM reported that the
phosphatase activity of PTEN was not associated with AKT
deactivation; wild-type PTEN protein expression in the cytoplasm
has been shown to be associated with decreased AKT phosphorylation,
which in turn reduces AKT activity and downstream signaling,
leading to suppressed cellular proliferation and survival (7). PTEN mutations or deletions are
commonly observed in several cancers, including prostate cancer,
endometrial cancer and GBM (8). A
total of ~40% of patients with GBM exhibit PTEN deficiencies, which
are associated with a poor prognosis (9). This poor prognosis includes shorter
overall survival times and lower response rates to conventional
therapies. PTEN acts as a tumor suppressor by negatively regulating
the AKT/mTOR signaling pathway, and its loss leads to uncontrolled
cell proliferation and survival. This makes PTEN a critical target
for therapeutic strategies aimed at treating GBM (10). Several studies have reported that
PTEN restoration in GBM cells can decrease cell proliferation and
increase apoptosis, suggesting a potential therapeutic strategy
(11–13). However, effective delivery systems
for PTEN gene therapy in the brain have not been fully developed.
Traditional delivery methods face significant obstacles owing to
the BBB, which limits the entry of therapeutic agents into the
brain. Innovative approaches to deliver therapeutic genes such as
PTEN to GBM cells are urgently required.
Newcastle disease virus (NDV) is an intrinsic
oncolytic virus that selectively replicates in tumor cells without
affecting normal cells (14). NDV
induces cancer cell death through mechanisms such as apoptosis,
autophagy and necroptosis and can stimulate the host immune
response against cancer by releasing cytokines and chemokines that
attract immune cells to the tumor site (14). This characteristic makes NDV a
promising candidate for oncolytic virotherapy, particularly for
tumors such as GBM, which are difficult to treat with conventional
therapies. NDV has shown promise in clinical trials for the
treatment of several cancers, including GBM (15,16).
In a previous study, intravenous administration of NDV in patients
with recurrent GBM resulted in a marked reduction in tumor size and
mild adverse reactions comparable with the symptoms of influenza
(17). However, the precise
mechanism by which NDV crosses the BBB remains unclear. NDV may
exploit pathways similar to those used by other viruses, such as
severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which
infect brain vascular endothelial cells and cross the BBB (18). Astrocytes, which are in direct
contact with the outer surface of brain blood vessel endothelial
cells, may serve as a conduit for viral entry into the brain
(19).
Our previous study constructed a recombinant NDV
expressing human PTEN (rNDV-PTEN) and demonstrated its ability to
inhibit GBM cell growth in vitro and in a xenograft animal
model (20). The recombinant virus
combined the tumor-selective replication properties of NDV with the
tumor-suppressive functions of PTEN, thereby providing an
increasing GBM cell death of GBM cells. Therefore, the present
study aimed to build on these findings by evaluating the
therapeutic potential of rNDV-PTEN in an orthotopic mouse model of
GBM, focusing on its ability to cross the BBB and deliver PTEN to
GBM cells. Through these comprehensive analyses, the present study
aimed to provide a detailed understanding of the potential use of
rNDV-PTEN as a therapeutic agent for GBM. Furthermore, the present
study aimed to develop an effective treatment strategy that
overcomes the limitations imposed by the BBB and improves the
prognosis of patients with this disease.
Materials and methods
Cell culture and cell growth
Human GBM cells, U87-MG (cat no. HTB-14; GBM of
unknown origin), U87-MG-luc2 (cat. no. HTB-14-LUC2; GBM of unknown
origin), T98G (cat. no. CRL-1690) and CCF-STTG1 (cat. no.
CRL-1718), were purchased from American Type Culture Collection
(ATCC). Proneural X01 (21) and
Mensenchymal 83 (22) cells were
donated by Professor Park Jong Bae's team at the Korea National
Cancer Center (23). The passage
number at which these cells were supplied was passage 11 and the
cells were experimentally used starting from passage 14.
U87-MG, U87-MG-luc2, T98G and CCF-STTG1 cells were
cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) containing 10%
heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA) and 1% penicillin-streptomycin (Gibco; Thermo Fisher
Scientific, Inc.) and maintained at 37°C in humidified air with 5%
CO2.
Proneural X01 and were cultured in DMEM/F12 (Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10 ng/ml
epidermal growth factor (cat. no. 236-EG; R&D Systems, Inc.),
basic fibroblast growth factor (cat. no. 4114-TC; 5 ng/ml for
Proneural X01 and 10 ng/ml for Mesenchymal 83; R&D Systems,
Inc.). B27 (Invitrogen™; Thermo Fisher Scientific, Inc.)
and 1% penicillin-streptomycin (Gibco; Thermo Fisher Scientific,
Inc.), and maintained at 37°C in humidified air with 5%
CO2.
rNDV-PTEN virus was previously constructed (20). The virus was propagated by infection
of vero cells (CCL-81; ATCC) at a multiplicity of infection (MOI)
of 0.5 for 2 days prior (20). The
virus titer was tested by the 50% tissue culture infective dose
(TCID50/ml) method of Spearman and Kärber (24,25).
Sample preparation
U87-MG and CCF-STTG1 cells (1×107 cells)
seeded in a 175T flask were cultured overnight at 37°C in
humidified air with 5% CO2. The cells were infected with
rNDV or rNDV-PTEN viruses at an MOI of 1.0 for 1 h, washed two
times with PBS and then incubated for 12–36 h at 37°C in humidified
air with 5% CO2, with DMEM containing 10% FBS and 1%
penicillin-streptomycin. The supernatant was removed and cells were
collected at 12, 24 and 36 h after virus infection and the cells
were subjected to three freezing/thawing cycles at −80°C and 4°C.
The cell lysates were used for quantitative (q)PCR and immunoblot
analysis.
Animal studies
Female BALB/c nu-/nu- mice (n=40; 5 weeks old),
weighing ~18–20 g, were purchased from Orient Bio, Inc. The mice
were housed under standard conditions with a 12-h light/dark cycle,
a temperature of 22±2°C and a humidity of 55±10%, with food and
water provided ad libitum. For anesthesia, each mouse was
weighed to calculate the appropriate dose of 2,2,2-tribromoethanol
(Avertin®) via intraperitoneal administration at 250
mg/kg.
U87-MG cells were cultured in DMEM (Gibco; Thermo
Fisher Scientific, Inc.) containing 10% heat-inactivated FBS
(Sigma-Aldrich; Merck KGaA) and 1% penicillin-streptomycin (Gibco;
Thermo Fisher Scientific, Inc.), and maintained at 37°C in
humidified air with 5% CO2. Before injection, U87-MG
cells were trypsinized, counted and resuspended in growing media.
The cell suspension was kept on ice until the time of injection.
Each mouse was injected with 5×104 U87-MG cells in 5 µl
(1×104 cells/µl) as follows: A midline incision of ~1.2
cm was made. A small hole was drilled in the skull at the point 0.2
mm back and 2.2 mm to the left of the bregma. Cell suspensions were
injected with a Hamilton syringe at a rate of 1 µl/min, and the
syringe was left in place for 5 min. The mice were screened using
an in vivo imaging system (IVIS) every 7 days. After 40
days, the mice were randomly divided them into three groups (n=4
per group): rNDV (100 µl 107 TCID50/dose,
intravenous), rNDV-PTEN (100 µl 107
TCID50/dose, intravenous) and PBS as a negative
control.
Immunoblotting
For immunoblotting, proteins were extracted using
RIPA buffer (Thermo Fisher Scientific, Inc.) containing 50 mM
Tris-HCl (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate
and 0.1% SDS. Protein concentration was determined using the
bicinchoninic acid method. Equal amounts of protein (30 µg) were
loaded per lane on a 15% SDS-PAGE gel. Proteins were then
transferred to a polyvinylidene difluoride membrane. The membrane
was blocked with 5% non-fat dry milk in TBS-T (0.1% Tween-20) for 1
h at room temperature. The membranes were incubated overnight at
4°C with the following primary antibodies: Anti-GAPDH (1:3,000;
cat. no. sc-32233; Santa Cruz Biotechnology, Inc.); anti-LC3
(1:1,000; cat. no. NB100-2220; Novus Biologicals, LLC); anti-matrix
metallopeptidase 9 (MMP9; 1:1,000; cat. no. MA5-15886; Thermo
Fisher Scientific, Inc.); anti-proliferating cell nuclear antigen
(PCNA; 1:500; cat. no. PC 10; Sigma-Aldrich; Merck KGaA);
anti-P-mTOR (Ser2448; 1:1,000; cat. no. 2971S; Cell Signaling
Technology, Inc.); anti-mTOR (1:1,000; cat. no. 2972S; Cell
Signaling Technology, Inc.); anti-P-Akt (Ser473; 1:1,000; cat. no.
9271S; Cell Signaling Technology, Inc.); anti-Akt (1:1,000; cat.
no. 9272S; Cell Signaling Technology, Inc.); anti-cleaved Caspase
(Cas)9 (1:1,000; cat. no. 9509S; Cell Signaling Technology, Inc.);
anti-cleaved Cas3 (1:1,000; cat. no. 9664S; Cell Signaling
Technology, Inc.); anti-cleaved Cas8 (1:1,000; cat. no. 9496S; Cell
Signaling Technology, Inc.); anti-B-cell lymphoma 2 (Bcl-2)
associated X protein (Bax; 1:1,000; cat. no. 2772S; Cell Signaling
Technology, Inc.); anti-p62 (1:1,000; cat. no. 5114S; Cell
Signaling Technology, Inc.); anti-Occludin (1:1,000; cat. no.
5506S; Cell Signaling Technology, Inc.); anti-zonula occludens
protein 1 (ZO-1; 1:1,000; cat. no. 5406S; Cell Signaling
Technology, Inc.); anti-Clauddin-5 (E8F3D; 1:1,000; cat. no. 49564;
Cell Signaling Technology, Inc.); and anti-PTEN (1:1,000; cat. no.
9552S; Cell Signaling Technology, Inc.). After washing with TBS-T
(0.1% Tween 20), the membranes were incubated with horseradish
peroxidase-conjugated secondary antibodies (Anti-Rabbit, cat. no.
7074S; and Anti-Mouse, cat. no. 7076S; 1:5,000; Cell Signaling
Technology, Inc.) for 1 h at room temperature. Proteins were
visualized using the Pierce™ ECL Western Blotting
Substrate (cat. no. 32106; Thermo Fisher Scientific, Inc.). Values
were normalized to GAPDH as loading controls. Protein levels were
semi-quantified using densitometric analysis using Image J software
(version 1.49; National Institutes of Health).
RNA extraction and reverse
transcription-qPCR
Total RNA was isolated using TRIzol™
Reagent (Invitrogen; Thermo Fisher Scientific, Inc.) based on the
acid guanidinium thiocyanate-phenol-chloroform method. Total RNA
concentration was determined using a spectrophotometer (Nano
Drop™ 2000/2000c Spectrophotometer; Thermo Fisher
Scientific, Inc.). Complementary DNA was prepared from total RNA (1
µg) using the RevertAid First Strand cDNA Synthesis Kit (cat. no.
K1622; Thermo Fisher Scientific, Inc.). The thermocycling
conditions for cDNA synthesis were as follows: 65°C for 5 min, 55°C
for 50 min and 85°C for 5 min. qPCR was then performed using the
StepOnePlus™ Real-Time PCR system (Bio-Rad Laboratories,
Inc.) with the SYBR® Premix Ex Taq™ kit (cat.
no. RR820A; Takara Bio, Inc.). The thermocycling conditions for
qPCR were as follows: Initial denaturation, 95°C for 5 sec;
followed by 35 cycles of denaturation at 94°C for 15 sec, annealing
at 55°C for 25 sec and extension at 70°C for 30 sec. The primers
used for human PTEN were as follows: Sense,
5′-CAAGATGATGTTTGAAACTAT-3′ and antisense,
5′-CCTTTAGCTGGCAGACCACAA-3′. The primers used for mouse Occludin
were as follows: Sense, 5′-ACTGGGTCAGGGAATATCCA-3′ and antisense,
5′-TCAGCAGCAGCCATGTACTC-3′. The primers used for mouse ZO-1 were as
follows: Sense, 5′-AGGCTACCTTTGTATTCTC-3′ and antisense,
5′-TAGGGCACAGTATTGTATC-3′. The primers used for mouse Claudin-5
were as follows: Sense, 5′-CTTCCTGGACCACAACATCGTG-3′ and antisense,
5′-CACGTCGGATCATAGAACTCG-3′. The primers for human 18s, used as the
internal control, were as follows: Sense,
5′-GTAACCCGTTGAACCCCATT-3′ and antisense,
5′-CCATCCAATCGGTAGTAGCG-3′. The primers for mouse 18s, used as the
internal control, were as follows: Sense,
5′-GAGCGACCAAAGGAACCATA-3′ and antisense,
5′-CGCTTCCTTACCTGGTTGAT-3′. Dissociation curves were monitored to
assess the aberrant formation of primer-dimers. The fold change in
the interest gene expression was calculated using the
2−ΔΔCq method (26).
Histological analysis
Brain tissues from the orthotopic GBM model were
fixed with 4% (w/v) paraformaldehyde at room temperature for 24 h.
The fixed tissues were then embedded in paraffin and sectioned into
5 µm-thick slices. The sections were deparaffinized using xylene,
followed by rehydration through a graded series of alcohols (100,
80 and 70%), and finally rinsed in PBS. Next, hematoxylin and eosin
staining was performed by incubating the sections in hematoxylin
for 5 min at room temperature, followed by eosin for 2 min at room
temperature. For immunohistochemistry staining, tumor tissue
sections were fixed with 10% neutral buffered formalin at room
temperature for 24 h. After fixation, the sections were embedded in
paraffin using standard procedures. Tumor sections of a 5-µm
thickness were cut and mounted on slides. For antigen retrieval,
sections were treated with sodium citrate buffer (pH 6.0; cat. no.
C999; MilliporeSigma) and heated in a microwave for 3 min at 95°C.
After retrieval, the sections were rehydrated through a descending
alcohol series (100, 80 and 70% ethanol) and washed in PBS. The
sections were blocked with 1% bovine serum albumin (cat. no. 4378;
MilliporeSigma) in PBS for 1 h at room temperature, and then
stained with the following primary antibodies: Anti-MMP9 (1:100;
cat. no. MA5-15886; Thermo Fisher Scientific, Inc.), anti-PTEN
(1:200; cat. no. 9559S; Cell Signaling Technology, Inc.) anti-NDV
hemagglutinin-neuraminidase protein (1:200; HN; cat. no. sc-53562;
Santa Cruz Biotechnology, Inc.) and anti-Ki-67 (1:100; cat. no.
MA5-14520; Thermo Fisher Scientific, Inc.) overnight at 4°C.
HRP-conjugated anti-rabbit or anti-mouse IgG secondary antibodies
(cat. nos. AP160P and 12–348; MilliporeSigma) were then applied for
60 min at room temperature. Color was developed for 30 sec by
incubation with DAB. Sections were counterstained with hematoxylin
at room temperature for 3 min and observed under a light microscope
(Motic Instruments) at ×100 magnification.
Cell Counting Kit-8 (CCK-8) cell
proliferation assay
CCF-STTG1, U87-MG, T98G, Mesenchymal 83 and
Proneural X01 cells were seeded at 1×104 cells/well in
96-well plates (cat. no. 34096; SPL Life Sciences). On the next
day, the cells were treated with rNDV-PTEN or rNDV (0.3, 1 or 3
MOI) for 24 h. Cell proliferation was measured using a CCK-8 kit
(cat. no. CK04-1000; Dojindo Laboratories, Inc.) according to the
manufacturer's instructions. Briefly, cells were washed with PBS
and suspended in growth medium including CCK-8 reagent added at
1/100 the media volume. Cells were then incubated at 37°C for 1 h
in the dark. Cell proliferation was measured at a wavelength of 450
nm.
TUNEL assay
A TUNEL assay was used to detect DNA fragmentation,
such as apoptosis. U87-MG cells were seeded at 1×105
cells/well in a 6-well plate (cat. no. 30006; SPL Life Sciences).
Cells were treated with rNDV-PTEN or rNDV (1 MOI) for 24 h. After
24 h of incubation at 37°C with 5% CO2, the cells were
washed twice with PBS, detached from the plate using trypsin and
collected in a 15 ml tube. These cells were fixed in 100% ethanol
overnight at 4°C. A TUNEL assay was performed according to the
manufacturer's instructions (TUNEL Assay Kit-FITC; cat. no.
ab66108; Abcam). Following fixation, the cells were permeabilized
with 0.1% Triton X-100 in PBS for 2 min on ice. The cells were then
incubated with FITC-labeled dUTP in the presence of terminal
deoxynucleotidyl transferase at 37°C for 1 h. Stained cells were
analyzed using flow cytometry and fluorescence for FITC using a
NovoCyte Quanteon flow cytometer (Agilent Technologies, Inc.) and
fluorescence microscope (Zeiss Axio Imager M1; Zeiss GmbH) as per
the manufacturer's instructions (Agilent Technologies, Inc.). Data
acquisition was performed using a flow cytometer (FACS; NovoCyte
Quanteon flow cytometer; Agilent Technologies, Inc.), measuring
PE-A fluorescence intensity, and ~1,000 cells per sample were
analyzed to determine the extent of apoptosis. Flow cytometry data
were analyzed using NovoExpress software (version 1.6.2; http://www.agilent.com/ko-kr/product/research-flow-cytometry/flow-cytometry-software/novocyte-novoexpress-software-1320805).
After completing the FACS experiment, 100 µl of the stained cells
were transferred onto a cover slide. The cells were then assessed
using fluorescence microscopy (ZEISS LSM 980; Zeiss GmbH) to
evaluate and visualize the expression and localization of the TUNEL
(FITC).
Transwell assay
A Transwell assay was used to assess cell migration.
U87-MG cells were seeded at 1×105 cells/well, with
uninfected cells (CON) or rNDV (1 MOI) or rNDV-PTEN (1 MOI), into
6-well tissue culture plates for 24 h, followed by transfer of
5×105/ml cells in the upper Transwell chamber (24-well
plate; Corning, Inc.) and cultured with FBS-free medium at 37°C,
with 5% CO2. Complete growth medium with 10% FBS (Merck
KGaA) was added to the lower chamber and incubated for another 24 h
at 37°C, with 5% CO2. Cells on the upper side
(non-migrating cells) were then removed and migrated cells on the
lower face were washed with PBS, fixed with 4% paraformaldehyde at
room temperature for 15 min, and stained with DAPI at room
temperature for 10 min. The cells were counted in 5 random
high-power fields (×200 magnification) under a microscope (ZEISS
LSM 980; Zeiss GmbH) and averaged.
IVIS
Mice were anesthetized with 2.5% isoflurane for
induction and maintained with 1.5% isoflurane until the completion
of IVIS imaging. Luciferase imaging was performed using an in
vivo optical imaging system (IVIS Lumina XR; PerkinElmer, Inc.)
15 min after intraperitoneal injection of 100 µl Luciferin (30
mg/ml). The images were captured and then the signal was displayed
as Radiant Efficiency (Photons/sec/cm2/steradium (sr) or
µW/cm2). Images of the region-of interest were analyzed
using the Living imaging 4.4 software (PerkinElmer, Inc.).
Magnetic resonance imaging (MRI)
Mice were transferred to the MRI unit using
individual portable cages within 30 min of anesthesia with 2.5%
isoflurane. Using a 32-channel phased array sensitivity encoding
head coil, MRI imaging was performed on the anesthetized mice with
a 7.0 Tesla Philips MR scanner (Ingenia; Philips Healthcare). Each
mouse was scanned in the upright position with a coil over its
head. Mice were mainlined under anesthesia with 1% isoflurane. The
following parameters were used for acquisition of the multi-shot
echo-planar imaging fast spin echo, with image reconstruction using
image-space sampling functions with b-values of 0 and 1,000
s/mm2, and 3 orthogonal directions of diffusion
gradients: Echo time, 45 msec; repetition time, 5,000 msec; slice
thickness, 8 mm; interslice gap, 1 mm; number of averaging, =2;
bandwidth, 936 Hz/pixel; echo train length, 35; field of view,
25.0×25.0 cm; and matrix size, 256×256 pixels.
Statistical analysis
Statistical analysis was performed using Prism 8
software (Dotmatics). Data are presented as mean ± standard
deviation. Differences between two groups were evaluated using
unpaired t-tests. For multiple comparisons, one-way analysis of
variance was performed followed by Tukey's multiple comparison
test. P<0.05 were considered to indicate a statistically
significant difference. Data are representative of at least three
independent experiments.
Results
Restoration of PTEN via NDV attenuates
the proliferation of U87-MG cells
A significant decrease in PTEN expression was
demonstrated in the U87-MG GBM cell line in comparison with normal
brain cells (astrocytes: CCF-STTG1). PTEN expression in other GBM
cell lines (T98G, Proneural X01 and Mesenchymal 83 cells) did not
demonstrate a significant decrease compared with astrocytes
(Fig. 1A and B). Therefore, U87-MG
cells were chosen as the primary focus for assessing the anticancer
effects of PTEN restoration.
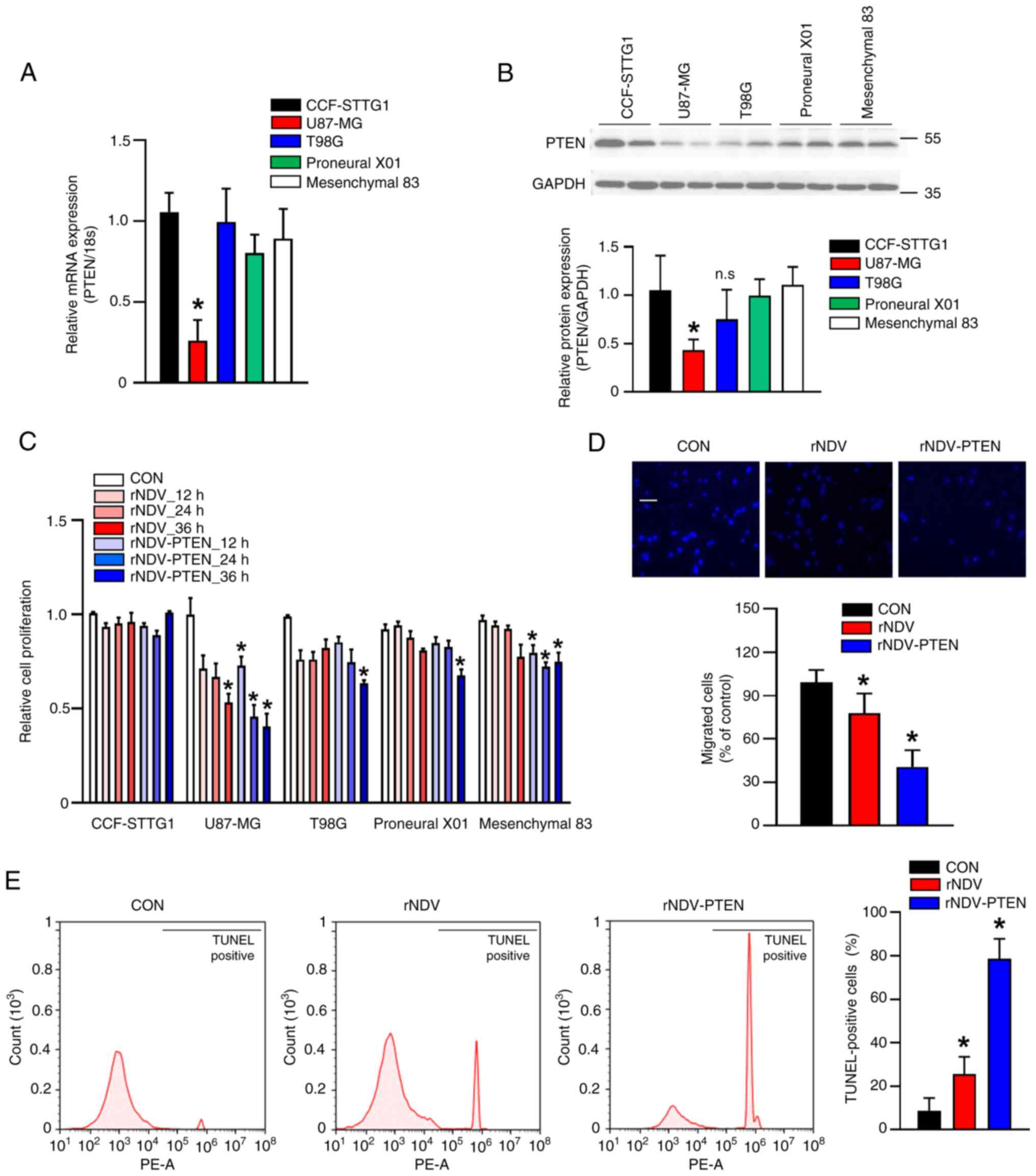 | Figure 1.mRNA expression and protein
expression of PTEN in normal astrocyte or GBM cell lines, and
inhibition by rNDV-PTEN of cell viability and migration by inducing
apoptotic cell death in U87-MG cells. PTEN (A) mRNA and (B) protein
expression in normal astrocyte (CCF-STTG1) and GBM cell lines
(U87-MG, T98G, Proneural X01 and Mesenchymal 83). CCF-STTG1 cells
and U87-MG cells were infected rNDV or rNDV-PTEN 1 MOI for 36 h.
(C) Cell viability assay performed using CCF-STTG1 cells and GBM
cell lines with rNDV or rNDV-PTEN 1 MOI treatment using a Cell
Counting Kit-8 Kit. (D) U87-MG cells were infected with rNDV or
rNDV-PTEN and a Transwell assay was performed to assess cell
migration. Cells migrated from the upper chamber to the lower
chamber were stained with DAPI. Scale bar, 50 µm. (E) Apoptosis
(DNA fragmentation) in U87-MG cells measured using TUNEL staining
after virus infection. *P<0.05 vs. CCF-STTG1 or CON. PTEN,
phosphatase and tensin homolog; GBM, glioblastoma; rNDV,
recombinant Newcastle disease virus; MOI, multiplicity of
infection; CON, control; n.s., not significant. |
The present study constructed rNDV-PTEN (20) to induce PTEN mRNA and protein
expression in the cytoplasm of U87-MG and CCF-STTG1 cells. In
normal cells, such as astrocytes, the Type I interferon (IFN)
pathway, particularly IFN-α, is active and effectively inhibits
viral replication by inducing an antiviral state, preventing the
proliferation of NDV. By contrast, cancer cells like those in GBM
often have a compromised IFN-α signaling pathway due to the
deletion of the cyclin-dependent kinase inhibitor 2A and Type I IFN
gene cluster (15). This impairment
allows NDV to replicate efficiently within cancer cells, leading to
selective oncolysis. Therefore, IFN-α serves a critical role in
maintaining antiviral defenses in normal cells, whereas its
dysfunction in cancer cells enables the oncolytic activity of the
virus (27). The results of the
present study demonstrate that in CCF-STTG1 cells, the virus did
not exhibit oncolytic activity, resulting in no significant change
in cell proliferation compared with GBM cell lines. However, in GBM
cell lines, there was a greater reduction in cell proliferation
with increased virus exposure time or when treated with rNDV-PTEN,
which contains the inserted PTEN gene. Statistical analysis
revealed a significant reduction in cell proliferation in GBM cell
lines compared with that in untreated cells (Fig. 1C). The reason for using the PTEN
gene is that 40% of patients with GBM have PTEN gene mutations, as
well as the U87-MG cell line we used. These mutations lead to the
dysfunction of the PTEN protein, which is associated with a worse
prognosis in GBM (28–30). Therefore, the present study aimed to
enhance the therapeutic effect on GBM by delivering and expressing
the PTEN gene through rNDV-PTEN. Treatment with rNDV-PTEN
significantly suppressed the proliferation of U87-MG cells in
comparison with its effects in the CCF-STTG1 and other GBM cell
lines (Fig. 1C). Furthermore, the
present study assessed the NDV-HN protein in astrocyte cells
(CCF-STTG1) and GBM cell lines in samples that were either
untreated (0 h, no treatment) or treated (36 h, rNDV-PTEN virus).
The results revealed that in astrocytes (CCF-STTG1), the expression
of the NDV-HN protein was significantly lower compared with that in
the other GBM cell lines (Fig.
S1). Taken together, U87-MG cells were selected for further
assessment of the anticancer effects of rNDV-PTEN.
A Transwell assay to evaluate U87-MG cell migration
revealed that most of the rNDV-PTEN-treated cells did not migrate
to the lower chamber containing the complete medium. Further
analysis, including DAPI staining of migrated cells, followed by
microscopic counting, demonstrated significantly decreased
migration (Fig. 1D). To assess the
mechanism through which PTEN restoration induces apoptosis in
U87-MG cells, a TUNEL assay was performed (Fig. 1E). TUNEL-positive cells were
observed in both rNDV and rNDV-PTEN, with rNDV-PTEN treatment
associated with ~2.5× more positive cells compared with rNDV. The
TUNEL assay (FITC) was performed using FACS and cell staining was
evaluated with a fluorescence microscope on a cover slide (Fig. S2). These findings indicate that
rNDV-PTEN treatment inhibited the migration of U87-MG cells and
induced DNA fragmentation, leading to apoptosis.
Restoration of PTEN via NDV regulates
AKT/mTOR pathway and increases apoptosis of U87-MG GBM cells
The deactivation (dephosphorylation) of AKT/mTOR and
apoptosis-associated signaling pathways in U87-MG cells were also
analyzed using immunoblot analysis. Specifically, these cells were
infected with rNDV or rNDV-PTEN at a MOI of 1 and collected for
analysis at 12, 24 and 36 h post-infection (hpi). The results
revealed that in comparison with the PTEN bands in rNDV-infected
cells, those in rNDV-PTEN-infected cells gradually increased
between 12 and 36 hpi and peaked at 36 hpi, indicating active virus
replication, with a significant increase in PTEN expression over
time (Fig. 2A). Additionally, the
levels of PCNA were assessed, a well-conserved protein in
eukaryotes and a proliferation marker expressed in cells undergoing
division (31). The level of MMP9
was also evaluated, which serves an essential role in local
proteolysis of the extracellular matrix and in leukocyte migration
(32,33). The results demonstrated decreased
PCNA and MMP9 expression levels in rNDV-PTEN-infected U87-MG cells
compared with those in uninfected cells (Fig. 2A). Additionally, the activation of
AKT/mTOR, as the endpoint of the PI3K pathway, contributes to the
malignant transformation of cells in several cancers (34). In the present study, AKT and mTOR
phosphorylation in cells infected with rNDV or rNDV-PTEN were
assessed. The results revealed that rNDV-PTEN treatment decreased
the levels of phosphorylated AKT and mTOR in a dose-dependent
manner (Fig. 2B). Furthermore, an
increase in the levels of Bax and cleaved caspases 3, 8 and 9 were
demonstrated, along with a decrease in the level of Bcl-2 in
rNDV-PTEN-infected cells, compared with that in uninfected cells,
indicating that this treatment induced apoptosis in GBM cells
(Fig. 2C).
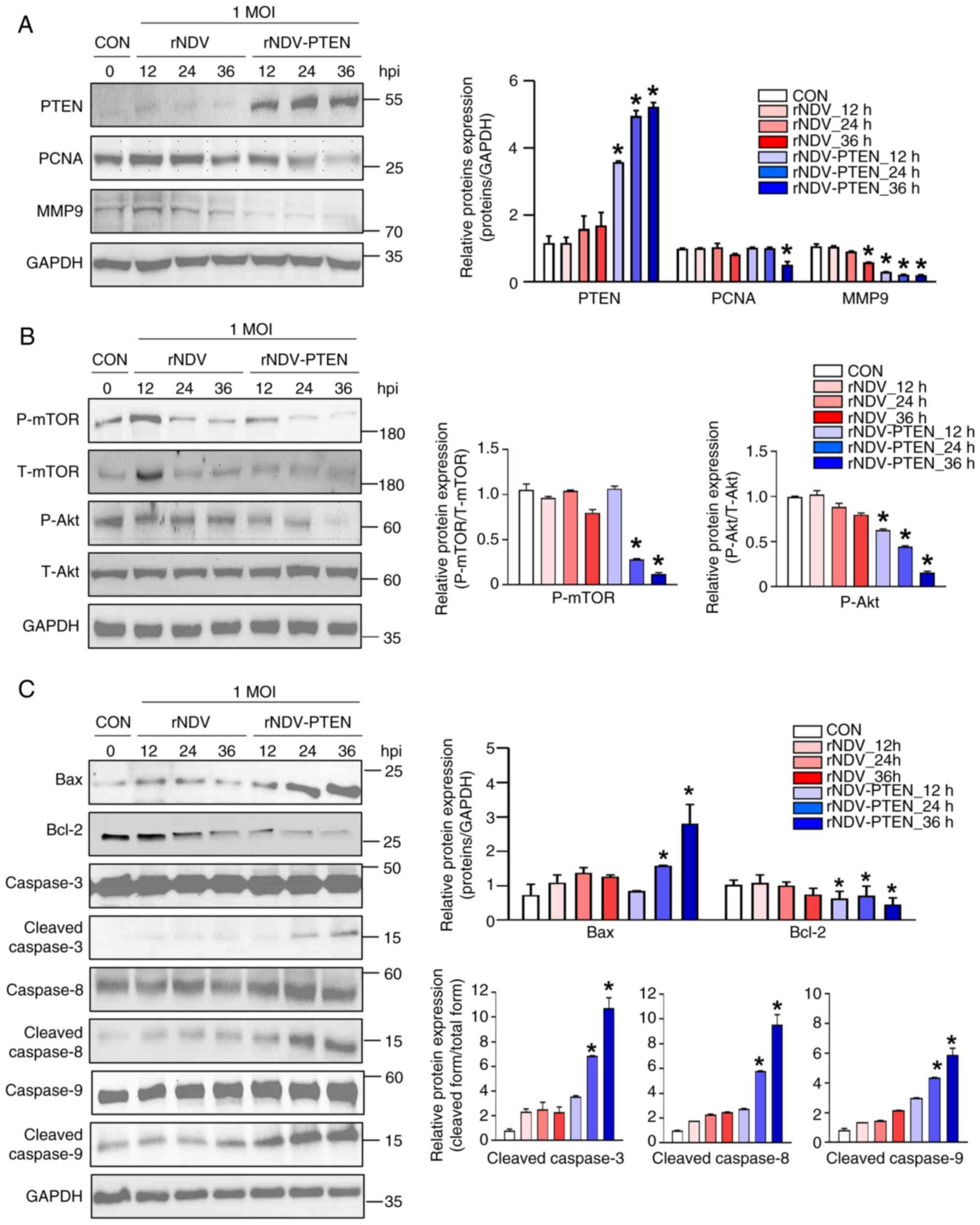 | Figure 2.Effect of rNDV-PTEN infection on
apoptotic cell death through imbalance of Akt/mTOR pathway. U87-MG
cells were infected with rNDV or rNDV-PTEN at an MOI of 1 for 12,
24 or 36 h. (A) Cell proliferation markers PCNA and MMP9, (B) mTOR
signaling-related proteins and autophagy-related proteins and (C)
pre-apoptotic cell death-related proteins were assessed using
immunoblotting analysis in U87-MG cells. GAPDH was used as an
internal control. *P<0.05 vs. CON. rNDV, recombinant Newcastle
disease virus; PTEN, phosphatase and tensin homolog; PCNA,
proliferating cell nuclear antigen; MMP9, matrix metallopeptidase
9; MOI, multiplicity of infection; CON, control; Bcl-2, B-cell
lymphoma 2; Bax, Bcl-2-associated X protein. |
rNDV-PTEN treatment suppresses cancer
growth in an orthotopic mouse model of GBM
To determine whether PTEN restoration significantly
suppresses U87-MG cell proliferation in an in vivo
preclinical mouse model and in vitro, U87-MG-Luc2 cells were
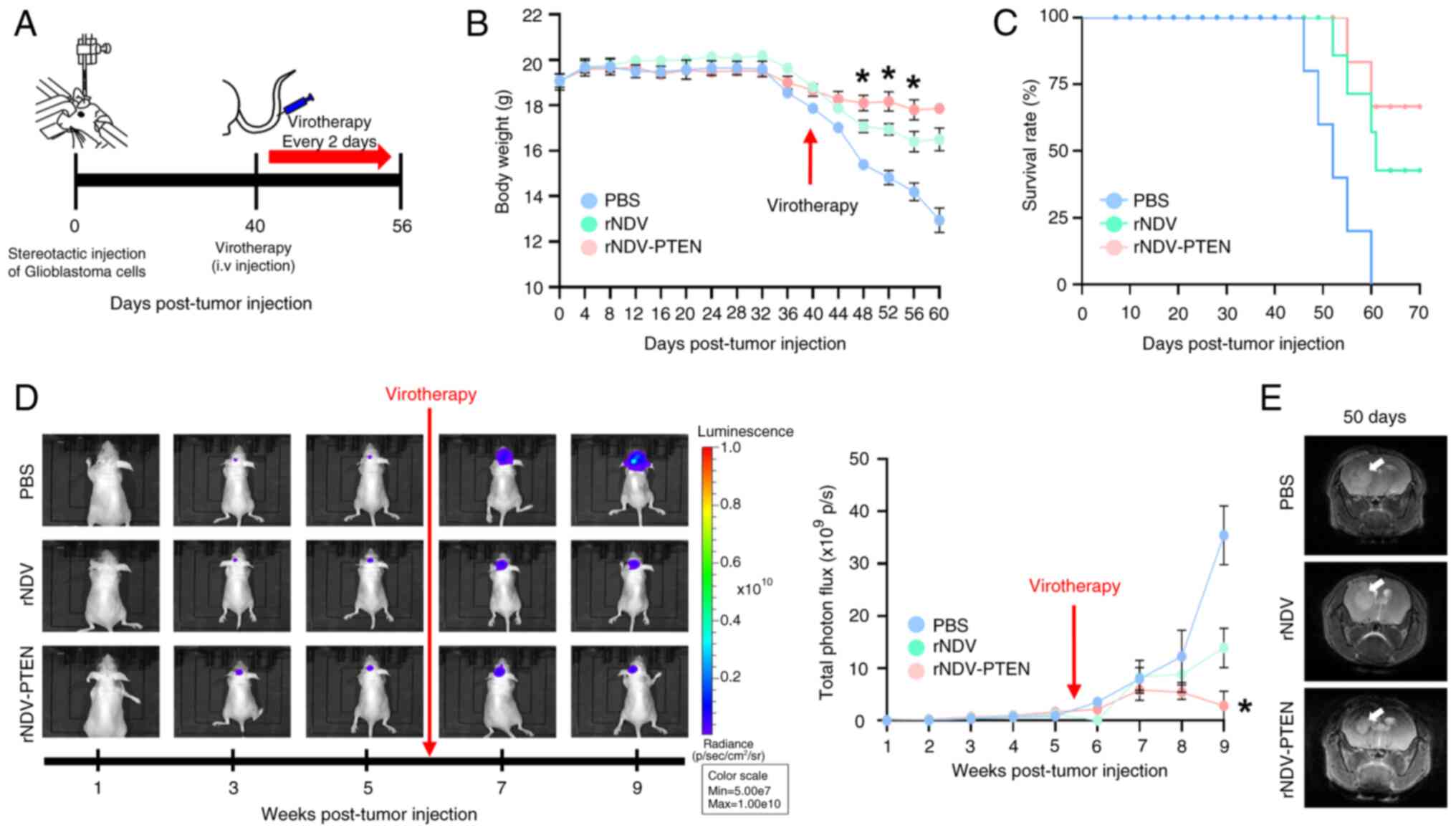
orthotopically xenografted into BALB/c nude mice (Fig. 3A). A total of 40 days after tumor
injection, virotherapy was initiated using intravenous injections
of rNDV-PTEN, rNDV or PBS (CON). The body weight (Fig. 3B) and survival rates (Fig. 3C) of the mice were also monitored
during the treatment period. To measure the in vivo efficacy
of PTEN restoration for tumor growth suppression in GBM mouse
models, the MRI findings and IVIS assessments of luciferase
activity in tumor scans were compared at 1–8 weeks after tumor
injection.
The results indicated weight loss was associated
with tumor progression rather than survival rate in the GBM
orthotopic mouse model (Fig. 3B).
The treatment group that received virotherapy experienced
significantly less weight loss compared with the control group mice
(PBS). Furthermore, whilst all control mice died by day 60
post-tumor establishment, the rNDV and rNDV-PTEN treated mice had
survival rates of 50 and 70%, respectively (Fig. 3C). MRI and IVIS assessments also
demonstrated markedly lower tumor growth in rNDV-PTEN-treated mice
than in rNDV- and PBS-treated mice (Fig. 3D and E). Moreover, the level of
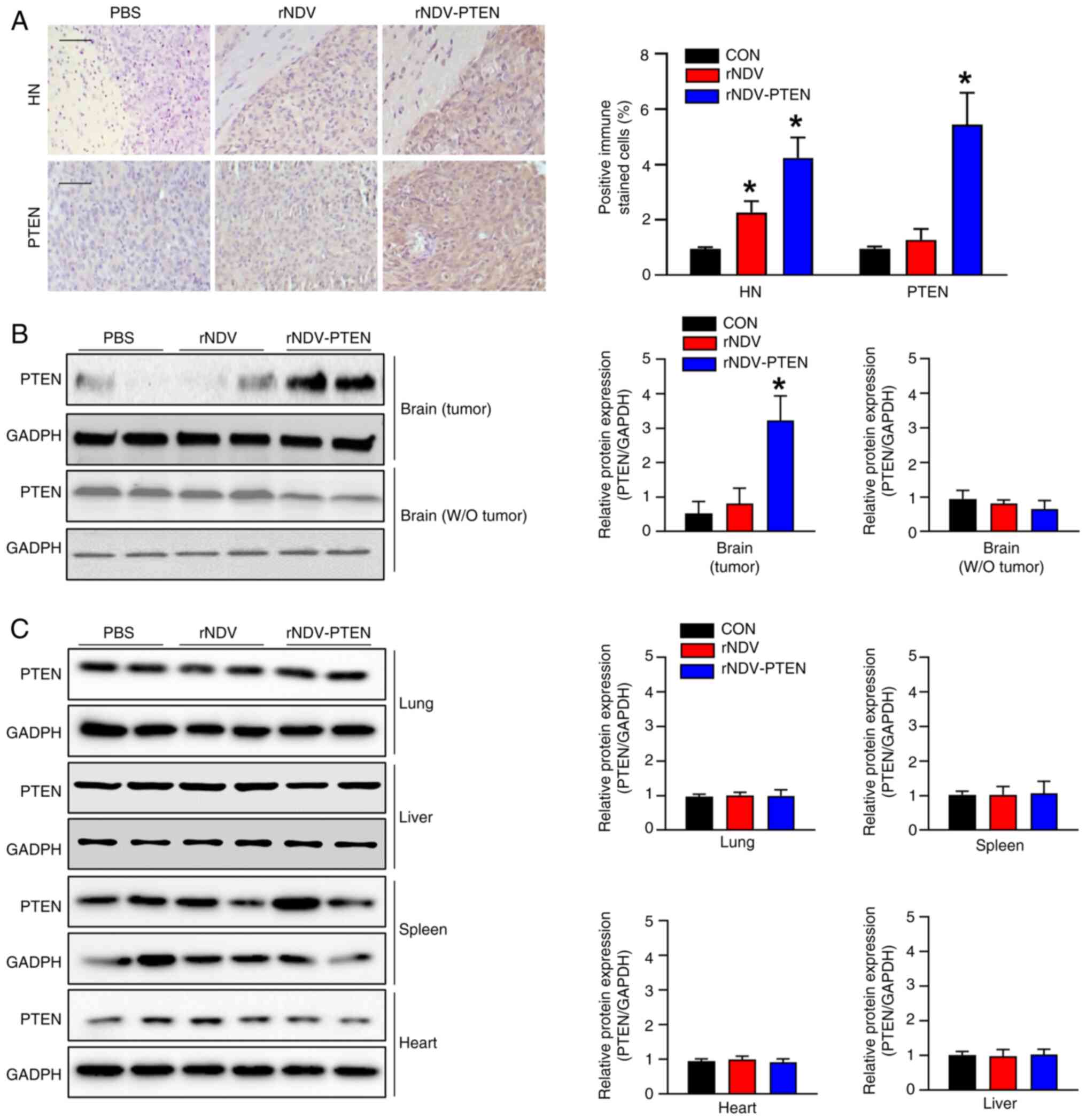
NDV-HN in the brain tumor tissues of rNDV- and rNDV-PTEN-treated
mice was significantly greater than that in the brain tissue of
PBS-treated mice (Fig. 4A). This
was further supported by the higher infection rate of rNDV-PTEN
observed through HN staining in Fig.
4A. The mRNA and protein expression levels of tight junction
proteins such as ZO-1, claudin-5 and occludin were also assessed;
however, the expression levels did not significantly change in the
brain tissue of mock mice models or tumor tissues in rNDV-PTEN,
rNDV and PBS-treated mice (Fig.
S3). This indicates that NDV crosses the BBB to reach the tumor
tissue without disrupting tight junctions.
Immunohistochemical analysis was used to assess
whether PTEN expression was upregulated in GBM tissues. The brains
of rNDV-PTEN-treated mice exhibited significantly higher PTEN
expression levels than those of PBS-treated mice (Fig. 4A). This finding indicates that the
treatment with rNDV and rNDV-PTEN successfully led to the presence
and activity of the NDV virus within the brain tumor tissues. The
increased levels of NDV-HN in rNDV- and rNDV-PTEN-treated mice
compared with PBS-treated mice is shown in Fig. 4A. These increases were statistically
significant, indicating a meaningful difference in NDV-HN and PTEN
levels between the treated and control groups, suggesting effective
viral targeting and replication in the tumor environment. This
indicates that the virus actively engaged with the tumor cells,
potentially leading to oncolytic effects, which were absent in the
PBS-treated group. The elevated presence of the viral protein in
the treated mice demonstrates the potential of rNDV and rNDV-PTEN
as therapeutic agents capable of reaching and affecting tumor sites
within the brain. Immunoblotting performed to assess PTEN
expression in cancerous and normal tissues in the brain and other
tissues revealed that NDV injected into the tail vein did not
affect PTEN expression in other organs compared with that in
PBS-treated mice, but significantly upregulated PTEN expression in
brain cancer cells (Fig. 4B and
C).
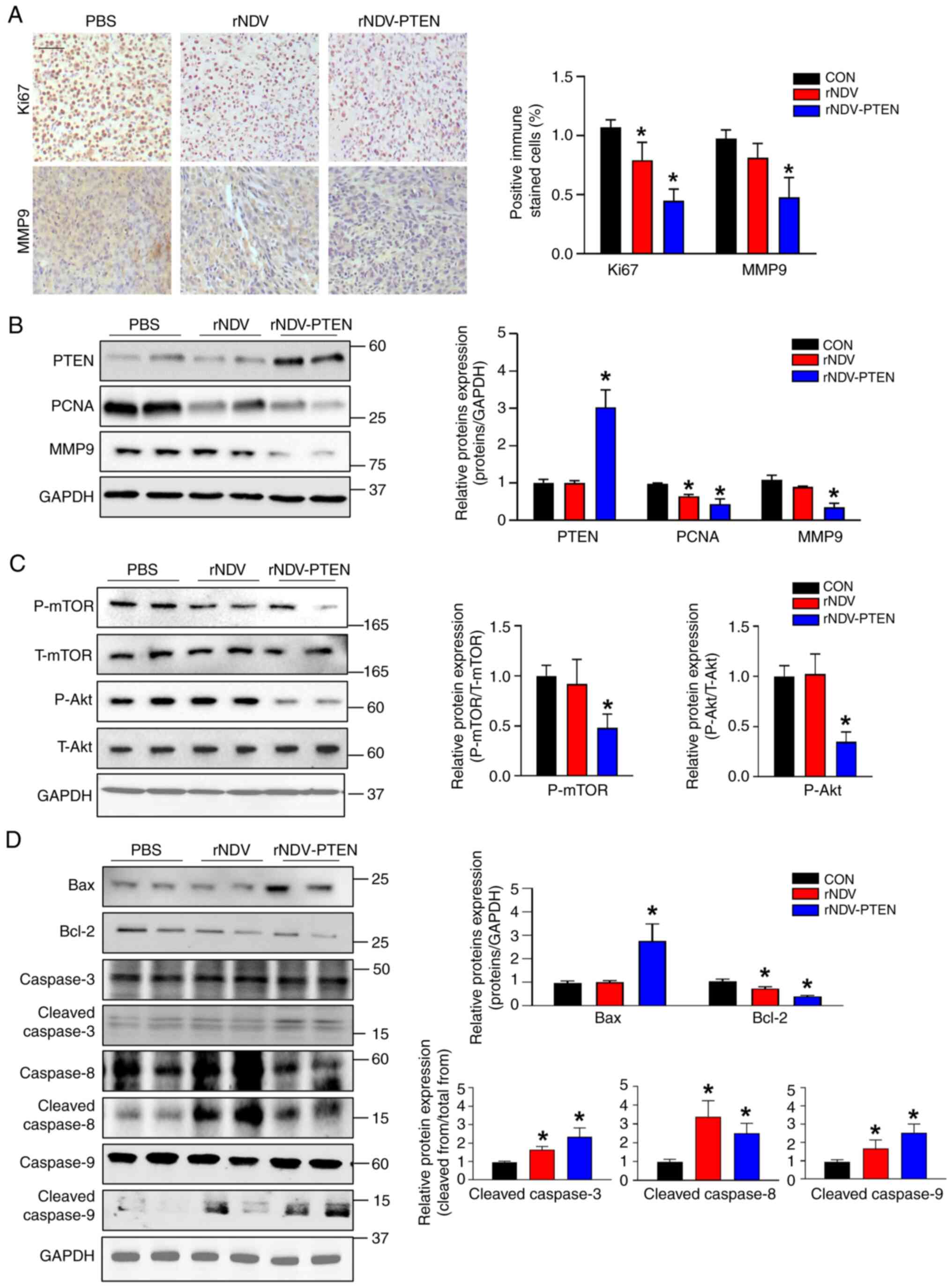
Staining of brain sections with antibodies against
Ki-67 and MMP9 significantly markedly lower cancer cell
proliferation and migration indices (brown) in rNDV-PTEN-treated
mice than in rNDV- and PBS-treated mice (Fig. 5A). Furthermore, analysis of PCNA and
MMP9 protein expression levels demonstrated similar results
(Fig. 5B). To elucidate the
mechanism by which rNDV-PTEN treatment induces apoptosis, the
regulation of the AKT/mTOR signaling pathway in GBM mouse models
was evaluated. The phosphorylation levels of AKT and mTOR were
significantly reduced in rNDV-PTEN-treated mice compared with those
in PBS-treated or rNDV-treated mice (Fig. 5C). Changes in apoptotic protein
markers were also assessed and significantly increased levels of
cleaved caspases 3, 8 and 9 and Bax, and decreased levels of Bcl-2
were demonstrated in rNDV-PTEN-treated mice compared with those in
PBS-treated mice. This observation indicates the activation of
apoptosis in the rNDV-PTEN-treated group (Fig. 5D). Taken together, the results
suggest that PTEN restoration induces apoptosis during GBM cell
proliferation and migration by disrupting the AKT/mTOR signaling
pathway.
Discussion
GBM remains one of the most challenging brain tumors
to treat due to its aggressive nature and the protective role of
the BBB, which limits the effectiveness of traditional therapies
such as surgery, radiation therapy and chemotherapy. Despite these
interventions, GBM often recurs, and patient prognosis remains
poor, with a median survival of ~15 months from diagnosis (35). This highlights the urgent need for
novel therapeutic strategies that can effectively target GBM cells
and overcome the BBB.
The current delivery system for NDV offers several
advantages over traditional delivery methods like those used for
herpes simplex virus (HSV) and adeno-associated virus (AAV)
(36). Unlike HSV and AAV, which
often require direct injection into tumors for effective delivery,
NDV can be administered intravenously (37,38).
This capability allows NDV to circulate through the bloodstream and
reach metastatic sites that are difficult to access with direct
injections, thereby enhancing its therapeutic reach and efficacy
(39). Furthermore, NDV
demonstrates a high level of tumor selectivity and low
immunogenicity, reducing the risk of adverse effects on normal
tissues (40). Clinical trials have
reported that NDV can be effectively delivered systemically,
offering a more versatile and patient-friendly approach compared
with localized injection strategies (41). These characteristics make NDV a
superior choice for treating cancers that are not easily accessible
by direct injection, highlighting its potential as an effective and
innovative viral therapy (42).
The present study assessed the therapeutic potential
of an rNDV-PTEN in treating GBM. The use of oncolytic viruses like
NDV offers a promising approach due to their ability to selectively
replicate in and kill tumor cells whilst sparing normal cells.
However, several limitations should be acknowledged in the present
study. Firstly, the study was conducted in a preclinical setting
using in vitro and animal models, which may not fully
replicate the complexity of human glioblastoma. Further clinical
trials are necessary to validate the efficacy and safety of
rNDV-PTEN in patients with GBM. Secondly, while rNDV-PTEN
demonstrated its ability to cross the blood-brain barrier in animal
models, the precise mechanisms facilitating this process remain
unclear and warrant further investigation. Lastly, the
heterogeneity of GBM tumors presents a challenge, as PTEN
deficiency is not uniform across all GBM cases. Therefore, the
therapeutic benefits of rNDV-PTEN may vary depending on the
molecular profile of the tumor, which highlights the need for
personalized treatment strategies based on genetic screening. NDV
induces cancer cell death through multiple mechanisms, including
apoptosis, autophagy and necroptosis, and has been reported to
enhance the antitumor immune response (43). A critical challenge in GBM treatment
is the BBB, a highly selective barrier that restricts the entry of
therapeutic agents into the brain. The BBB is composed of BMECs,
astrocytes and pericytes, forming tight junctions that prevent
non-selective entry of substances (19). The present study aimed to determine
whether rNDV-PTEN could cross the BBB and deliver PTEN to GBM cells
effectively.
The present study selected U87-MG cells as it has
been reported that 30–40% of GBM patients with PTEN gene mutations
exhibit low PTEN protein expression, which is associated with a
poorer prognosis compared with that in patients without PTEN
mutations (44,45). Furthermore, mutation or depletion of
PTEN leads to an increase in GBM progression. Low expression level
of PTEN mediates poor prognosis in GBM and by increasing
proliferation and invasion, it eventually promotes the malignancy
of tumor cells (11,46). Moreover, the loss of PTEN function
is associated with more aggressive tumors and resistance to
conventional therapies (47). Given
the results of the present study with this significant subset of
GBM cases, the findings suggest that rNDV-PTEN could serve as a
promising therapeutic strategy for patients with PTEN-deficient
tumors. The ability of rNDV-PTEN to restore PTEN expression in
U87-MG cells and inhibit the AKT/mTOR signaling pathway, leading to
the inhibition of cell migration and apoptosis, emphasizes its
potential efficacy in the GBM cells. Therefore, the present study
highlights the relevance and potential impact of rNDV-PTEN as a
targeted therapy for PTEN-deficient tumors.
Furthermore, the results of the present study
demonstrated that rNDV-PTEN treatment significantly restored PTEN
expression in U87-MG cells or the orthotopic mouse model, leading
to decreased cell proliferation and migration, and induced
apoptosis through the inhibition of the AKT/mTOR signaling pathway
(Figs. 2B and 5B). rNDV is an intrinsic oncolytic virus
that has been reported to have tumor-selective replication
capabilities, resulting in lysis and apoptosis in several cancer
cell types (43,48). Indeed, the results of the present
study also confirmed that treatment with rNDV alone increased
apoptosis compared with no treatment in U87-MG cells or PBS-treated
mice. However, despite the apoptosis-inducing effects of rNDV, the
present study aimed to further enhance its antitumor efficacy by
modifying rNDV to deliver the tumor suppressor gene PTEN, thereby
restoring PTEN protein levels. This modification not only amplified
the apoptosis signaling pathways but also led to a significant
reduction in the expression of cancer metastasis factors.
Specifically, in cells treated with rNDV-PTEN, the present study
observed a more pronounced increase in pro-apoptotic markers such
as Bax and cleaved caspases 3, 8 and 9 (Figs. 2C and 5D) compared with treatment with rNDV
alone. These findings are consistent with previous studies that
have reported PTEN restoration can inhibit tumor growth and enhance
apoptosis in several cancer models (49,50).
In vivo imaging and histological analysis of
the orthotopic GBM mouse model demonstrated that rNDV-PTEN
treatment resulted in a significant reduction in tumor size
compared with controls. MRI and IVIS imaging confirmed that
rNDV-PTEN could effectively target and reduce GBM growth in the
brain (Figs. 3E and 4A). Notably, the present study did not
detect any disruption of the BBB tight junctions following
rNDV-PTEN treatment, as demonstrated by MRI and protein expression
analyses. This indicates that rNDV-PTEN can cross the BBB without
compromising its integrity, likely through a transcellular pathway
similar to that used by SARS-CoV-2 (18). The ability of rNDV-PTEN to cross the
BBB and deliver PTEN directly to GBM cells is a significant finding
(Figs. 4A and S3). Traditional gene therapy vectors,
such as adenoviruses or adeno-associated viruses, cannot
effectively cross the BBB and are limited by potential
immunogenicity and host genome integration issues (51). In contrast, NDV offers a safer and
potentially more effective delivery mechanism, particularly for
brain tumors.
PTEN serves a crucial role in regulating cell
proliferation and migration, and its loss or mutation is associated
with a poor prognosis in patients with GBM (52). By restoring PTEN expression,
rNDV-PTEN treatment inhibits the AKT/mTOR signaling pathway,
reducing tumor growth and enhancing the effectiveness of
conventional therapies (53). The
mechanism by which rNDV-PTEN induces apoptosis in GBM cells
involves the inhibition of the AKT/mTOR pathway. The AKT/mTOR
pathway is a critical regulator of cell survival, proliferation and
metabolism, and its dysregulation is common in many cancers,
including GBM (54). PTEN
negatively regulates this pathway by dephosphorylating
phosphatidylinositol-3,4,5-trisphosphate, thereby preventing AKT
activation. In the present study, rNDV-PTEN treatment restored PTEN
expression, leading to decreased phosphorylation of AKT and mTOR,
increased levels of pro-apoptotic markers, and reduced cell
proliferation and migration.
Additionally, the findings of the present study
suggest that rNDV-PTEN treatment can modulate the tumor
microenvironment by reducing angiogenesis and metastasis.
Histological analysis revealed decreased expression of angiogenic
markers and metastasis markers in rNDV-PTEN-treated tumors
(Fig. 5A and B). Moreover, the data
indirectly indicated that rNDV-PTEN could cross the BBB, as
demonstrated by the expression of the NDV-HN and PTEN protein
(transgene) in brain tumor tissues detected through
immunohistochemistry analysis (Fig. 4A
and B). No changes in the tight junction proteins and mRNA
(Fig. S2A and B) suggests that the
virus could infect a transcellular, rather than paracellular,
mechanism to cross the BBB, a hypothesis supported by similar
findings in SARS-CoV-2 research (18). However, the present study did not
directly observe the movement of viruses using fluorescent markers
such as luciferase, indicating that the conclusions of the present
study are based on indirect evidence. Despite these promising
results, further research is needed to fully understand the
mechanism by which NDV crosses the BBB and optimize the delivery
system for clinical applications. Future studies should focus on
elucidating the pathways involved in NDV transcytosis across the
BBB and exploring the potential of combining rNDV-PTEN with other
therapeutic modalities to enhance its efficacy. Additionally,
long-term studies are needed to evaluate the safety and efficacy of
rNDV-PTEN in clinical settings and to determine the potential for
resistance development to this therapeutic approach.
In conclusion, the findings of the present study
demonstrate that rNDV-PTEN is a potent oncolytic virus capable of
crossing the BBB and delivering the PTEN gene to GBM cells, thereby
inducing apoptosis and inhibiting tumor growth. Inhibition of the
AKT/mTOR pathway and subsequent activation of pro-apoptotic markers
indicates the potential of rNDV-PTEN as a novel therapeutic agent
for treating GBM, particularly in patients with PTEN mutations or
low PTEN expression. These results provide a strong foundation for
further development and clinical testing of rNDV-PTEN as a
promising treatment for GBM.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present research was supported by the research fund of
Chungnam National University and by the Technology Development
Program, funded by the Ministry of SMEs and Startups (grant no.
S3271268).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HJ initiated and designed the study. SK and BKJ
performed most of the experiments. SK, BKJ and HJ wrote the
manuscript. JK and MK performed certain parts of the experiments.
SHJ, JHJ and CSK were responsible for managing and processing data
and performing quality checks to ensure data accuracy and
consistency. HJ supervised the study. All authors have read and
approved the final manuscript. HJ and SK confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
All animal studies were approved by and performed
and in the animal facility following the guidelines of the
Institutional Animal Use and Care Committee of Chungnam National
University Hospital (Daejeon, Republic of Korea; approval no.
CNUH-021-A0042). The animal experiments (in vivo experiment)
complied with the Animal Research: Reporting of In Vivo
Experiments guidelines (55).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ostrom QT, Gittleman H, Fulop J, Liu M,
Blanda R, Kromer C, Wolinsky Y, Kruchko C and Barnholtz-Sloan JS:
CBTRUS statistical report: primary brain and central nervous system
tumors diagnosed in the United States in 2008–2012. Neuro Oncol. 17
(Suppl 4):iv1–iv62. 2015. View Article : Google Scholar
|
|
2
|
Nelson CP, Bloom DA, Kinast R, Wei JT and
Park JM: Long-term patient reported outcome and satisfaction after
oral mucosa graft urethroplasty for hypospadias. J Urol.
174:1075–1078. 2005. View Article : Google Scholar
|
|
3
|
Luchsinger JA: Type 2 diabetes, related
conditions, in relation and dementia: An opportunity for
prevention? J Alzheimers Dis. 20:723–736. 2010. View Article : Google Scholar
|
|
4
|
Koide S, Koide A and Lipovšek D:
Target-binding proteins based on the 10th human fibronectin type
III domain (10Fn3). Methods Enzymol. 503:135–156. 2012.
View Article : Google Scholar
|
|
5
|
Maehama T and Dixon JE: The tumor
suppressor, PTEN/MMAC1, dephosphorylates the lipid second
messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem.
273:13375–13378. 1998. View Article : Google Scholar
|
|
6
|
Chen JK, Taipale J, Cooper MK and Beachy
PA: Inhibition of Hedgehog signaling by direct binding of
cyclopamine to Smoothened. Genes Dev. 16:2743–2748. 2002.
View Article : Google Scholar
|
|
7
|
Choi SW, Lee Y, Shin K, Koo H, Kim D, Sa
JK, Cho HJ, Shin HM, Lee SJ, Kim H, et al: Mutation-specific
non-canonical pathway of PTEN as a distinct therapeutic target for
glioblastoma. Cell Death Dis. 12:3742021. View Article : Google Scholar
|
|
8
|
Golovina VA and Blaustein MP: Spatially
and functionally distinct Ca2+ stores in sarcoplasmic and
endoplasmic reticulum. Science. 275:1643–1648. 1997. View Article : Google Scholar
|
|
9
|
van Noort J, Verbrugge S, Goosen N, Dekker
C and Dame RT: Dual architectural roles of HU: Formation of
flexible hinges and rigid filaments. Proc Natl Acad Sci USA.
101:6969–6974. 2004. View Article : Google Scholar
|
|
10
|
Chen C, Zhu S, Zhang X, Zhou T, Gu J, Xu
Y, Wan Q, Qi X, Chai Y, Liu X, et al: Targeting the synthetic
vulnerability of PTEN-deficient glioblastoma cells with MCL1
inhibitors. Mol Cancer Ther. 19:2001–2011. 2020. View Article : Google Scholar
|
|
11
|
Du L, Zhang Q, Li Y, Li T, Deng Q, Jia Y,
Lei K, Kan D, Xie F and Huang S: Research progress on the role of
PTEN deletion or mutation in the immune microenvironment of
glioblastoma. Front Oncol. 14:14095192024. View Article : Google Scholar
|
|
12
|
Chen H, Mei L, Zhou L, Shen X, Guo C,
Zheng Y, Zhu H, Zhu Y and Huang L: PTEN restoration and PIK3CB
knockdown synergistically suppress glioblastoma growth in vitro and
in xenografts. J Neurooncol. 104:155–167. 2011. View Article : Google Scholar
|
|
13
|
Chen Z, Varney ML, Backora MW, Cowan K,
Solheim JC, Talmadge JE and Singh RK: Down-regulation of vascular
endothelial cell growth factor-C expression using small interfering
RNA vectors in mammary tumors inhibits tumor lymphangiogenesis and
spontaneous metastasis and enhances survival. Cancer Res.
65:9004–9011. 2005. View Article : Google Scholar
|
|
14
|
Jiang K, Song C, Kong L, Hu L, Lin G, Ye
T, Yao G, Wang Y, Chen H, Cheng W, et al: Recombinant oncolytic
Newcastle disease virus displays antitumor activities in anaplastic
thyroid cancer cells. BMC Cancer. 18:7462018. View Article : Google Scholar
|
|
15
|
García-Romero N, Palacín-Aliana I,
Esteban-Rubio S, Madurga R, Rius-Rocabert S, Carrión-Navarro J,
Presa J, Cuadrado-Castano S, Sánchez-Gómez P, García-Sastre A, et
al: Newcastle disease virus (NDV) oncolytic activity in human
glioma tumors is dependent on CDKN2A-type I IFN gene cluster
codeletion. Cells. 9:14052020. View Article : Google Scholar
|
|
16
|
Burman B, Pesci G and Zamarin D: Newcastle
disease virus at the forefront of cancer immunotherapy. Cancers
(Basel). 12:35522020. View Article : Google Scholar
|
|
17
|
Csatary LK, Gosztonyi G, Szeberenyi J,
Fabian Z, Liszka V, Bodey B and Csatary CM: MTH-68/H oncolytic
viral treatment in human high-grade gliomas. J Neurooncol.
67:83–93. 2004. View Article : Google Scholar
|
|
18
|
Zhang L, Zhou L, Bao L, Liu J, Zhu H, Lv
Q, Liu R, Chen W, Tong W, Wei Q, et al: SARS-CoV-2 crosses the
blood-brain barrier accompanied with basement membrane disruption
without tight junctions alteration. Signal Transduct Target Ther.
6:3372021. View Article : Google Scholar
|
|
19
|
Abbott NJ, Rönnbäck L and Hansson E:
Astrocyte-endothelial interactions at the blood-brain barrier. Nat
Rev Neurosci. 7:41–53. 2006. View Article : Google Scholar
|
|
20
|
Jang SH, Jung BK, An YH and Jang H: The
phosphatase and tensin homolog gene inserted between NP and P gene
of recombinant New castle disease virus oncolytic effect test to
glioblastoma cell and xenograft mouse model. Virol J. 19:212022.
View Article : Google Scholar
|
|
21
|
Oka N, Soeda A, Inagaki A, Onodera M,
Maruyama H, Hara A, Kunisada T, Mori H and Iwama T: VEGF promotes
tumorigenesis and angiogenesis of human glioblastoma stem cells.
Biochem Biophys Res Commun. 360:553–559. 2007. View Article : Google Scholar
|
|
22
|
Mao P, Joshi K, Li J, Kim SH, Li P,
Santana-Santos L, Luthra S, Chandran UR, Beno PV, Smith L, et al:
Mesenchymal glioma stem cells are maintained by activated
glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc
Natl Acad Sci USA. 110:8644–8649. 2013. View Article : Google Scholar
|
|
23
|
Lin W, Niu R, Park SM, Zou Y, Kim SS, Xia
X, Xing S, Yang Q, Sun X, Yuan Z, et al: IGFBP5 is an ROR1 ligand
promoting glioblastoma invasion via ROR1/HER2-CREB signaling axis.
Nat Commun. 14:15782023. View Article : Google Scholar
|
|
24
|
Spearman C: The method of ‘right and wrong
cases’ (constant stimuli) without gauss's formula. Br J Psychol.
2:227–242. 1908.
|
|
25
|
Kärber G: Beitrag zur kollektiven
behandlung pharmakologischer reihenversuche. Naunyn Schmiedebergs
Arch Exp Pathol Pharmakol. 162:480–483. 1931. View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Raftery N and Stevenson NJ: Advances in
anti-viral immune defence: Revealing the importance of the IFN
JAK/STAT pathway. Cell Mol Life Sci. 74:2525–2535. 2017. View Article : Google Scholar
|
|
28
|
Zhou S, Wang H, Huang Y, Wu Y and Lin Z:
The global change of gene expression pattern caused by PTEN
mutation affects the prognosis of glioblastoma. Front Oncol.
12:9525212022. View Article : Google Scholar
|
|
29
|
Li Y, Liang Y, Sun Z, Xu K, Fan X, Li S,
Zhang Z, Jiang T, Liu X and Wang Y: Radiogenomic analysis of PTEN
mutation in glioblastoma using preoperative multi-parametric
magnetic resonance imaging. Neuroradiology. 61:1229–1237. 2019.
View Article : Google Scholar
|
|
30
|
Hernandez J, Bonnedahl J, Eliasson I,
Wallensten A, Comstedt P, Johansson A, Granholm S, Melhus A, Olsen
B and Drobni M: Globally disseminated human pathogenic Escherichia
coli of O25b-ST131 clone, harbouring blaCTX-M-15, found in
Glaucous-winged gull at remote Commander Islands, Russia. Environ
Microbiol Rep. 2:329–332. 2010. View Article : Google Scholar
|
|
31
|
Strzalka W and Ziemienowicz A:
Proliferating cell nuclear antigen (PCNA): A key factor in DNA
replication and cell cycle regulation. Ann Bot. 107:1127–1140.
2011. View Article : Google Scholar
|
|
32
|
Wilhelm SM, Collier IE, Marmer BL, Eisen
AZ, Grant GA and Goldberg GI: SV40-transformed human lung
fibroblasts secrete a 92-kDa type IV collagenase which is identical
to that secreted by normal human macrophages. J Biol Chem.
264:17213–17221. 1989. View Article : Google Scholar
|
|
33
|
Yang JM, Schiapparelli P, Nguyen HN,
Igarashi A, Zhang Q, Abbadi S, Amzel LM, Sesaki H,
Quiñones-Hinojosa A and Iijima M: Characterization of PTEN
mutations in brain cancer reveals that pten mono-ubiquitination
promotes protein stability and nuclear localization. Oncogene.
36:3673–3685. 2017. View Article : Google Scholar
|
|
34
|
Cheung M and Testa JR: Diverse mechanisms
of AKT pathway activation in human malignancy. Curr Cancer Drug
Targets. 13:234–244. 2013. View Article : Google Scholar
|
|
35
|
Fine HA: Radiotherapy plus adjuvant
temozolomide for the treatment of glioblastoma-a paradigm shift.
Nat Clin Pract Oncol. 2:334–335. 2005. View Article : Google Scholar
|
|
36
|
Colón-Thillet R, Jerome KR and Stone D:
Optimization of AAV vectors to target persistent viral reservoirs.
Virol J. 18:852021. View Article : Google Scholar
|
|
37
|
Ferguson MS, Lemoine NR and Wang Y:
Systemic delivery of oncolytic viruses: Hopes and hurdles. Adv
Virol. 2012:8056292012. View Article : Google Scholar
|
|
38
|
Belete TM: The current status of gene
therapy for the treatment of cancer. Biologics. 15:67–77. 2021.
|
|
39
|
Freeman AI, Zakay-Rones Z, Gomori JM,
Linetsky E, Rasooly L, Greenbaum E, Rozenman-Yair S, Panet A,
Libson E, Irving CS, et al: Phase I/II trial of intravenous NDV-HUJ
oncolytic virus in recurrent glioblastoma multiforme. Mol Ther.
13:221–228. 2006. View Article : Google Scholar
|
|
40
|
Lemos de Matos A, Franco LS and McFadden
G: Oncolytic viruses and the immune system: The dynamic duo. Mol
Ther Methods Clin Dev. 17:349–358. 2020. View Article : Google Scholar
|
|
41
|
Li X, Sun X, Wang B, Li Y and Tong J:
Oncolytic virus-based hepatocellular carcinoma treatment: Current
status, intravenous delivery strategies, and emerging combination
therapeutic solutions. Asian J Pharm Sci. 18:1007712023. View Article : Google Scholar
|
|
42
|
Lin D, Shen Y and Liang T: Oncolytic
virotherapy: Basic principles, recent advances and future
directions. Signal Transduct Target Ther. 8:1562023. View Article : Google Scholar
|
|
43
|
Zamarin D and Palese P: Oncolytic
Newcastle disease virus for cancer therapy: Old challenges and new
directions. Future Microbiol. 7:347–367. 2012. View Article : Google Scholar
|
|
44
|
Hashemi M, Etemad S, Rezaei S, Ziaolhagh
S, Rajabi R, Rahmanian P, Abdi S, Koohpar ZK, Rafiei R, Raei B, et
al: Progress in targeting PTEN/PI3K/Akt axis in glioblastoma
therapy: Revisiting molecular interactions. Biomed Pharmacother.
158:1142042023. View Article : Google Scholar
|
|
45
|
Zhang P, Meng X, Liu L, Li S, Li Y, Ali S,
Li S, Xiong J, Liu X, Li S, et al: Identification of the prognostic
signatures of glioma with different PTEN status. Front Oncol.
11:6333572021. View Article : Google Scholar
|
|
46
|
Fraser MM, Zhu X, Kwon CH, Uhlmann EJ,
Gutmann DH and Baker SJ: Pten loss causes hypertrophy and increased
proliferation of astrocytes in vivo. Cancer Res. 64:7773–7779.
2004. View Article : Google Scholar
|
|
47
|
Han F, Hu R, Yang H, Liu J, Sui J, Xiang
X, Wang F, Chu L and Song S: PTEN gene mutations correlate to poor
prognosis in glioma patients: A meta-analysis. Onco Targets Ther.
9:3485–3492. 2016.
|
|
48
|
Reichard KW, Lorence RM, Cascino CJ,
Peeples ME, Walter RJ, Fernando MB, Reyes HM and Greager JA:
Newcastle disease virus selectively kills human tumor cells. J Surg
Res. 52:448–453. 1992. View Article : Google Scholar
|
|
49
|
Islam MA, Xu Y, Tao W, Ubellacker JM, Lim
M, Aum D, Lee GY, Zhou K, Zope H, Yu M, et al: Author correction:
Restoration of tumour-growth suppression in vivo via systemic
nanoparticle-mediated delivery of PTEN mRNA. Nat Biomed Eng.
2:9682018. View Article : Google Scholar
|
|
50
|
Li Y, Zhang P, Qiu F, Chen L, Miao C, Li
J, Xiao W and Ma E: Inactivation of PI3K/Akt signaling mediates
proliferation inhibition and G2/M phase arrest induced by
andrographolide in human glioblastoma cells. Life Sci. 90:962–967.
2012. View Article : Google Scholar
|
|
51
|
Pardridge WM: Drug transport across the
blood-brain barrier. J Cereb Blood Flow Metab. 32:1959–1972. 2012.
View Article : Google Scholar
|
|
52
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar
|
|
53
|
Huang X, You L, Nepovimova E, Psotka M,
Malinak D, Valko M, Sivak L, Korabecny J, Heger Z, Adam V, et al:
Inhibitors of phosphoinositide 3-kinase (PI3K) and phosphoinositide
3-kinase-related protein kinase family (PIKK). J Enzyme Inhib Med
Chem. 38:22372092023. View Article : Google Scholar
|
|
54
|
Fruman DA, Chiu H, Hopkins BD, Bagrodia S,
Cantley LC and Abraham RT: The PI3K pathway in human disease. Cell.
170:605–635. 2017. View Article : Google Scholar
|
|
55
|
Kilkenny C, Browne WJ, Cuthi I, Emerson M
and Altman DG: Improving bioscience research reporting: The ARRIVE
guidelines for reporting animal research. Vet Clin Pathol.
41:27–31. 2012. View Article : Google Scholar
|