Introduction
Metabolic rewiring in support of the energetic and
biosynthetic needs required for untethered cell proliferation is a
hallmark observed in the majority of human cancers (1). This encompasses not only changes in
glycolytic metabolism but alterations in fatty acid
synthesis/oxidation and amino acid utilization. Recently, activity
within the de novo serine synthesis pathway (SSP) has been
observed to be elevated in several tumor types, including NSCLC
(2–4). The SSP originates from the glycolytic
intermediate, 3-phosphoglycerate, which is converted to serine
through enzymatic activities of phosphoglycerate dehydrogenase
(PHGDH), phosphoserine aminotransferase (PSAT1), and phosphoserine
phosphatase (PSPH). Serine can then be utilized in protein
synthesis and as one-carbon units for the folate and methionine
cycles. Interestingly, serine is a non-essential amino acid (NEAA)
that can be imported from the extracellular space. Metabolism
through the SSP also generates NADH and α-ketoglutarate (α-KG), the
latter of which is a key Kreb cycle intermediate and epigenetic
regulator important for tumorigenic growth. Further, manipulation
of SSP enzymes has been previously demonstrated to alter cell
proliferation and migration/invasion in multiple cancer cell types
both in vitro and in vivo (5–7).
PSAT1 catalyzes the interconversion of
phosphohydroxypyruvate and glutamate to phosphoserine and α-KG.
Genetic knockdown studies have identified a requirement for PSAT1
in ovarian, colorectal, glioblastoma, and subtypes of both NSCLC
and breast cancer (8–11). Loss of PSAT1 not only suppresses
cell proliferation and metastatic potential but also promotes
chemosensitivity to several clinically used agents, such as
platinum-based chemotherapies and TKIs (12,13).
Functional analysis has identified multiple cellular effectors
affected by PSAT1 suppression, including E2F-CyclinD1 and β-catenin
(7,14,15).
However, a broad analysis of gene expression changes in response to
PSAT1 manipulation has not been examined, particularly in the
context of NSCLC.
Disruption of key metabolic mediators not only
hampers nutrient utilization but can also lead to specific
transcriptional changes (16,17).
Transcriptomic profiling, such as RNA-seq, under these conditions,
allows the interrogation of genome-wide changes controlled by these
metabolic activities. Bioinformatics analysis of these datasets can
then identify impacted pathways or altered cellular processes,
which can be further extended to define expression differences
between patient cohorts with respect to tumor staging, response to
therapy, and/or survival outcomes (18–20).
In our previous study, it was demonstrated that EGFR activation
promoted PSAT1 nuclear translocation, which was required for proper
nuclear localization of pyruvate kinase M2 (PKM2) (11). It was thus hypothesized that PSAT1
loss may yield a robust transcriptional response due to this
selective compartmentalization and the known transcriptional
activity of PKM2 (16). Given this
specific signal-dependent nuclear trafficking, the cellular
response to PSAT1 knockdown in EGFR mutant NSCLC was examined here.
Transcriptomic analysis detailed multiple affected pathways, such
as actin-cytoskeleton arrangement and β-catenin activity, which
were functionally verified in EGFR mutant NSCLC cells. In addition,
a comparative analysis of these differentially expressed genes with
transcriptomic changes observed in EGFR mutant NSCLC patient tumors
identified a gene signature with prognostic potential for RFS that
was able to distinguish high-risk patients with stage 1
disease.
Materials and methods
Reagents and antibodies
Antibodies against PKM2 (cat. no. 4053), β-catenin
(cat. no. 8480), OCT1 (cat. no. 8157), and α-tubulin (cat. no.
3873) were obtained from Cell Signaling Technology, Inc. The
anti-PSAT1 (cat. no. 10501-1-AP) antibody was purchased from
ProteinTech Group, Inc. β-actin (cat. no. A2228), 100× EmbryoMax
Nucleosides (cat. no. ES-008-D), and Dimethyl 2-oxoglutarate (cat.
no. 349631) were obtained from MilliporeSigma.
pGL4.49[luc2P/TCF-LEF/Hygro] Vector (E4611) and the Dual-Luciferase
Reporter Assay System (cat. no. E1960) were purchased from Promega
Corporation. Difco Noble Agar (cat. no. 214220) was purchased from
BD Biosciences and 100× NEAA (cat. no. 25-025) was obtained from
Corning Inc.
Cell culture
Generation of stably transfected PC9 cells (Control
and shPSAT1 PC9, Control-EV, shPSAT1-EV, shPSAT1-FLAG-PSAT1, and
shPSAT1-PKM2NLS-K433Q) were established from parental
PC9 cells provided by Dr Levi Beverly (University of Louisville)
after STR profiling, as described previously (11). The PSAT1 shRNA (TRCN0000291729:
target sequence: GCACTCAGTGTTGTTAGAGAT; pLKO-puro plasmid backbone)
and the pLKO-puro non-mammalian shRNA control (SHC202:
CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT) plasmids
used to establish the shPSAT1 and Control PC9 cells, respectively,
were purchased from Millipore Sigma. Control and shPSAT1 PC9 cells
were maintained in RPMI media (Gibco) supplemented with 10% FBS, 50
µg/ml gentamicin (Gibco; Thermo Fisher Scientific, Inc.), and 1
µg/ml puromycin (Gibco; Thermo Fisher Scientific, Inc.).
Control-EV, shPSAT1-EV, shPSAT1-FLAG-PSAT1, and
shPSAT1-PKM2NLS-K433Q PC9 cells were maintained in RPMI
media (Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
FBS, 50 µg/ml gentamicin (Gibco; Thermo Fisher Scientific, Inc.), 1
µg/ml puromycin, and 200 µg/ml geneticin (Gibco; Thermo Fisher
Scientific, Inc.). InvivoGen Mycostrips (rep-mys-50) were used to
continuously assess for mycoplasma contamination, and all cells
were cultured in humidified incubators at 37°C and 5%
CO2.
RNA-seq transcriptomic profiling and
analysis
Data acquisition and pre-processing
A total of three distinct sets of RNA for RNA-seq
profiling were prepared from 24-h serum-starved Control and shPSAT1
PC9 cells. Samples were submitted to the University of Louisville
Genomics Facility, which performed the library preparation and
sequencing reactions. Sequencing was performed on an Illumina
NextSeq 500 Platform using the High Output Kit v2 with 75 cycles
(cat. no. FC-40402005, Illumina, Inc.). Sequencing data has been
submitted to the Gene Expression Omnibus (GEO; accession no.
GSE173270). The KYINBRE Bioinformatics core was used to perform the
initial data analysis. Raw fastq files were mapped to the human
hg38 reference genome using tophat2 (version 2.0.13) (21). Differentially expressed genes were
determined for each pairwise comparison using the tuxedo suite
(cufflinks-cuffdiff2) (version 2.2.1) (22) with Ensembl v82 annotations.
Normalized Fragments Per Kilobase of transcript per Million mapped
reads (FPKM) expression values and statistical analysis results
from cuffdiff2, including P- and q-value with ENSEMBL gene ID, were
downloaded for further investigation.
Identification of differentially
expressed genes (DEGs)
The following parameters served as the selection
criterion for DEGs: the absolute value of
log2(shPSAT1/Control) ≥0.48, FPKM value (Control or
shPSAT1) ≥5, and q-value ≤0.05. Genes were divided into two groups,
down-regulated genes (termed shPSAT1-down-regulated) and
up-regulated genes (termed shPSAT1-up-regulated), based on the
comparison between shPSAT1 and control PC9 cells. For hierarchical
cluster analysis, the normalized log-transformed expression txt
files were imported to Cluster 3 software and clustered based on
the average linkage (23). The
output was visualized using Java Treeview software (24).
Functional analysis
Functional and KEGG pathway analyses were conducted
by uploading the ENSEMBL IDs to MSigDB version 7.4 (https://www.gsea-msigdb.org/gsea/msigdb). The top 20
enriched pathways and datasets for KEGG pathways, chemically and
genetically perturbed data sets (CGP), and positional analysis with
FDR ≤0.05 were considered significant, and the top 50 enriched gene
ontology (GO) terms with FDR ≤0.05 for GO-biological process (BP)
and GO-cellular component (CC) analysis were filtered.
Transcription factor analysis was conducted using the MetaCore
Transcription Regulation algorithm with the default settings
(MetaCore™ version 22.1, build 70800, http://portal.genego.com/) by uploading the ENSEMBL
IDs of the DEGs with their fold-changes and q-values. The UCSC
human genome browser was used to map the genes enriched on specific
cytogenic bands (https://genome.ucsc.edu).
RNA isolation and reverse
transcription-quantitative (RT-q) PCR
Total RNA was extracted using a RNeasy Mini Kit
according to the manufacturer's instructions (Qiagen GmbH, cat. no.
74106). RNA quality and concentration were measured using a
Nanodrop RNA 6000 nano-assay (for RT-qPCR). A total of 2 µg total
RNA was reverse transcribed using a High-Capacity RNA-to-cDNA kit
according to the manufacturer's instructions (Thermo Fisher
Scientific, Inc; cat. no. 4387406) The cDNA sample was diluted by
adding 60 µl nuclease-free water to make an estimated final
concentration of 25 ng/µl. Then, 10 µl reaction mix was prepared by
adding 1 µl cDNA, 0.5 µl target probe (FAM conjugated), 0.5 µl ACTB
(VIC), 3 µl nuclease-free water, and 5 µl TaqMan Fast Advanced
Master Mix (Thermo Fisher Scientific, Inc., cat. no. 4444557) and
reactions were performed in accordance with the TaqMan Fast
Reaction Protocol on an AB StepOnePlus Real-Time PCR System
(Applied Biosystems; Thermo Fisher Scientific, Inc.). Data were
analyzed using the DDCq method (25) and β-actin was used as the reference
gene. The TaqMan probes for Real-time PCR were as follows: ACTB
(Hs01060665_g1), USP14 (Hs00193036_m1), VAPA (Hs00427749_m1),
NDUFV2 (Hs00221478_m1), TYMS (Hs00426586_m1), METTL4
(Hs01559838_m1), SEH1L (Hs01031566_m1), IMPA2 (Hs00274110_m1),
MYL12B (Hs01050560_m1), S100A4 (Hs00243202_m1), TMSB4X
(Hs03407480_gH), and FHOD1 (Hs01077922_m1).
Anchorage-independent growth
Metabolite rescue
A bottom layer of 0.6% noble agar in complete medium
was prepared in 6 cm dishes. In metabolite rescue experiments,
1×103 Control and shPSAT1 PC9 cells/dish were plated in
0.3% agar in RPMI-complete media supplemented with or without the
indicated metabolite/s: No supplement (−); single metabolite, NEAA,
Nucleoside, or 500 µM α-KG; double metabolites, (NEAA + 500 µM
α-KG), (NEAA + Nucleoside), (Nucleoside + 500 µM α-KG); and all
metabolites (NEAA+ Nucleoside+ 500 µM α-KG). Colonies were fed with
0.25% agar in RPMI complete medium with or without metabolite/s
every 3–4 days during the 21-day incubation. At the end of the
study, whole plate images were captured, and colonies were counted
using ImageJ version 1.53 (National Institutes of Health). Dimethyl
2-oxoglutarate was used as the α-KG supplement.
Rescue with PSAT1 or nuclear
acetyl-mimetic PKM2 expression
A bottom layer of 0.6% noble agar in a complete
medium was prepared in 6 cm dishes. A total of 1×103 PC9
cells/dish (Control-EV, shPSAT1-EV, and shPSAT1-FLAG-PSAT1 or
shPSAT1-PKM2NLS-K433Q) were plated in a 0.3% agar in
RPMI complete media solution. Colonies were fed with 0.25% agar in
RPMI complete medium every 3–4 days during the 21-day incubation.
At the end of the study, whole plate images were captured, and
colonies were counted using ImageJ.
Whole-cell protein extracts and
subcellular fractionation
Total protein was extracted using Pierce IP lysis
buffer supplemented with protease and phosphatase inhibitor
cocktail according to the manufacturer's protocol (Thermo Fisher
Scientific, Inc.; cat. no. 87787). Cytosolic and nuclear proteins
were isolated using the NE-PER kit (Thermo Fisher Scientific, Inc.
cat. no. 78835). A total of 15 µg cytoplasmic protein and 25 µg
nuclear protein was used for immunoblotting analyses.
Immunoblotting
Proteins within the whole-cell lysates, cytosolic
fraction, and nuclear fraction were resolved by SDS-PAGE and
transferred to PVDF membranes. Membranes were blocked and then
incubated with the indicated primary antibodies [PSAT1 1:1,000,
β-catenin 1:1,000, PKM2 1:1,000, β-actin 1:5,000] overnight at 4°.
Protein detection was performed using the appropriate
HRP-conjugated secondary anti-mouse or anti-rabbit antibody
(1:10,000) and visualized using a chemiluminescence reagent (ECL
Prime, MilliporeSigma).
Luciferase reporter assay
Control-EV, shPSAT1-EV, and shPSAT1-FLAG-PSAT1 PC9
cells were plated into 6-well plates and transfected with 2 µg
pGL4.49[luc2P/TCF-LEF/Hygro] using jetPEI with overnight incubation
(media changed after 24 h). A total of 48 h post-transfection,
stably transfected cells were selected using 200 µg/ml hygromycin
(TCF-LEF vector), 200 µg/ml geneticin (pcDNA3.1 vector), and 1
µg/ml puromycin (shRNA vector). For each study, 4×105
stable cells were plated into each well of a 6-well plate (3
replicates/condition). The following day, the media was replaced
with serum-free media and maintained for 24 h. Cells were then
harvested according to the Dual-Luciferase Reporter Assay protocol.
Firefly luciferase activity was determined using a 96-well plate
luminometer. Protein concentration was measured using a BCA Protein
assay and used to normalize the luciferase activity.
Phalloidin staining
Cells were plated into 4-well chamber slides
(Lab-Tek II Chamber slides, cat. no. 154526) and incubated in
serum-free media for 24 h. Samples were then fixed with 3.7%
paraformaldehyde in PBS solution for 10 min at room temperature and
washed three times with PBS. Then, the cells were permeabilized
with 0.1% Triton X-100 in PBS for 3 min and washed again. For
visualization, cells were incubated with a Rhodamine Phalloidin
(Invitrogen; Thermo Fisher Scientific, Inc., cat. no. R415) working
solution (5 µl stock/200 µl PBS) for 20 min in the dark at room
temperature. After three additional washes with PBS, slides were
covered with SlowFade Diamond Antifade Mount with DAPI (cat. no.
S36964) reagent. Images were captured using an Olympus FV-3000
confocal microscope equipped with Fluoview software (Olympus
Corporation) at ×40 magnifications.
Public microarray datasets
analysis
Data search and import
The EGFR mutant lung cancer datasets were chosen
based on the number of EGFR mutant tumor samples (n>10) with
paired or unpaired normal tissue samples and the availability of
relevant clinical information. According to these selection
criteria, GSE31210, GSE27262, GSE31547, GSE31548, GSE32863, and
GSE75037 datasets were imported to BRB-ArrayTool [version
4.6.1-Stable (June 2020)] using the NCBI GEO Series tool (26).
Identification of common gene
sets
shPSAT1-mediated down-regulated and up-regulated
gene lists obtained from the RNA-seq analysis were prepared
separately as txt files and saved under the user gene list folder
within the program files of ArrayTool. Expression of these genes
was filtered using the ArrayTool- re-filter option and normalized.
Genes whose expression was <20% of the expression data and
<1.5-fold change in either direction from the gene's median
value were excluded. DEGs from the RNA-seq profiling were directly
compared to those gene changes between EGFR mutant tumor and normal
lung samples using the ArrayTool-Class Comparison plugin and the
significance threshold of univariate analysis with P≤0.05 served as
the statistical threshold for significance. This was done to
identify which PSAT1-regulated genes from the RNA-seq screen were
also differentially expressed in EGFR mutant NSCLC. Importantly,
up-regulated genes in EGFR tumors should be down-regulated by
shPSAT1 and vice-versa. Genes with fold-changes (EGFR mutant
tumor/normal lung) ≥1.4 from the shPSAT1-down-regulated genes list
and genes with fold-changes (EGFR mutant tumor/normal lung) ≤0.71
from the shPSAT1-up-regulated genes list were considered
PSAT1-associated genes linked with EGFR mutant lung cancer. This
procedure was repeated for each dataset (GSE31210, GSE27262,
GSE31547, GSE31548, GSE32863, and GSE75037). Then, the common gene
sets were determined using a Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/).
The fold-change of common genes with the P-values and FDR-values
from each dataset were extracted using the ArrayTool-Class
Comparison tool.
Survival risk prediction with the
common gene sets
Among the datasets, GSE31210 was the only set
encompassing all the following clinical information on defined
NSCLC genotypes: KRAS mutant and EGFR/KRAS wild-type tumor data in
addition to EGFR mutant lung tumors and their pathological stage,
relapse-free survival (RFS) and overall survival (OS) data
[clinicopathologic characteristics of patient cohort described in
(27)]. Survival predictions for OS
and RFS using the expression data of the PSAT1-associated common
gene lists were performed using the BRB-ArrayTool survival risk
prediction function (28).
Principal component analysis with leave-one-out cross-validation
with 100 permutation tests was used to calculate prognostic indices
and classified the patients as high-risk and low-risk groups. This
analysis was then used to generate the permutated Kaplan-Meier
survival plots, time-dependent receiver-operating characteristics
(ROC) curves with area under the curve (AUC) values, and a table
containing the predicted genes associated with survival with their
cross-validated (CV) support % and covariant (wi) used in this
study.
Identification of a potential
PSAT1-associated metastatic gene signature
GSE14107 was imported as described above due to the
presence of a genome-wide expression profile of both parental PC9
cells and its metastatic brain subline of PC9-BrM3 (29). The ArrayTool-Class comparison plugin
determined DEGs with a significance threshold of univariate
analysis of P≤0.05. Down-regulated and up-regulated gene lists were
assigned based on fold-change (PC9-BrM3/PC9-Parental) ≥0.71 and
≤1.4, respectively. The GSE14107 gene list was compared with the
differential expression gene list from the PC9-shPSAT1 RNA
sequencing analysis using Venn diagrams to find common genes. A
heatmap was generated using Cluster 3 (23) and Java Treeview (24).
Statistical analysis
Comparisons were performed based on the number of
groups with one or more independent variables. A repeated-measures
one-way ANOVA with a post-hoc Tukey's multiple comparisons test was
used for comparisons between three groups (PSAT1 and nuclear PKM2
rescue studies). This data is presented in their respective figures
as the mean ± SEM. For analysis of the soft agar assay with
metabolite supplementation, a two-step analytical approach was
used: First, a two-way ANOVA with a post hoc Dunnett's multiple
comparison test was performed with raw data to examine the effect
of metabolite supplementation on both control and shPSAT1 cells.
Then, the ratio of colony numbers (shPSAT1/Control) within each
treatment was used for the repeated measures one-way ANOVA with a
post hoc Dunnett's multiple comparisons test to assess the effect
of rescue. This data is presented as a box & whisker blot with
error bars represented as 5–95%. All protein rescue and metabolite
supplementation studies were statistically analyzed using Prism
version 9 (GraphPad Software, Inc.). The number of experimental
replicates for each analysis is stated within the figure legends.
P≤0.05 was considered to indicate a statistically significant
difference. All statistical analysis of the publicly available
microarray data was performed with BRB-Array Tool. The class
comparison tool was used to perform two-sample T-test for GSE27262,
GSE31547, GSE31548, GSE32863, GSE75037, and GSE14107 and F-test
with pairwise analysis (EGFR mutant Stage I/Normal and EGFR mutant
Stage II/Normal) for GSE31210 for each gene. P≤0.05 served as the
statistical threshold for significance. Statistical analysis for
the survival predictions for OS and RFS using the expression data
of the PSAT1-associated common gene lists was performed using the
BRB-ArrayTool survival risk prediction tool function (28). Risk groups were generated through
the supervised principal component method described in (30). The leave-one-out cross-validation
(LOOCV) method was chosen to determine the survival risk groups and
used to generate the cross-validated Kaplan-Meier survival curve
and the estimation of cross-validated time-dependent
receiver-operating characteristic (ROC) curves. Values with
P<0.05 based on 100 permutations of the cross-validated log-rank
statistics for the Kaplan-Meier survival curves and the area under
the cross-validated ROC curves (AUC) were reported as statistically
significant.
Results
Determination of DEG in PSAT1 silenced
PC9 cells
Several reports have shown that metabolic enzymes
translocate into different cellular compartments, particularly the
nucleus, to exert non-canonical functions (17,31,32).
In our previous study, it was demonstrated that PSAT1 undergoes
nuclear translocation in an EGFR activation-dependent manner in
NSCLC cells (11). As a global
transcript analysis with respect to PSAT1 depletion had yet to be
reported in EGFR mutant NSCLC, a genome-wide gene expression
profiling was performed using RNA-seq technology to uncover
potential novel cellular processes altered by PSAT1 loss. A total
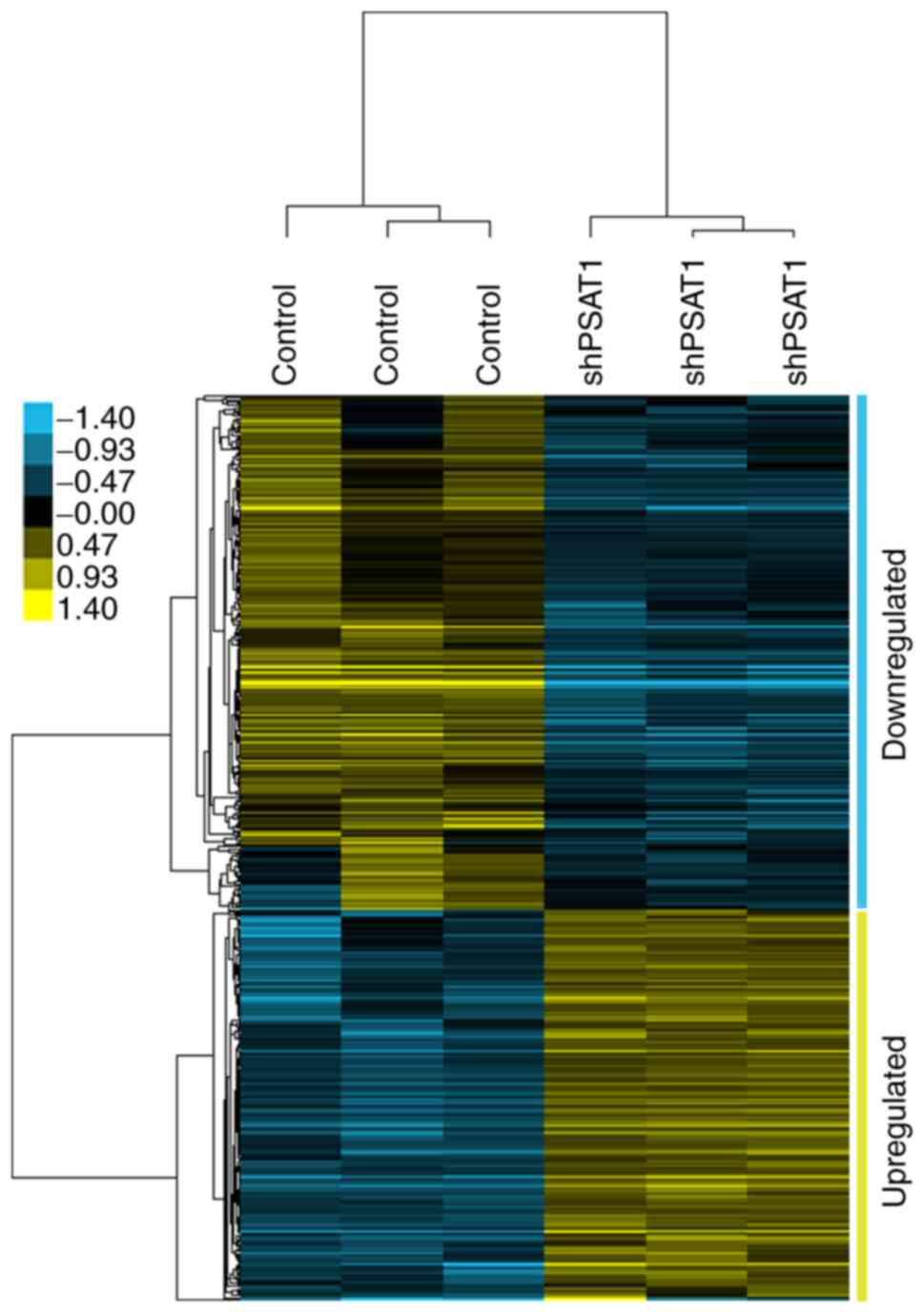
of 279 down-regulated and 211 up-regulated genes were identified
following PSAT1 silencing (Fig. 1
and Tables SI and II).
Pathways and biological processes
affected by PSAT1 suppression
These PSAT1-related DEGs were interrogated using
MSigDB (33). Both down-regulated
and up-regulated genes were assessed separately by MSigDB and the
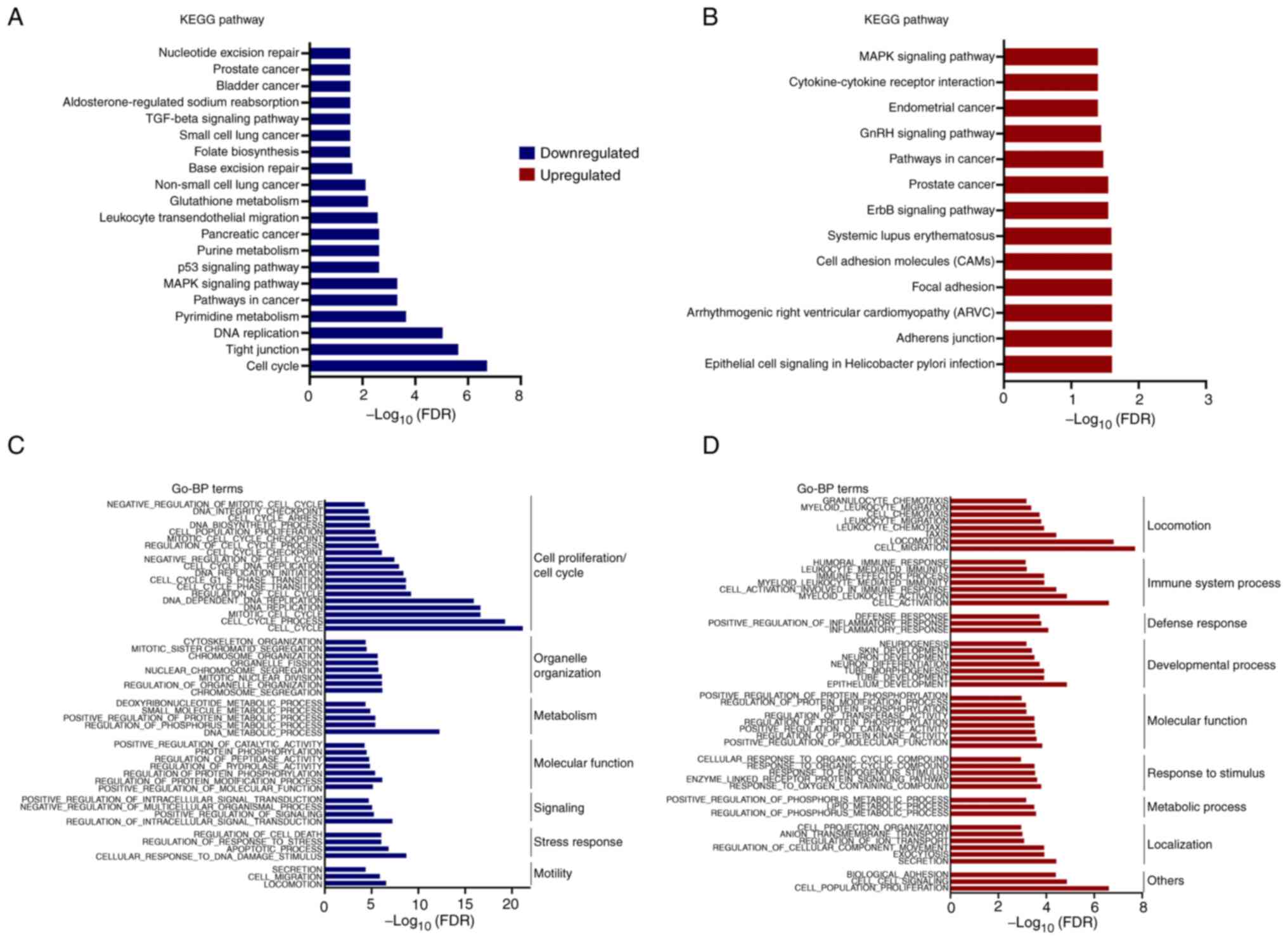
top 20 KEGG pathways were plotted as log10(FDR) in the
indicated pathways (Fig. 2A and B).
This analysis identified genes associated with folate biosynthesis,
glutathione metabolism, and purine and pyrimidine metabolism
pathways impacted by PSAT1 silencing (Fig. 2A), suggesting that decreased SSP
activity transcriptionally influences the serine biosynthetic
pathway. In addition, down-regulated genes were enriched in
well-known oncogenic pathways, including MAPK, P53, and TGFβ
signaling pathways, base excision repair, and the cell cycle
(Fig. 2A). Paradoxically, KEGG
pathway analysis of up-regulated genes also found cancer-related
pathways involving the MAPK signaling pathway, ERBB signaling
pathway, pathways in cancer, and endometrial cancer (Fig. 2B). As the DEGs enriched in KEGG
pathway analysis represented a small portion of DEGs (25/211 up,
58/279 down), further exploration was required to obtain a broader
understanding of processes impacted by PSAT1 loss.
GO analysis was performed using the MSigDB- GO-BP
(Biological Process) and GO-CC (Cellular Component) tools with the
top 50 signatures (FDR ≤0.05; Figs. 2C,
D, S1A and B). In line with
the KEGG pathways, down-regulated genes were enriched in the GO-BP
headings of cell cycle, cell proliferation, nucleotide-related
metabolism, and migration (Fig.
2C), while the protein products of these genes primarily
function within the nucleus or are associated with the cytoskeleton
(Fig. S1A). Interestingly, GO-BP
processes related to immune response, such as locomotion, immune
system processes, and defense response, were enriched in GO
analysis of up-regulated genes (Fig.
2D) and found to function within Golgi-ER trafficking and
secretion-related pathways (Fig.
S1B). These observations implicate the involvement of PSAT1 in
various cellular processes and highlight a requirement for
functional studies to elucidate the contribution of PSAT1 to these
pathways.
PSAT1 contributes to
anchorage-independent growth, in part, by providing metabolites for
serine-glycine-one carbon metabolism
The KEGG analysis of shPSAT1-mediated down-regulated
genes identified metabolic pathways necessary for cell division,
such as purine/pyrimidine metabolism and folate biosynthesis
(Fig. 2A). Similarly, GO analysis
of down-regulated genes were implicated in DNA metabolism and cell
proliferation (Fig. 2C). As a
result, loss of PSAT1 appears to impact key metabolic
macromolecules that contribute to the oncogenic capacity of EGFR
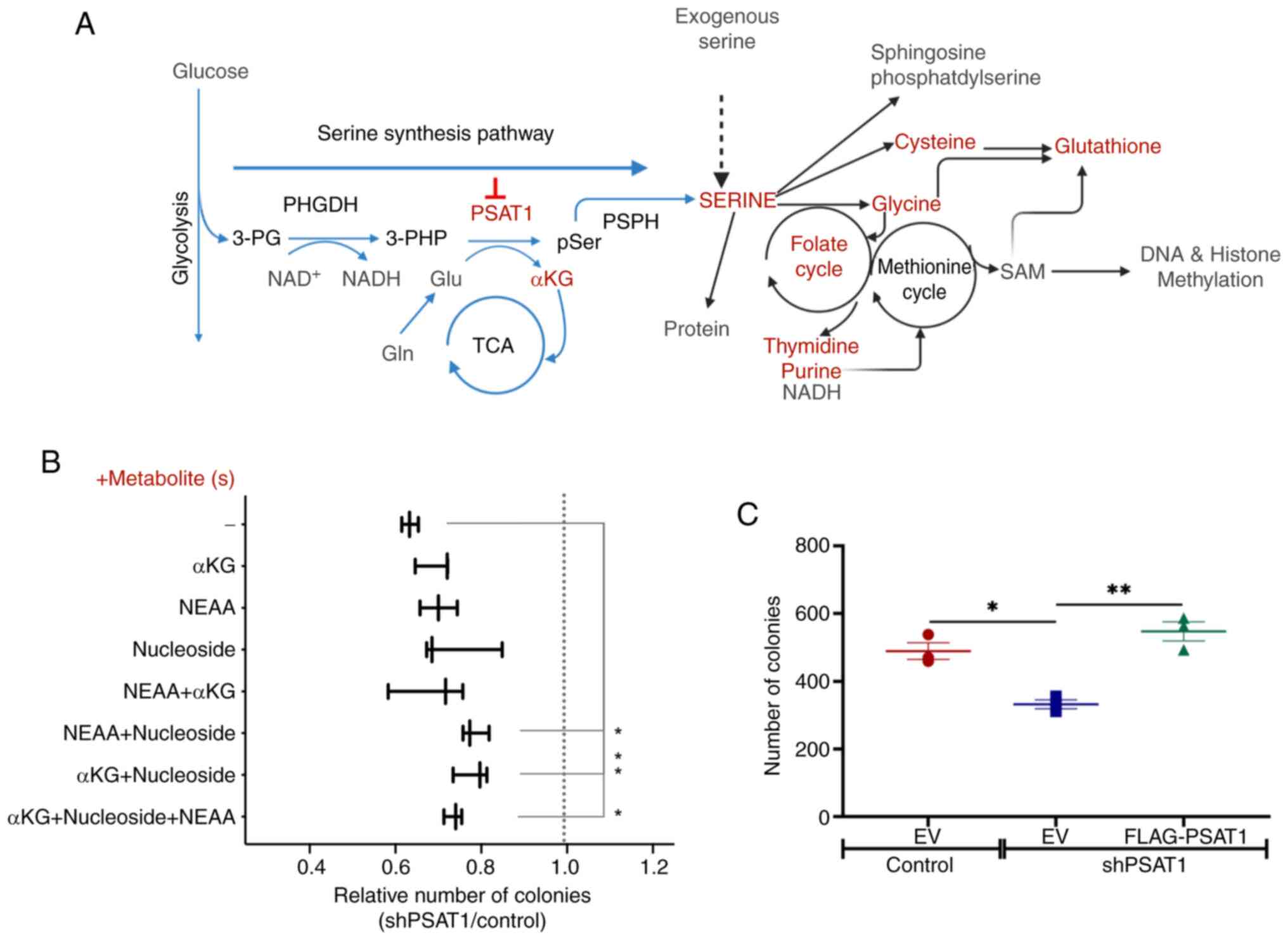
mutant NSCLC cells (Fig. 3A). To
interrogate this, soft agar assays were performed in the absence of
PSAT1 with or without supplementation of SSP-downstream
metabolites, including NEAA, nucleosides, and α-KG. Initially, it
was observed that depletion of PSAT1 resulted in a 40% reduction in
colony formation compared to control cells (Fig. 3B) and that this defect was specific
to PSAT1 depletion since the restoration of PSAT1 expression was
able to fully rescue anchorage-independent growth (Fig. 3C). Yet, the addition of individual
downstream metabolites alone did not affect the loss of colony
formation upon PSAT1 suppression. However, combining any
metabolite(s) with nucleosides significantly increased colony
number in the absence of PSAT1 compared with media without
supplementation (Fig. 3B).
Together, these results are consistent with a metabolic requirement
for PSAT1 for anchorage-independent growth.
Our previous study demonstrated that loss of PSAT1
inhibited the nuclear localization of PKM2 and that expression of a
nuclear acetyl-mimetic form of PKM2 partially rescued cell motility
in PSAT1-silenced cells (11).
Thus, whether this nuclear acetyl-mimetic form of PKM2
(PKM2NLS-K433Q) also contributed to PSAT1-driven
anchorage-independent growth was assessed here. It was found that
PKM2NLS-K433Q expression failed to restore soft agar
colony formation in the absence of PSAT1 (Fig. S2). These findings suggest that
nuclear PKM2, while necessary for cell motility, is dispensable for
PSAT1-mediated anchorage-independent growth.
PSAT1 loss modulates the expression of
actin-binding proteins and rearranges the actin-cytoskeleton
In our previous study it was demonstrated that
reduced cell migration upon PSAT1 silencing could be partially
rescued by nuclear acetyl-mimetic PKM2 (11), the lack of complete rescue prompted
exploration of other cell migratory processes that may be
influenced by PSAT1. GO analysis found enrichment of
shPSAT1-down-regulated genes that are involved in actin
cytoskeletal organization (Figs. 2C
and S1A). It is well-established
that the actin cytoskeleton not only determines cellular morphology
but plays key roles in migration and invasion due to the
requirement for cell movement (34). To interrogate this, the
PSAT1-mediated DEGs were assessed to identify genes involved in
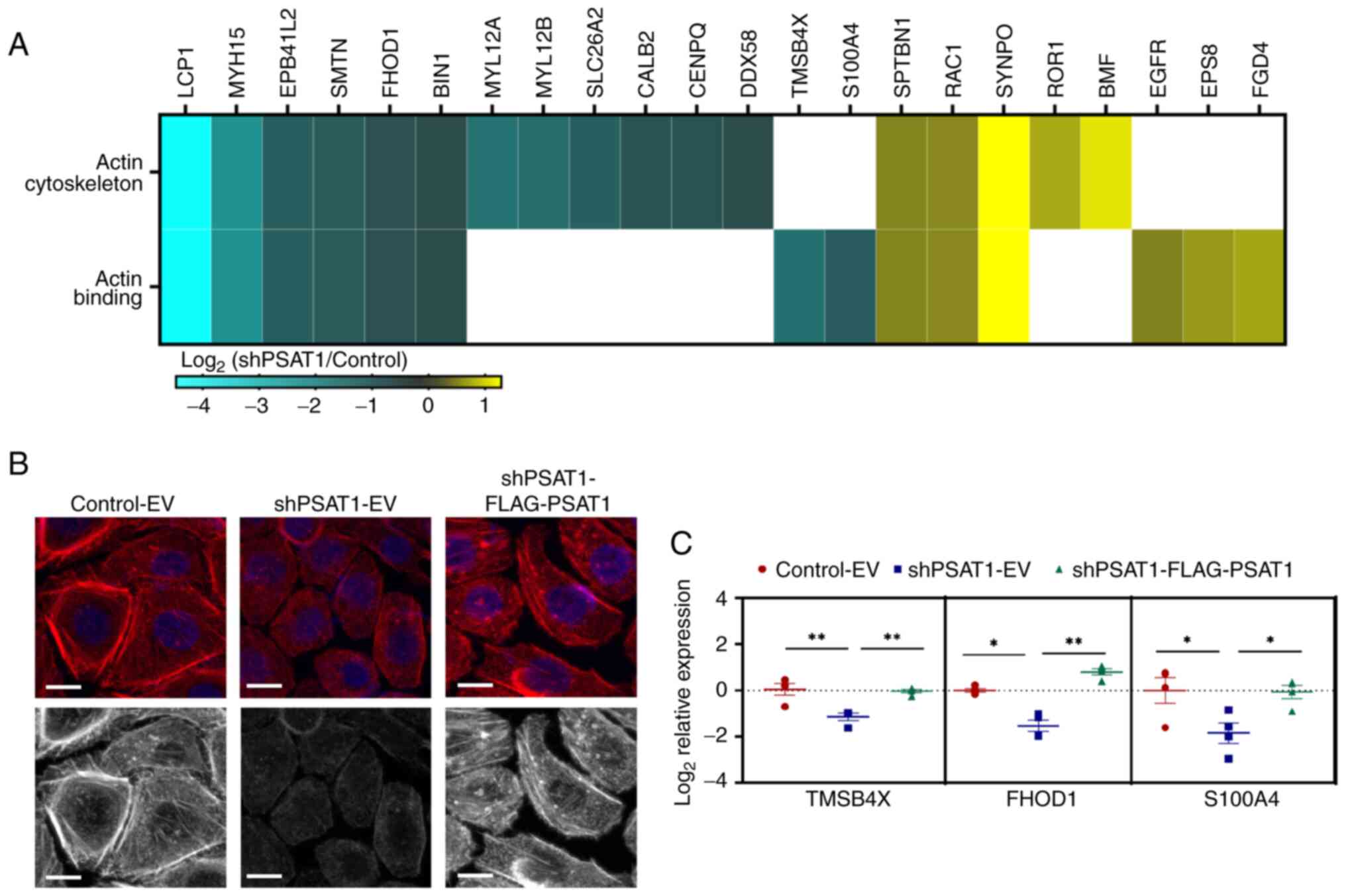
actin-related processes. Fig. 4A
shows the DEGs related to actin binding or the actin cytoskeleton.
To assess a PSAT1 functional requirement for cytoskeletal
arrangement, phalloidin staining was performed to monitor
filamentous actin (F-actin) formation in the presence or absence of
PSAT1 expression. Immunofluorescence microscopy found that PC9
cells exhibited structured actin fibers spanning the whole cell
body, while cells devoid of PSAT1 displayed loss of these actin
stress fibers (Fig. 4B). Yet,
re-expression of PSAT1 in silenced cells rescued long fiber
formation, thereby validating the on-target effects of PSAT1 and
confirming a role for PSAT1 in actin cytoskeletal organization.
Next, PSAT1-regulation of actin-related genes
directly involved in F-actin formation was verified (Fig. 4C). Transcript analysis found that
PSAT1 silencing reduced FHOD1, TMSB4X, and S100A4
levels, which were rescued via PSAT1 re-expression (Fig. 4C). Coupled with the cytoskeletal
analysis, these results validate our transcriptomic findings and
implicate a new role for PSAT1 in cell migration through regulating
the expression of actin-related factors and influencing
cytoskeletal rearrangement.
Transcriptional analysis validates a
link between PSAT1 and the RB/E2F pathway
To further understand the effect of PSAT1 on gene
expression, MSigDB CGP analysis was performed based on the gene set
analysis of the shPSAT1-DEGs (Fig.
S3). Genes down-regulated upon PSAT1 loss were significantly
enriched in the ‘FISCHER_ G1_S_CELL _CYCLE’ and
‘CHICAS_RB1_TARGETS_SENESCENT’ datasets (Fig. S4A) and MetaCore transcription
factor network analysis found E2F1 as a major regulator of these
DEGs (Table SIII and Fig. S4B). Despite the presence of certain
up-regulated genes, most genes within the E2F1 transcriptional
network, including E2F1, were decreased upon PSAT1 suppression
(Fig. S4B). Taken together, the
in-silico analysis indicated that PSAT1 regulated E2F1
transcriptional activity.
PSAT1 levels impact β-catenin
expression and transactivation
EGFR activation promotes the nuclear localization of
β-catenin through various mechanisms (35). While phosphorylation of membranous
β-catenin by EGFR or AKT leads to migration of β-catenin away from
the membrane, EGFR activation inhibits the proteasomal degradation
of cytoplasmic β-catenin protein by GSK3β inactivation. Thus, both
EGFR-mediated pathways result in the accumulation of β-catenin in
the nucleus and seem to contribute to every step within EGFR-driven
tumor progression (36–39). While previous studies have linked
PSAT1 to inhibition of GSK3β in multiple tumor types (7,14,15),
the association between β-catenin and PSAT1 remains elusive in EGFR
mutant NSCLC cells. Furthermore, nuclear PKM2 requires EGF-induced
β-catenin transactivation in EGFR-driven tumor growth of GBM and
EGF-induced epithelial-to-mesenchymal transition and invasion in
HCC cells (31,40). Considering these prior reports and
our results showing the link between PSAT1 and nuclear PKM2
(11), it was speculated that loss
of PSAT1 may result in altered β-catenin transactivation.
Interrogation of the CGP analysis found differential
expression of genes within the ‘FEVR_CTNNB1_ TARGETS_ DN’ and
‘WANG_RESPONSE_TO_GSK3B_INHBITOR_ SB216763_DN’ gene sets, which
indicated a possible regulatory role for PSAT1 on β-catenin
function (Fig. S5A). β-catenin
induces transcription via interacting with TCF (T-cell specific
transcription factor)/LEF1 transcription factor family (41,42).
Notably, TCF7L1 (TCF3) and TCF7L2 (TCF4) were found to be changed
by PSAT1 loss in the MetaCore transcription analysis (Table SIII). As TCF7L1 is a known
repressor (42) and is up-regulated
by PSAT1 silencing (Fig. S5B, red
circle), it was hypothesized that there would be reduced β-catenin
transactivation. However, up- and down-regulated genes were
identified within the TCF3 and TCF4 networks (Fig. S5B and C). In short, while
supportive, these analyses were unable to provide clear insights
into whether β-catenin transactivation may change upon PSAT1
silencing.
To better understand the association between PSAT1
and β-catenin, whether β-catenin protein expression was altered in
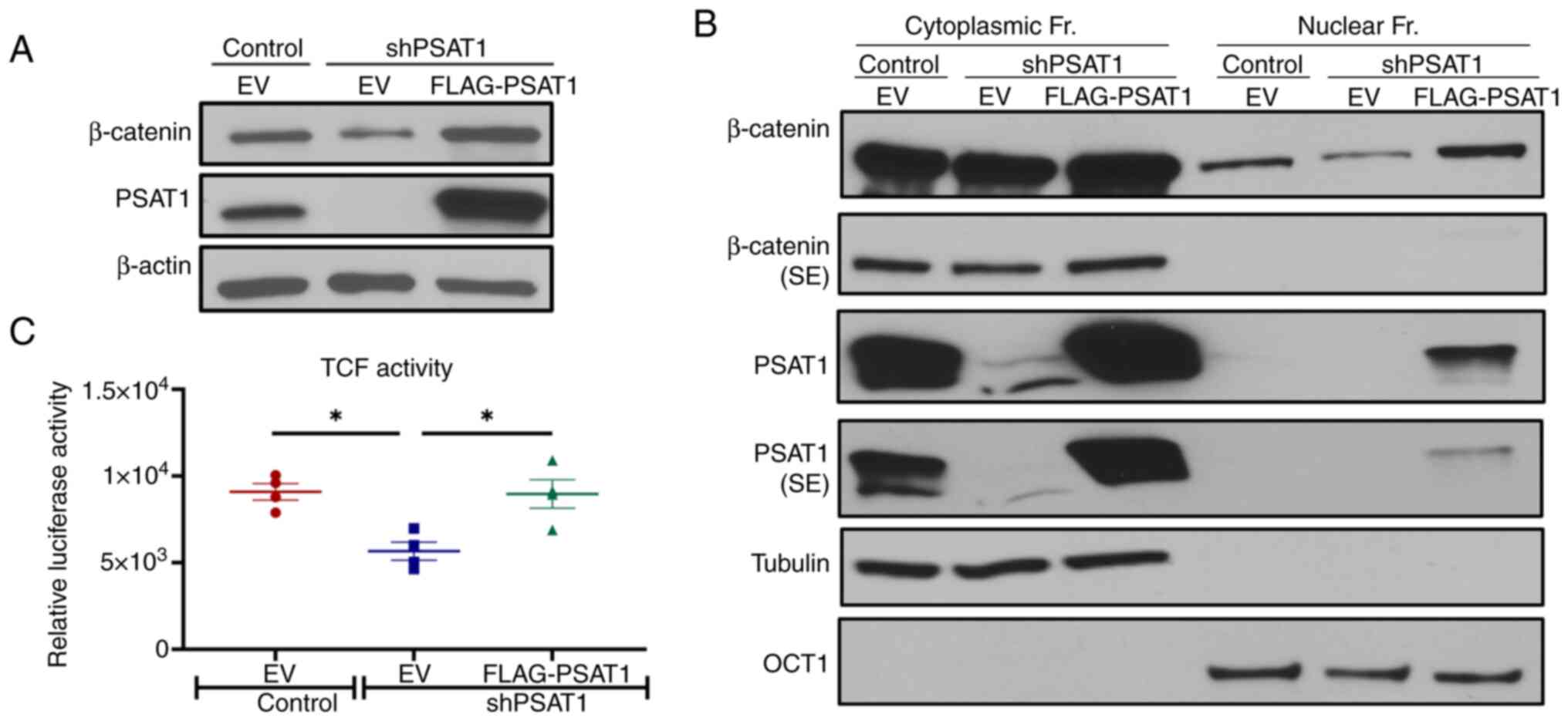
the absence of PSAT1 was next assessed. Immunoblot analysis found
that β-catenin expression decreased upon PSAT1 silencing, which can
be rescued by re-expression of PSAT1 (Fig. 5A). Yet, nuclear PKM2 expression
(PKM2NLS-K433Q) had no effect on β-catenin expression
under PSAT1 silencing, indicating that regulation of β-catenin
expression is independent of nuclear PKM2 in the context of PSAT1
loss (Fig. S6).
Accumulation of cytoplasmic β-catenin due to
inhibited proteasomal degradation leads to its nuclear localization
and transactivation (43,44). Since a reduction in total β-catenin
levels was observed here, its cellular distribution upon PSAT1
silencing was examined. Subcellular fractionation found that
nuclear β-catenin expression decreased in PSAT1 silenced cells in
comparison with control cells, which could be rescued upon
re-expression of PSAT1 (Fig.
5B).
The same pattern of β-catenin expression was also
observed in the cytoplasmic fraction. According to the RNA-Seq
analysis, loss of PSAT1 did not alter the mRNA expression of
β-catenin (data not shown), which implies that PSAT1 potentially
contributes to β-catenin stability in PC9 cells, possibly through
increasing phospho-GSK3β levels.
Next, the β-catenin transcriptional activity was
measured directly using a luciferase reporter assay
(luc2p/TCF-LEF). Loss of PSAT1 in serum-starved cells led to
significantly reduced β-catenin activity, which was rescued upon
PSAT1 restoration (Fig. 5C). Taken
together, these results suggest that PSAT1 increases β-catenin
expression and transactivation, most likely through regulating
protein stability.
Identification of differentially
expressed PSAT1-associated genes in primary EGFR mutant NSCLC
PC9 cells have frequently been used as an in
vitro model for EGFR mutant lung cancer due to the presence of
an activation mutation (exon19del) in the EGFR tyrosine kinase
domain and their responsiveness to EGFR tyrosine kinase inhibitor
treatment (45). Within the above
transcriptomic analysis, RNA was collected from serum-starved PC9
cells to assess the EGFR-dependent gene expression alterations
while minimizing the contribution of other serum factors from the
media. Therefore, it was hypothesized that a subset of
PSAT1-mediated genes would be observed that have been independently
implicated in EGFR-driven lung tumorigenesis. To identify these
genes, a bioinformatics approach was used through comparative
analysis between the differentially regulated genes in our RNA-seq
analysis and publicly available microarray datasets obtained from
EGFR mutant patient tumors (Fig.
S7).
The GEO database was searched for datasets
containing transcriptional analysis of EGFR mutant lung tumors and
normal lung tissues (n≥10, each) derived from untreated patients.
Based on these criteria, GSE31210, GSE31547, GSE31548, GSE27262,
GSE32863, and GSE75037 datasets were chosen for subsequent
analysis. Gene lists derived from the GSE31547 and GSE31548
datasets were combined as ‘GSE31547-48’ since expression profiles
were obtained from the same patients but neither
Affymetrix-HG-U133A nor Affymetrix-HG-U133B can cover all genes
from our RNA-seq list (Table SIV)
(46). Class comparison analysis
was then conducted using the BRB-ArrayTool to determine the DEGs
from our PSAT1-DEG list (26).
PSAT1-associated genes were defined as those up-regulated in tumor
tissues compared to normal lung that are correspondingly
down-regulated upon PSAT1 silencing in our RNA-seq profiling
(shPSAT1-down-regulated) and, conversely, down-regulated in tumor
tissue that are correspondingly up-regulated upon PSAT1 loss
(shPSAT1-up-regulated).
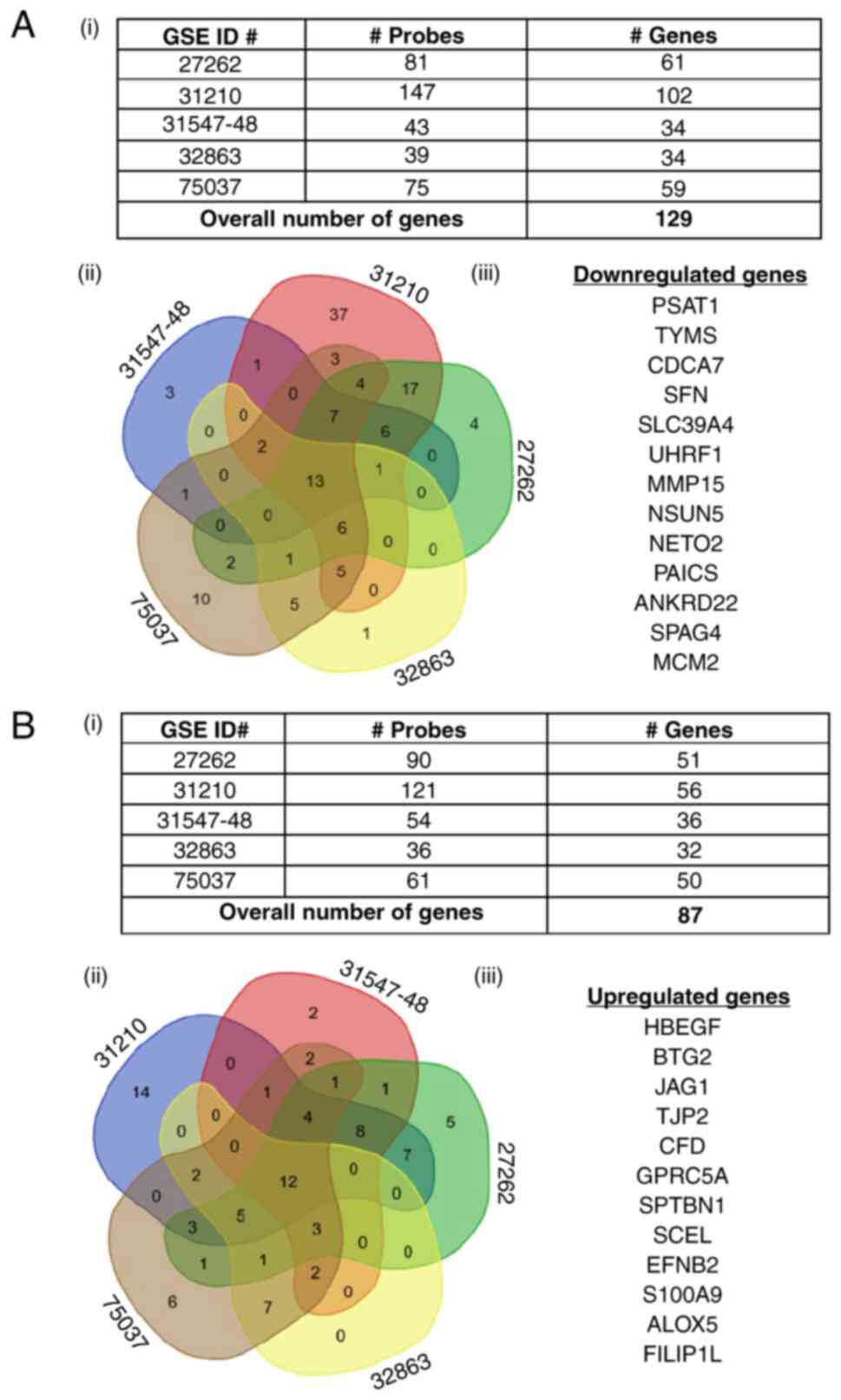
The PSAT1-associated gene list from each dataset was
then compared to obtain ‘common genes’ altered across all datasets.
A total of 13 genes from the shPSAT1-down-regulated gene list and
12 genes from the shPSAT1-up-regulated gene list that were
differentially expressed in EGFR mutant tumors compared to normal
lung tissue were identified (Fig. 6A
and B, respectively). Together, these were designated as a
PSAT1-associated gene signature in EGFR mutant lung tumors
(Table SV and SVI). This bioinformatics approach was
able to identify common genes linked through PSAT1 regulation in
EGFR mutant lung tumors.
A PSAT1-associated gene expression
signature correlates with poorer outcomes in patients with EGFR
mutant lung cancer
Next, the prognostic value of this PSAT1-associated
gene signature in EGFR mutant lung cancer was assessed. Among the
five publicly available datasets, the GSE31210 was used as it
included all relevant clinical information such as NSCLC genotype
(127 EGFR mutant tumors), staging (EGFR mutant tumors: IA: n=77,
IIB: n=26, II: n=24), and patient outcomes (RFS and OS) (Table SIV), whereas the other datasets had
a limited number of EGFR mutant lung cancer samples for survival
analysis and could not be combined due to platform
incompatibility.
The BRB-ArrayTool survival risk prediction tool was
utilized to perform OS and RFS analysis as previously described
(28). To validate this 25-gene
signature in patient samples, principal component analysis with
leave-one-out cross-validation and log-rank statistics with 100
permutation tests was used to calculate the prognostic indices and
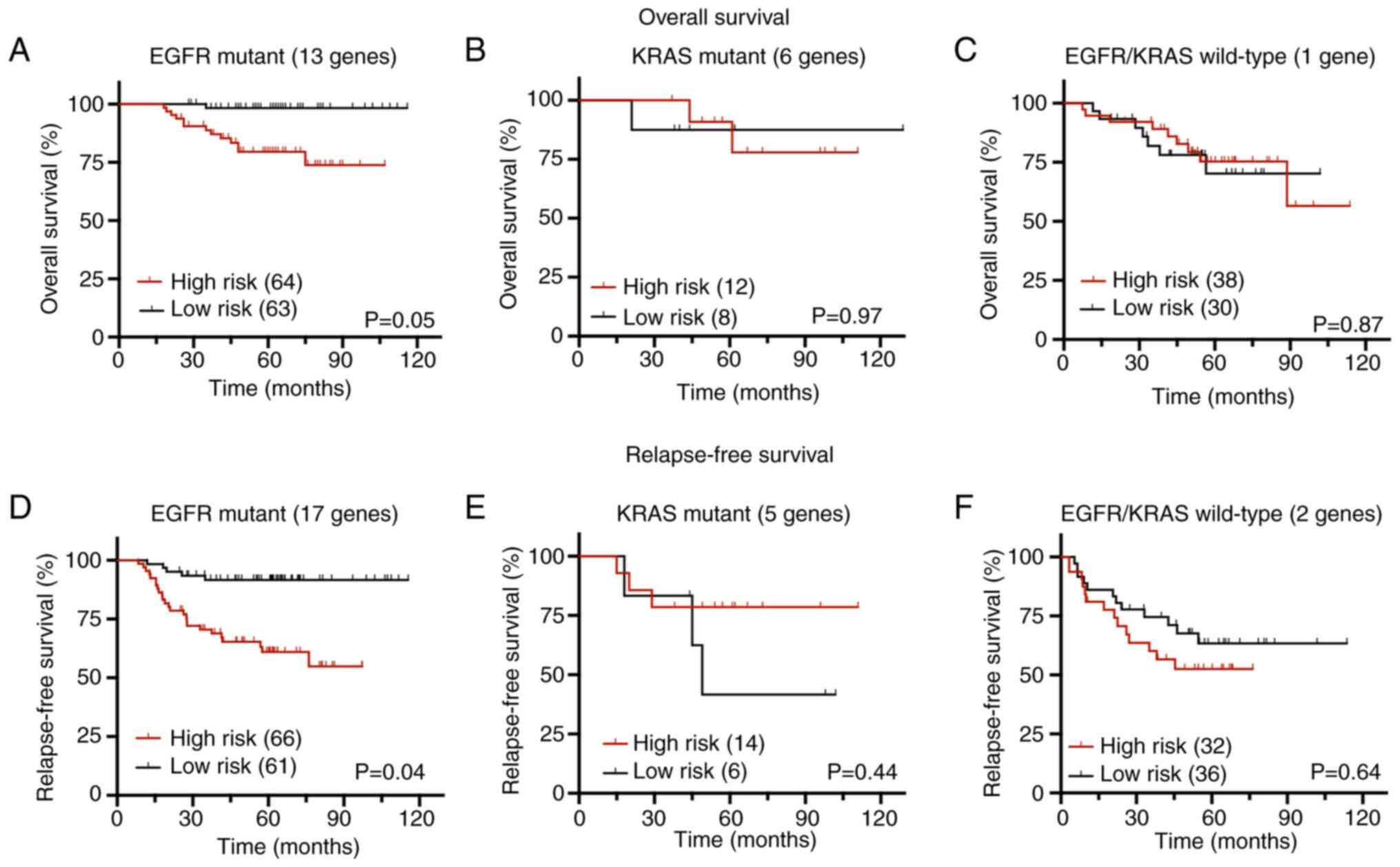
classify the patients as high-risk and low-risk groups. According
to the KM analysis for OS, the high-risk group (defined by 13 genes
out of 25; Table SVII) exhibited a
significantly shorter OS than the low-risk group with a prediction
accuracy AUC value of 0.77 (Figs.
7A and S8A). Survival risk
prediction analysis found additional genes (17 genes out of 25;
Table SVII) that contributed to
RFS. The KM plot demonstrated that the high-risk group correlated
with a worse RFS with a prediction accuracy AUC value of 0.72
(Figs. 7D and S8D). Corresponding genes involved in RFS
and OS prediction are summarized in Table SVII and VIII, which detail their corresponding
relevant statistics used in survival risk score calculation and
known roles in lung tumorigenesis, respectively. Probes/genes with
positive coefficients (wi) in Table
SVII indicate that higher expression correlates with shorter
survival, whereas negative coefficients imply that higher
expression is associated with longer survival. Down-regulated genes
upon PSAT1 silencing possessed positive coefficients and
up-regulated genes had negative coefficients, corroborating the
findings above that the PSAT1-associated gene signature in EGFR
mutant lung cancer is associated with worse outcomes.
In addition to EGFR mutant lung tumors, the GSE31210
microarray dataset also harbors ALK-fusion positive, KRAS mutant,
and EGFR/KRAS/ALK wild-type tumor samples with their corresponding
clinical information. Therefore, whether the predictive ability of
PSAT1-associated gene signature applied to NSCLC tumors with other
oncogenic drivers was assessed. Survival analysis for ALK-fusion
positive tumors was excluded due to the limited sample size (n=10)
and EGFR/KRAS/ALK wild-type tumors were defined as EGFR/KRAS
wild-type. Survival risk prediction analysis for KRAS mutant (n=20)
and EGFR/KRAS wild-type (n=68) tumors was then conducted as
described above. Genes within the defined PSAT1-associated
signature were unable to significantly separate high-risk and
low-risk groups for both OS and RFS in either the KRAS mutant
(Figs. 7B, E, 8B and E) or EGFR/KRAS wild-type tumors
(Figs. 7C, F, S8C and F).
RFS-related genes can distinguish the
high-risk group in patients with EGFR mutant Stage I NSCLC
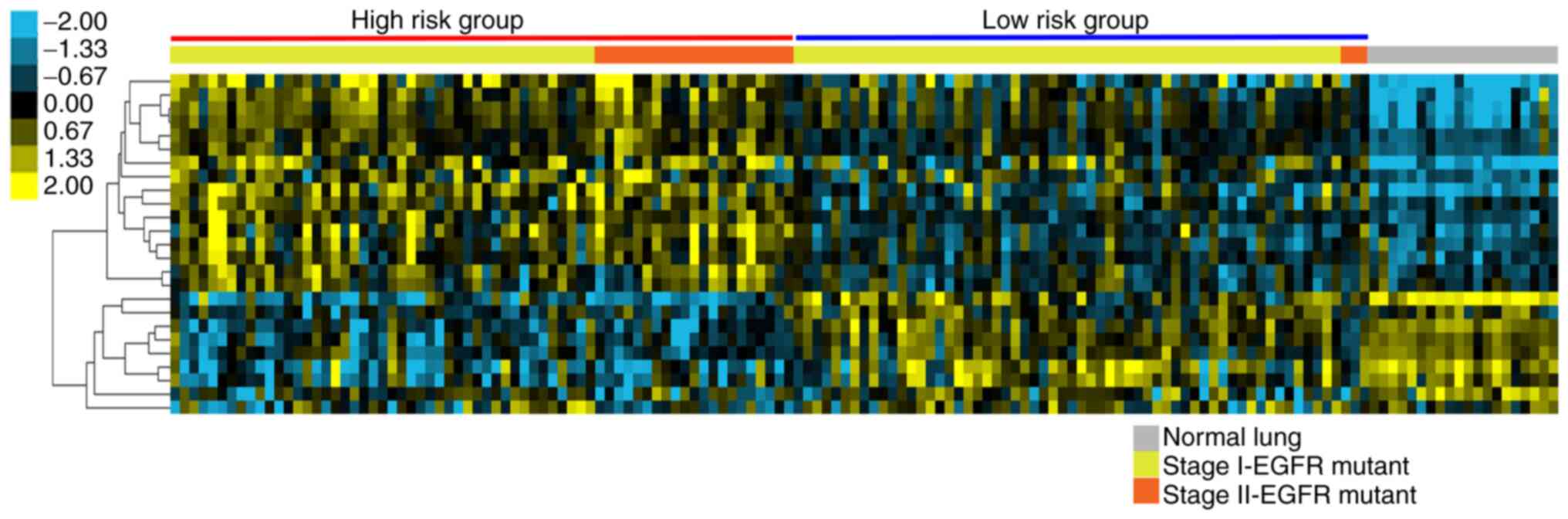
The expression of the 17 PSAT1-signature RFS genes
(Table SVII) from EGFR mutant
tumors and normal lung tissue in the GSE31210 dataset were used for
cluster analysis. As illustrated in Fig. 8, the high-risk group identified by
RFS analysis clustered together and exhibited an opposite
expression profile compared to that from the normal lung tissue.
More notably, this analysis could selectively distinguish between
high and low-risk groups even among clinical stage I patient
samples (Stage 1, n=103; Stage 2, n=24). This implies that this
PSAT1-associated gene signature may be predictive for high-risk
groups within patients with stage I EGFR mutant NSCLC.
Discussion
PSAT1 expression is increased in several types of
cancer, including NSCLC, and is associated with poor patient
outcomes (15,47,48).
While its metabolic function within SSP activity contributes to
cell proliferation and tumor growth, oncogenic signals may result
in the gain of alternative functions that promote tumor
progression; particularly as nuclear localization of PSAT1 in
EGFR-activated lung cancer cells was observed in our previous study
(11). To gain a better insight
into the role of PSAT1 in tumorigenesis, genome-wide expression
profiling by RNA-seq technology was performed. DEGs were
interrogated using bioinformatics-based tools for comparison with
other gene expression datasets.
Inhibition of serine biosynthetic pathways blocks
the production of precursors for folate, glutathione, and
nucleotide biosynthesis, resulting in tumor growth arrest (5,6,49,50).
The present study found down-regulation of genes within these
pathways following PSAT1 silencing, adding another layer of
regulation of these pathways by SSP. Impaired anchorage-independent
growth by PSAT1 silencing was partially restored by downstream
metabolite supplementation, supporting the metabolic function of
PSAT1 within the serine biosynthetic pathway. PSAT1 is also
implicated in inhibiting GSK3β-dependent phosphorylation and
proteasomal degradation of target proteins (14,15,51).
PSAT1-mediated stabilization of cyclin D1 promotes E2F
transactivation in NSCLC cells, resulting in cell cycle progression
and proliferation (15).
Furthermore, transcriptomic analysis from patients with NSCLC
identified the enrichment of E2F target expression in PSAT1-high
tumors compared with PSAT1-low tumors. The in-silico
analysis further supported this by demonstrating a reduction of E2F
target genes following PSAT1 silencing. β-catenin was another
potential target for the PSAT1/GSK3β pathway and is implicated in
EGFR mutant lung tumorigenesis (14,36,37).
Both bioinformatics analysis and functional results in the present
study corroborated these previous findings that PSAT1 may be
involved in the regulation of β-catenin stability and activity.
These observed gene expression changes upon PSAT1 silencing support
the known tumorigenic functions of PSAT1.
Myocardin-related transcription
factors/serum-response factor (MRTF/SRF) signaling is a
well-established pathway that promotes cell motility via
transcriptionally regulating the expression of actin
cytoskeleton-related genes (52,53).
As MRTFs are actin-binding proteins, a higher monomeric/polymeric
actin ratio results in the sequestration of MRTFs in the cytoplasm,
thereby reducing SRF-dependent gene expression. Formin Homology 2
Domain Containing 1 (FHOD1) functions as an actin filament capping
and bundling protein and enhances cell migration by inducing the
formation and stabilization of F-actin at the leading edge
(54–58). Furthermore, the observation of
elevated expression at the invasive front of squamous cell
carcinoma further supports its role in cancer metastasis (57). Thymosin β4, encoded by TMSB4X, is
another actin-binding protein that exhibits a G-actin sequestering
function that inhibits spontaneous actin polymerization (59). It contributes to cell motility by
localizing the monomeric G-actin at the leading edge of
lamellipodia for actin polymerization, leading to membrane
protrusions (60). Thymosin β4 has
also been reported as a prognostic factor for poor survival and
metastasis in patients with early-stage NSCLC (61). In addition, S100A4 is a
well-recognized metastasis-associated protein that functions as a
binding partner for actin-related factors such as actin, myosin,
and tropomyosin (34,62). A recent report showed that
FHOD1-loss-driven MRTFA accumulation in the cytoplasm impacted cell
motility in melanoma cells (63).
Another study found that TGF-β-induced thymosin β4 expression
enhanced MRTF/SRF transcriptional activity, potentially through
sequestering monomeric actin binding to MRTFs (64,65).
As the analysis found that PSAT1 silencing altered the expression
of several actin cytoskeleton-related genes, including FHOD1 and
TMSBX4 (thymosin β4), it will be intriguing to investigate the
involvement of the MRTF/SRF pathway in PSAT1-mediated cytoskeleton
rearrangement and cell migration.
Genes involved in immune response and leukocyte
migration/chemotaxis were upregulated upon PSAT1 silencing
(Fig. 2D). In addition, the protein
products of these genes are localized in the Golgi, within the
membrane and lumen of vesicles, and secretory membranes, suggesting
a change in vesicle-mediated transport and secretion (Fig. S1B). As tumor-secreted factors
contribute to immune cell infiltration into the tumor
microenvironment, the results of the present study suggested that
intratumoral PSAT1 may influence reprogramming within the tumor
microenvironment (66,67). While immune checkpoint inhibitors
(ICI) have been adopted as a therapeutic option for patients with
NSCLC, patients with EGFR mutant lung cancer are excluded from this
option since these patients have shown limited responses to ICI
treatment (68). Thus, it is
intriguing to investigate how tumoral PSAT1 may modulate the tumor
microenvironment and whether targeting PSAT1 activity may sensitize
EGFR mutant lung tumors to ICI treatment (69).
A literature search for the reported function of the
identified survival genes in lung cancer was thus performed, and
the findings are summarized in Table
SVIII. It was observed that shPSAT1-down-regulated genes (which
are conversely increased in tumors) were associated with a poor
patient outcome and tumor progression and were involved in various
oncogenic processes, including cell cycle progression,
proliferation, migration, and invasion. Conversely,
shPSAT1-up-regulated genes (which are conversely decreased in
tumors) have been linked to a better prognosis and played roles in
inhibiting cell proliferation, migration, and invasion. Among these
genes, B-cell translocation gene 2 and G Protein-Coupled Receptor
Class C Group 5 Member A (GPRC5A) have already been identified as
tumor suppressors and GPRC5A acts as a negative regulator of EGFR
signaling in NSCLC cells [3-5]. However, a relationship between
these genes and EGFR mutant lung tumors in the current literature
was not found, implying the novelty of the PSAT1-associated genes
in EGFR mutant lung cancer.
The PSAT1-associated gene signature was primarily
dominated by early-stage EGFR mutant lung cancer transcriptomic
profiles due to the presence of a high number of stage I patients
in GSE31210 (n=103) and GSE27262 (all stage I tumors). Due to the
lack of advanced tumor samples, PSAT1-associated genes involved in
late-stage tumor progression and metastasis may be lost. Through an
examination of distinct datasets (GSE14107), which encompass
transcriptomic profiles of the parental PC9 cell line (PC9-P) and a
brain metastatic subline (PC9-BrM3) (29), a total of 81 common genes that were
differently expressed following PSAT1 depletion and brain selective
metastatic potential were found, which were considered as potential
PSAT1-associated pro-metastatic genes (Fig. S9, Table SIX). Yet, the functional
connection between PSAT1 and these putative pro-metastatic genes
requires further investigation within an EGFR mutant NSCLC
metastatic model.
Nuclear localized metabolic enzymes, including PDC,
ACLY, and α-KGDH are involved in epigenetic regulation by providing
a substrate for histone modifications (17,70,71).
Accordingly, in our previous study, it was demonstrated that PSAT1
localizes to the nucleus in EGFR-activated NSCLC cells (11). Recent studies have found a link
between PSAT1 and epigenetic alteration/remodeling/landscaping.
Particularly, PSAT1 contributed to S-adenosylmethionine production
for DNA retrotransposon methylation in Kras-mutant-Lkb1 loss
pancreatic adenocarcinoma mouse models (72). Another study revealed that PSAT1
contributed to the maintenance of pluripotency of embryonic stem
cells by supplying α-KG for enzymes that account for histone and
DNA demethylation (73). Expression
of adjacent genes can be regulated by epigenetics and is known as
long-range epigenetic silencing or activation (74,75).
Therefore, it is hypothesized that PSAT1 could epigenetically
regulate the expression of genes located throughout the same
chromosomal region. For this, positional gene set analysis was
performed in MSigDB and found that 10% of the
shPSAT1-down-regulated genes were enriched on the chr18p11
cytogenic band, while the chr7p21 cytogenic band harbored various
shPSAT1-up-regulated genes (Fig.
S10A). A previous report demonstrating the association of
chr18p11 with non-smoker lung cancer susceptibility in a Korean
population, which has a higher proportion of EGFR mutant NSCLC,
prompted the examination of these genes localized in chr18p11 in
response to differential PSAT1 expression in our previous study
(76). The down-regulated genes
identified were not restricted to a localized region but spanned a
large area within chr18p11 (Fig.
S10B). PSAT1-regulation of genes within this locus was
confirmed using qPCR (Fig. S10C).
The results suggested putative long-range gene expression
regulation by PSAT1 within this genetic locus, yet further
investigation is required to determine how PSAT1 may contribute to
epigenetic regulation in this region.
In summary, these experiments examined genome-wide
expression changes upon PSAT1 silencing using gene profiling and
bioinformatics approaches. The analysis corroborated previous
findings on the role of PSAT1 within the serine biosynthetic
pathway in regulating E2F activity and β-catenin protein
expression/transcription activity. In addition, rescue of F-actin
stress fibers and expression of actin-related genes by restored
PSAT1 validated a functional role for PSAT1 on cytoskeletal
structure. A PSAT1-dependent gene signature that may have
prognostic value regarding patient outcomes in EGFR mutant NSCLC
was also identified. Together, these findings suggest that
targeting PSAT1 may have clinical utility in this patient
population. To date, no PSAT1 inhibitors have been described, but
several PHGDH antagonists have been pre-clinically evaluated
against multiple tumor types (6).
This approach assumes that the changes observed upon PSAT1
silencing in this context are solely related to the metabolic
activity of PSAT1 and that targeting other SSP enzymes would yield
identical results. In our previous study, it was demonstrated that
PSAT1 exhibits differential compartmentalization under EGFR
activation and is necessary for nuclear PKM2 translocation
(11). Whether this activity is
SSP-independent and/or significantly contributes to the overall
pro-tumorigenic function of PSAT1 in EGFR mutant NSCLC is a focus
of ongoing work and will ultimately inform future putative
strategies for pharmacologically targeting PSAT1.
Supplementary Material
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Kentucky Lung Cancer Research
Program and the Office of the Assistant Secretary of Defense for
Health Affairs and the Defense Health Agency J9, Research and
Development Directorate, through the Lung Cancer Research Program
(grant no. W81XWH-19-1-0445). Sequencing and bioinformatics support
for this work was provided by the National Institutes of Health
(grant nos. P20GM103436 and P30GM106396).
Availability of data and materials
The data generated in the present study may be
found in the GEO database under accession number GSE173270 or at
the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173270.
Authors' contributions
RBS contributed to the conception and design of the
study, performed all the in vitro studies, the
bioinformatics comparative analysis and assisted in writing the
manuscript. SW performed the RNA-seq and assisted with the
analysis. KA and ER performed the bioinformatics analysis related
to the RNA sequencing. BFC contributed to the conception and design
of the study, performed the data interpretation and assisted in
writing and editing the manuscript. RBS and BFC confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
α-KG
|
α-ketoglutarate
|
|
AUC
|
area under the curve
|
|
DEG
|
differentially expressed gene
|
|
EGFR
|
epidermal growth factor receptor
|
|
KM
|
Kaplan Meier
|
|
NEAA
|
non-essential amino acids
|
|
NSCLC
|
non-small cell lung cancer
|
|
OS
|
overall survival
|
|
PHGDH
|
phosphoglycerate dehydrogenase
|
|
PKM2
|
pyruvate kinase M2
|
|
PSAT1
|
phosphoserine aminotransferase 1
|
|
PSPH
|
phosphoserine phosphatase
|
|
RFS
|
relapse-free survival
|
|
SSP
|
serine synthesis pathway
|
|
TKI
|
tyrosine kinase inhibitor
|
References
|
1
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhou X, Tian C, Cao Y, Zhao M and Wang K:
The role of serine metabolism in lung cancer: From oncogenesis to
tumor treatment. Front Genet. 13:10846092023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim SK, Jung WH and Koo JS: Differential
expression of enzymes associated with serine/glycine metabolism in
different breast cancer subtypes. PLoS One. 9:e1010042014.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun WY, Kim HM, Jung WH and Koo JS:
Expression of serine/glycine metabolism-related proteins is
different according to the thyroid cancer subtype. J Transl Med.
14:1682016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mullarky E, Lucki NC, Beheshti Zavareh R,
Anglin JL, Gomes AP, Nicolay BN, Wong JC, Christen S, Takahashi H,
Singh PK, et al: Identification of a small molecule inhibitor of
3-phosphoglycerate dehydrogenase to target serine biosynthesis in
cancers. Proc Natl Acad Sci USA. 113:1778–1783. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pacold ME, Brimacombe KR, Chan SH, Rohde
JM, Lewis CA, Swier LJ, Possemato R, Chen WW, Sullivan LB, Fiske
BP, et al: A PHGDH inhibitor reveals coordination of serine
synthesis and one-carbon unit fate. Nat Chem Biol. 12:452–458.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu S, Wang X, Liu L and Ren G:
Stabilization of Notch1 and β-catenin in response to ER-breast
cancer-specific up-regulation of PSAT1 mediates distant metastasis.
Transl Oncol. 20:1013992022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang Y, Li J, Dong X, Meng D, Zhi X, Yuan
L and Yao L: PSAT1 regulated oxidation-reduction balance affects
the growth and prognosis of epithelial ovarian cancer. Onco Targets
Ther. 13:5443–5453. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fang Y, Liang X, Xu J and Cai X: miR-424
targets AKT3 and PSAT1 and has a tumor-suppressive role in human
colorectal cancer. Cancer Manag Res. 10:6537–6547. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang H, Cui L, Li D, Fan M, Liu Z, Liu C,
Pan S, Zhang L, Zhang H and Zhao Y: Overexpression of PSAT1
regulated by G9A sustains cell proliferation in colorectal cancer.
Signal Transduct Target Ther. 5:472020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Biyik-Sit R, Kruer T, Dougherty S, Bradley
JA, Wilkey DW, Merchant ML, Trent JO and Clem BF: Nuclear pyruvate
kinase M2 (PKM2) contributes to phosphoserine aminotransferase 1
(PSAT1)-mediated cell migration in EGFR-activated lung cancer
cells. Cancers (Basel). 13:39382021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Luo MY, Zhou Y, Gu WM, Wang C, Shen NX,
Dong JK, Lei HM, Tang YB, Liang Q, Zou JH, et al: Metabolic and
nonmetabolic functions of PSAT1 coordinate signaling cascades to
confer EGFR inhibitor resistance and drive progression in lung
adenocarcinoma. Cancer Res. 82:3516–3531. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duan W and Liu X: PSAT1 upregulation
contributes to cell growth and cisplatin resistance in cervical
cancer cells via regulating PI3K/AKT signaling pathway. Ann Clin
Lab Sci. 50:512–518. 2020.PubMed/NCBI
|
|
14
|
Gao S, Ge A, Xu S, You Z, Ning S, Zhao Y
and Pang D: PSAT1 is regulated by ATF4 and enhances cell
proliferation via the GSK3β/β-catenin/cyclin D1 signaling pathway
in ER-negative breast cancer. J Exp Clin Cancer Res. 36:1792017.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Y, Wu J, Cai J, He Z, Yuan J, Zhu X,
Li Y, Li M and Guan H: PSAT1 regulates cyclin D1 degradation and
sustains proliferation of non-small cell lung cancer cells. Int J
Cancer. 136:E39–E50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wu S and Le H: Dual roles of PKM2 in
cancer metabolism. Acta Biochim Biophys Sin (Shanghai). 45:27–35.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sutendra G, Kinnaird A, Dromparis P,
Paulin R, Stenson TH, Haromy A, Hashimoto K, Zhang N, Flaim E and
Michelakis ED: A nuclear pyruvate dehydrogenase complex is
important for the generation of acetyl-CoA and histone acetylation.
Cell. 158:84–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
O'Cathail SM, Wu CH, Lewis A, Holmes C,
Hawkins MA and Maughan T: NRF2 metagene signature is a novel
prognostic biomarker in colorectal cancer. Cancer Genet. 248–249.
1–10. 2020.
|
|
19
|
Wang X, Yu Q, Ghareeb WM, Zhang Y, Lu X,
Huang Y, Huang S, Sun Y, Lin J, Liu J and Chi P: Downregulated
SPINK4 is associated with poor survival in colorectal cancer. BMC
Cancer. 19:12582019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
You GR, Cheng AJ, Lee LY, Huang YC, Liu H,
Chen YJ and Chang JT: Prognostic signature associated with
radioresistance in head and neck cancer via transcriptomic and
bioinformatic analyses. BMC Cancer. 19:642019. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim D, Pertea G, Trapnell C, Pimentel H,
Kelley R and Salzberg SL: TopHat2: Accurate alignment of
transcriptomes in the presence of insertions, deletions and gene
fusions. Genome Biol. 14:R362013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Trapnell C, Roberts A, Goff L, Pertea G,
Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL and Pachter L:
Differential gene and transcript expression analysis of RNA-seq
experiments with TopHat and cufflinks. Nat Protoc. 7:562–578. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
de Hoon MJL, Imoto S, Nolan J and Miyano
S: Open source clustering software. Bioinformatics. 20:1453–1454.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saldanha AJ: Java Treeview-extensible
visualization of microarray data. Bioinformatics. 20:3246–3248.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Simon R, Lam A, Li MC, Ngan M, Menenzes S
and Zhao Y: Analysis of gene expression data using BRB-ArrayTools.
Cancer Inform. 3:11–17. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okayama H, Kohno T, Ishii Y, Shimada Y,
Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S,
et al: Identification of genes upregulated in ALK-positive and
EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer Res.
72:100–111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Simon RM, Subramanian J, Li MC and Menezes
S: Using cross-validation to evaluate predictive accuracy of
survival risk classifiers based on high-dimensional data. Brief
Bioinform. 12:203–214. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Nguyen DX, Chiang AC, Zhang XHF, Kim JY,
Kris MG, Ladanyi M, Gerald WL and Massagué J: WNT/TCF signaling
through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis.
Cell. 138:51–62. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bair E and Tibshirani R: Semi-supervised
methods to predict patient survival from gene expression data. PLoS
Biol. 2:E1082004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang W, Xia Y, Ji H, Zheng Y, Liang J,
Huang W, Gao X, Aldape K and Lu Z: Nuclear PKM2 regulates β-catenin
transactivation upon EGFR activation. Nature. 480:118–122. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Snaebjornsson MT and Schulze A:
Non-canonical functions of enzymes facilitate cross-talk between
cell metabolic and regulatory pathways. Exp Mol Med. 50:1–16. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liberzon A, Subramanian A, Pinchback R,
Thorvaldsdóttir H, Tamayo P and Mesirov JP: Molecular signatures
database (MSigDB) 3.0. Bioinformatics. 27:1739–1740. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gross SR: Actin binding proteins: Their
ups and downs in metastatic life. Cell Adh Migr. 7:199–213. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Beurel E, Grieco SF and Jope RS: Glycogen
synthase kinase-3 (GSK3): Regulation, actions, and diseases.
Pharmacol Ther. 148:114–131. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nakata A, Yoshida R, Yamaguchi R, Yamauchi
M, Tamada Y, Fujita A, Shimamura T, Imoto S, Higuchi T, Nomura M,
et al: Elevated β-catenin pathway as a novel target for patients
with resistance to EGF receptor targeting drugs. Sci Rep.
5:130762015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakayama S, Sng N, Carretero J, Welner R,
Hayashi Y, Yamamoto M, Tan AJ, Yamaguchi N, Yasuda H, Li D, et al:
β-catenin contributes to lung tumor development induced by EGFR
mutations. Cancer Res. 74:5891–5902. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang F, Li Y, Liu B, You J and Zhou Q:
Cancer stem cell-like population is preferentially suppressed by
EGFR-TKIs in EGFR-mutated PC-9 tumor models. Exp Cell Res.
362:195–202. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang F, Xu J, Li H, Tan M, Xiong X and Sun
Y: FBXW2 suppresses migration and invasion of lung cancer cells via
promoting β-catenin ubiquitylation and degradation. Nat Commun.
10:13822019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Fan FT, Shen CS, Tao L, Tian C, Liu ZG,
Zhu ZJ, Liu YP, Pei CS, Wu HY, Zhang L, et al: PKM2 regulates
hepatocellular carcinoma cell epithelial-mesenchymal transition and
migration upon EGFR activation. Asian Pac J Cancer Prev.
15:1961–1970. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Aktary Z, Bertrand JU and Larue L: The
WNT-less wonder: WNT-independent β-catenin signaling. Pigment Cell
Melanoma Res. 29:524–540. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Arce L, Yokoyama NN and Waterman ML:
Diversity of LEF/TCF action in development and disease. Oncogene.
25:7492–7504. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Robertson H, Hayes JD and Sutherland C: A
partnership with the proteasome; the destructive nature of GSK3.
Biochem Pharmacol. 147:77–92. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Valenta T, Hausmann G and Basler K: The
many faces and functions of β-catenin. EMBO J. 31:2714–2736. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Arao T, Fukumoto H, Takeda M, Tamura T,
Saijo N and Nishio K: Small in-frame deletion in the epidermal
growth factor receptor as a target for ZD6474. Cancer Res.
64:9101–9104. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhou W, Han L and Altman RB: Imputing gene
expression to maximize platform compatibility. Bioinformatics.
33:522–528. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Amelio I, Markert EK, Rufini A, Antonov
AV, Sayan BS, Tucci P, Agostini M, Mineo TC, Levine AJ and Melino
G: p73 regulates serine biosynthesis in cancer. Oncogene.
33:5039–5046. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chan YC, Chang YC, Chuang HH, Yang YC, Lin
YF, Huang MS, Hsiao M, Yang CJ and Hua KT: Overexpression of PSAT1
promotes metastasis of lung adenocarcinoma by suppressing the
IRF1-IFNγ axis. Oncogene. 39:2509–2522. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mattaini KR, Sullivan MR and Vander Heiden
MG: The importance of serine metabolism in cancer. J Cell Biol.
214:249–257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
DeNicola GM, Chen PH, Mullarky E, Sudderth
JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al: NRF2
regulates serine biosynthesis in non-small cell lung cancer. Nat
Genet. 47:1475–1481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu B, Jia Y, Cao Y, Wu S, Jiang H, Sun X,
Ma J, Yin X, Mao A and Shang M: Overexpression of phosphoserine
aminotransferase 1 (PSAT1) predicts poor prognosis and associates
with tumor progression in human esophageal squamous cell carcinoma.
Cell Physiol Biochem. 39:395–406. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Morita T, Mayanagi T and Sobue K:
Reorganization of the actin cytoskeleton via transcriptional
regulation of cytoskeletal/focal adhesion genes by
myocardin-related transcription factors (MRTFs/MAL/MKLs). Exp Cell
Res. 313:3432–3445. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Gau D and Roy P: SRF'ing and SAP'ing-the
role of MRTF proteins in cell migration. J Cell Sci.
131:jcs2182222018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Shi X, Zhao S, Cai J, Wong G and Jiu Y:
Active FHOD1 promotes the formation of functional actin stress
fibers. Biochem J. 476:2953–2963. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Schönichen A, Mannherz HG, Behrmann E,
Mazur AJ, Kühn S, Silván U, Schoenenberger CA, Fackler OT, Raunser
S, Dehmelt L and Geyer M: FHOD1 is a combined actin filament
capping and bundling factor that selectively associates with actin
arcs and stress fibers. J Cell Sci. 126:1891–1901. 2013.PubMed/NCBI
|
|
56
|
Heuser VD, Mansuri N, Mogg J, Kurki S,
Repo H, Kronqvist P, Carpén O and Gardberg M: Formin proteins FHOD1
and INF2 in triple-negative breast cancer: Association with basal
markers and functional activities. Breast Cancer (Auckl).
12:11782234187922472018. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Gardberg M, Kaipio K, Lehtinen L, Mikkonen
P, Heuser VD, Talvinen K, Iljin K, Kampf C, Uhlen M, Grénman R, et
al: FHOD1, a formin upregulated in epithelial-mesenchymal
transition, participates in cancer cell migration and invasion.
PLoS One. 8:e749232013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Koka S, Neudauer CL, Li X, Lewis RE,
McCarthy JB and Westendorf JJ: The
formin-homology-domain-containing protein FHOD1 enhances cell
migration. J Cell Sci. 116:1745–1755. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rottner K, Faix J, Bogdan S, Linder S and
Kerkhoff E: Actin assembly mechanisms at a glance. J Cell Sci.
130:3427–3435. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee CW, Vitriol EA, Shim S, Wise AL,
Velayutham RP and Zheng JQ: Dynamic localization of G-actin during
membrane protrusion in neuronal motility. Curr Biol. 23:1046–1056.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Ji P, Diederichs S, Wang W, Böing S,
Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, et
al: MALAT-1, a novel noncoding RNA, and thymosin beta4 predict
metastasis and survival in early-stage non-small cell lung cancer.
Oncogene. 22:8031–8041. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Fei F, Qu J, Zhang M, Li Y and Zhang S:
S100A4 in cancer progression and metastasis: A systematic review.
Oncotarget. 8:73219–73239. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Peippo M, Gardberg M, Lamminen T, Kaipio
K, Carpén O and Heuser VD: FHOD1 formin is upregulated in melanomas
and modifies proliferation and tumor growth. Exp Cell Res.
350:267–278. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Morita T and Hayashi K: Tumor progression
is mediated by thymosin-β4 through a TGFβ/MRTF signaling axis. Mol
Cancer Res. 16:880–893. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Morita T and Hayashi K: G-actin
sequestering protein thymosin-β4 regulates the activity of
myocardin-related transcription factor. Biochem Biophys Res Commun.
437:331–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
da Cunha BR, Domingos C, Stefanini ACB,
Henrique T, Polachini GM, Castelo-Branco P and Tajara EH: Cellular
interactions in the tumor microenvironment: The role of secretome.
J Cancer. 10:4574–4587. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Karagiannis GS, Pavlou MP and Diamandis
EP: Cancer secretomics reveal pathophysiological pathways in cancer
molecular oncology. Mol Oncol. 4:496–510. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Lin A, Wei T, Meng H, Luo P and Zhang J:
Role of the dynamic tumor microenvironment in controversies
regarding immune checkpoint inhibitors for the treatment of
non-small cell lung cancer (NSCLC) with EGFR mutations. Mol Cancer.
18:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li H, Wu C, Chang W, Zhong L, Gao W, Zeng
M, Wen Z, Mai S and Chen Y: Overexpression of PSAT1 is correlated
with poor prognosis and immune infiltration in non-small cell lung
cancer. Front Biosci (Landmark Ed). 28:2432023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sivanand S, Rhoades S, Jiang Q, Lee JV,
Benci J, Zhang J, Yuan S, Viney I, Zhao S, Carrer A, et al: Nuclear
Acetyl-CoA production by ACLY promotes homologous recombination.
Mol Cell. 67:252–265.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia
Y, Tan L, Yang P, Lee JH, Li XJ, et al: KAT2A coupled with the
α-KGDH complex acts as a histone H3 succinyltransferase. Nature.
552:273–277. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kottakis F, Nicolay BN, Roumane A, Karnik
R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, et al:
LKB1 loss links serine metabolism to DNA methylation and
tumorigenesis. Nature. 539:390–395. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hwang IY, Kwak S, Lee S, Kim H, Lee SE,
Kim JH, Kim YA, Jeon YK, Chung DH, Jin X, et al: Psat1-dependent
fluctuations in α-ketoglutarate affect the timing of ESC
differentiation. Cell Metab. 24:494–501. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Harmston N and Lenhard B: Chromatin and
epigenetic features of long-range gene regulation. Nucleic Acids
Res. 41:7185–7199. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Clark SJ: Action at a distance: Epigenetic
silencing of large chromosomal regions in carcinogenesis. Hum Mol
Genet. 16:Spec No 1. R88–R95. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ahn MJ, Won HH, Lee J, Lee ST, Sun JM,
Park YH, Ahn JS, Kwon OJ, Kim H, Shim YM, et al: The 18p11.22 locus
is associated with never smoker non-small cell lung cancer
susceptibility in Korean populations. Hum Genet. 131:365–372. 2012.
View Article : Google Scholar : PubMed/NCBI
|