Introduction
Cholangiocarcinoma (CC), primary bile duct cancer,
divided into intrahepatic and extrahepatic tumors according to its
location, is a rare and highly malignant tumor (1). Risk factors include hepatitis B and C,
cirrhosis and alcohol consumption. The incidence of CC is
increasing globally (2–4). Recently, an increasing number of
studies have focused on a new class of small non-coding regulatory
RNA molecules, termed microRNAs (miRs, miRNAs). miRs are highly
conserved among species and regulate the expression of specific
target genes by binding to the 3′-untranslated regions (3′-UTRs) of
messenger RNA (mRNA). miR dysregulation has been implicated in
tumor initiation and progression in many types of cancer, including
CC (5–7). Studies have shown that miR-421
(8), miR-26a (9), miR-494 (10), miR-25 (11), miR-200c (12), miR-21 (13–15),
miR-214 (16), miR-373 (17–19),
miR-494 (20) and miR-370 (21) are involved in the regulation of cell
growth, cell cycle distribution, apoptosis and metastasis in
CC.
The downregulation of miR-138 is frequently observed
in a variety of tumors (22–26).
miR-138 is able to target a variety of genes, such as G protein α
inhibiting activity polypeptide 2 in tongue squamous cell carcinoma
(27), cyclin D3 in hepatocellular
carcinoma (28), cyclin D1 in
nasopharyngeal carcinoma (29),
Rho-associated, coiled-coil-containing protein kinase 2 (ROCK2) and
Ras homolog gene family, member C (RhoC) in tongue squamous cell
carcinoma (23), as well as others.
It is well known that Ras homolog (Rho) family GTPases, including
RhoC play a pivotal role in tumor malignant transformation and
metastasis (30,31). Knockdown of the RhoC gene suppresses
the proliferative and invasive capacity of human QBC939 CC cells
(32). However, the precise
regulatory effects of miR-138 on a gene [for example, Rho guanine
nucleotide exchange factor (GEF)3 ARHGEF3] may vary depending on
the cell type (23). Thus far, the
expression pattern of miR-138 and RhoC in CC tissues, and whether
miR-138 is involved in the progression of CC remain unknown.
In the present study, the expression levels, the
functional roles and the correlation between miR-138 and RhoC in CC
were investigated. It was confirmed that the miR-138/RhoC-mediated
progression of CC involves the upregulation of the expression of
extracellular signal-regulated kinase (ERK)/matrix
metalloproteinase (MMP)-2/MMP-9.
Materials and methods
Cell culture and transfection
RBE and CQB939 CC cell lines were purchased from the
China Center for Type Culture Collection (CCTCC, Wuhan, China) and
were maintained in RPMI-1640 (HyClone) containing 10% fetal bovine
serum (Hangzhou Sijiqing) in a humidified atmosphere of 5%
CO2 at 37°C. For functional analysis, RBE and CQB939
cells were transfected with scrambled control miRNA (negative
control, NC), miR-138 mimics, or miR-138 inhibitor (Ambion) using
Lipofectamine 2000 (Invitrogen) according to the manufacturer’s
recommendations.
Tumor tissue sample preparation
A total of 9 patients diagnosed with stage Ib, IIa
or IIb CC were recruited in our study. The experimental protocols
were approved by the Ethics Committee of the Xiangya Hospital,
Central South University, Changsha, China. RNA or protein samples
prepared from the tumor tissues and their matched adjacent
non-tumor tissues were then subjected to quantitative real-time
RT-PCR and western blot analysis.
Real-time RT-PCR
Total RNA was extracted from the cells or tissue
samples with TRIzol reagent (Invitrogen) following the
manufacturer’s instructions. The relative expression level of
miR-138 was determined by quantitative real-time RT-PCR using the
mirVana™ qRT-PCR microRNA Detection kit (Ambion) following the
manufacturer’s instructions. Specific primer sets for miR-138 and
U6 (used as an internal reference) were obtained from Ambion. The
expression of RohC mRNA was detected by real-time RT-PCR using the
standard SYBR-Green RT-PCR Kit (Takara, Otsu, Japan) following the
manufacturer’s instructions. The specific primer pairs were as
follows: RhoC (161 bp) sense, 5′-AAGGACCTGAGGCAAGACGAGC-3′ and
antisense, 5′-TCAAACACCTCCCGCACTCCC-3′; β-actin (202 bp; used as an
internal control) sense, 5′-AGGGGCCGGACTCGTCATACT-3′ and antisense,
5′-GGCGGCACCACCATGTACCCT-3′. The relative expression of RhoC mRNA
or miR-138 was quantified using GraphPad Prism 4.0 software
(GraphPad Software, San Diego, CA, USA) and the 2−ΔΔCt
method (33).
Dual luciferase reporter assay
The 3′-UTR of RhoC (position 1210 to 1271,
NM_175744) containing the miRNA-138 binding sites and its
corresponding mutated sequence were cloned into the psi-CHECK2
luciferase reporter vector (Promega) downstream of Renilla
luciferase, termed 3′-UTR RhoC and 3′-UTR Mut RhoC, respectively.
Using Lipofectamine 2000 (Invitrogen), human embryonic kidney
(HEK)293T cells were co-transfected with the reporter constructs
and miR-138 mimics, negative control or miR-138 inhibitor (Ambion).
Luciferase activity was determined after 48 h using the Dual-Glo
substrate system (Promega) and a Beckman Coulter LD400 luminometer.
Data are presented as the ratio of experimental (Renilla)
luciferase to control (firefly) luciferase.
Cell viability assay
Cells in exponential growth were plated at a final
concentration of 2×103 cells/well in 96-well plates.
Cell viability was assessed by MTT assay after 24, 48, 72 and 96 h
of seeding. The optical density at 570 nm (OD570) of each well was
measured with an ELISA reader (ELx-800 type; BioTek).
Cell cycle analysis by flow cytometry
(FCM)
The cells were digested and collected at 48 h
post-transfection and washed with PBS twice. The cells were
resuspended in PBS and then fixed in 70% ethanol at 4°C for 18 h.
The cells were washed with PBS and resuspended in staining solution
(50 μg/ml of propidium iodide, 1 mg/ml of RNase A, 0.1% Triton
X-100 in PBS). The stained cells (1×105) were then
analyzed with a flow cytometer (Beckman Coulter).
Cell migration assay
Cell migration was measured using a wound healing
assay as described by Saadoun et al(34). In brief, cells transfected with
miRNAs or inhibitor as indicated, were cultured to confluence.
Wounds of ~1 mm width were created with a plastic scriber, and the
cells were washed and incubated in serum-free medium. After
wounding for 24 h, cells were incubated in medium including 10%
fetal bovine serum. Cultures at 0, 24 and 48 h were fixed and
observed under a microscope.
Cell invasion assay
The cell invasion assay was performed using a Cell
Invasion Assay kit (Chemicon International, Temecula, CA, USA)
according to the manufacturer’s instruction as previously described
by Aspenstrom et al(35).
Briefly, the cells were placed in the upper compartment of the
chambers, and RPMI-1640 containing 10% fetal bovine serum was added
to the lower chambers. After 24 h of incubation at 37°C, cells on
the upper face of the membrane were scraped using a cotton swab and
cells on the lower face were fixed, stained and observed under a
microscope. The dye on the membrane was then dissolved with 10%
acetic acid, dispensed into 96-well plates (150 μl/well), and the
OD570 of each well was measured with an ELISA reader (ELx-800 type;
BioTek).
Western blot analysis
Cells were lysed in cell lysis buffer containing 7 M
Urea, 2 M Thiourea, 60 mM DTT, 4% CHAPS, 2% pharmalyte 3–10, 1.4
mg/ml PMSF, then centrifuged at 12,000 × g for 20 min at 4°C. The
supernatant was collected and denatured. Proteins were separated in
10% SDS-PAGE and blotted onto polyvinylidene difluoride membranes
(PVDF). The PVDF membranes were treated with TBST containing 50 g/l
skimmed milk at room temperature for 4 h, followed by incubation
with the primary antibodies, anti-RhoC, anti-phosphorylated ERK
(p-ERK), anti-total-ERK, anti-MMP-2, anti-MMP-9 and anti-β-actin
(Santa Cruz Biotechnology, Santa Cruz, CA, USA), at 37°C for 1 h.
The membranes were rinsed and incubated for 1 h with the
correspondent peroxidase-conjugated secondary antibodies.
Chemiluminescent detection was performed using the ECL kit (Pierce
Chemical Co., Rockford, IL, USA). The amount of the protein of
interest, expressed as arbitrary densitometric units, was
normalized to the densitometric units of β-actin.
Statistical analysis
Data are expressed as the means ± SD from at least 3
separate experiments. Statistical analysis was carried out using
SPSS 11.0 software. Differences between 2 groups were analyzed by
the Student’s t-test. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
Expression of miR-138 and RhoC in CC
tissues or cells
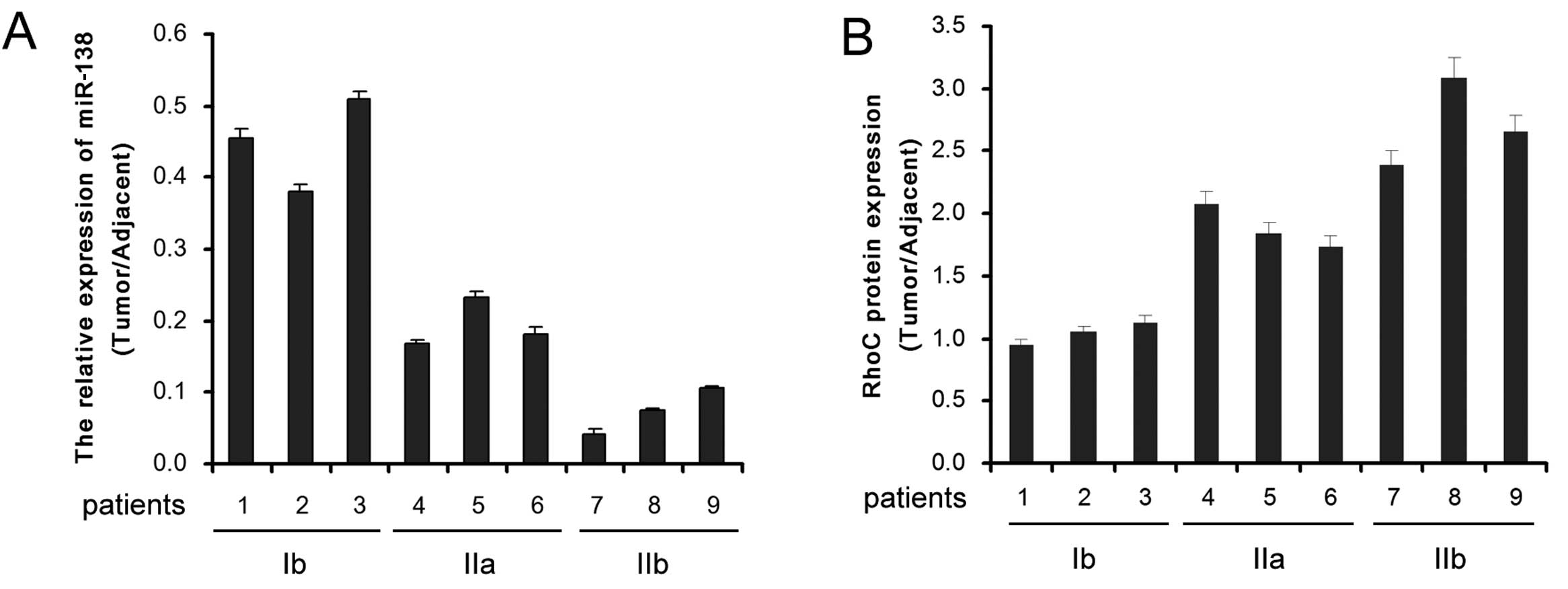
Real-time quantitative RT-PCR was performed to
detect the expression of miR-138 in CC (stage Ib, IIa or IIb) and
adjacent tissues. Compared to the adjacent non-tumor tissues, the
expression level of miR-138 in CC tissues was significantly reduced
and decreased progressively along with malignant progression
(Fig. 1A). Inversely, the
expression level of RhoC was significantly enhanced in CC tissues
and increased progressively along with malignant progression
(Fig. 1B). Thus, a negative
correlation may exist between miR-138 and RhoC in CC progression.
These results indicate that miR-138 upregulation and RhoC
downregulation are involved in the malignant progression of CC.
RhoC is a direct target of miR-138 in
CC
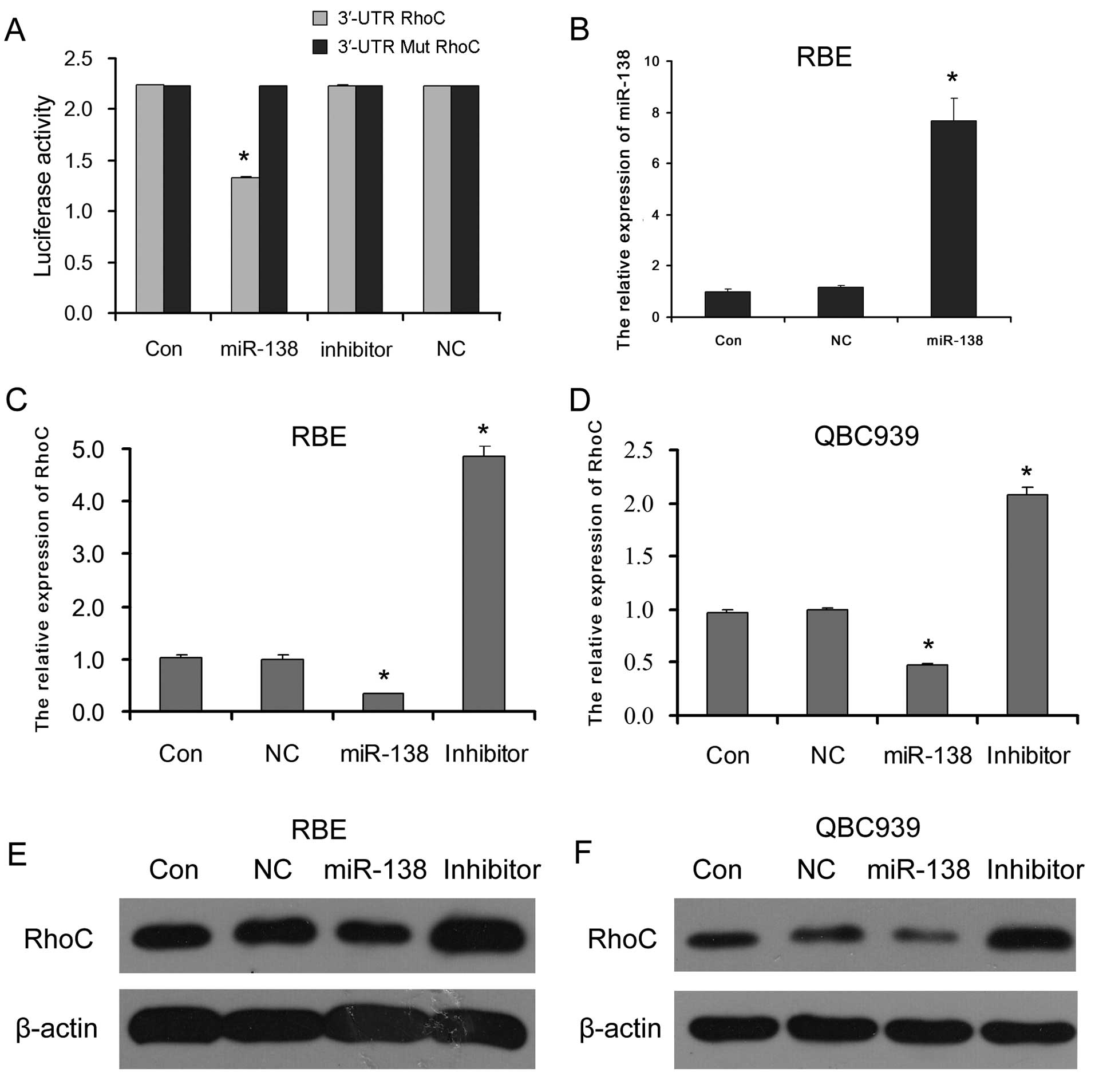
Our data from dual luciferase reporter assay showed
that the luciferase activity was significantly inhibited only in
the HEK293T cells co-transfected with miR-138 mimics and the 3′-UTR
RhoC reporter vector. There were no distinct differences in
luciferase activity between the other co-transfected cells and the
control cells transfected only with 3′-UTR or 3′-UTR mutation RhoC
reporter vector (Fig. 2A). These
results demonstrated that miR-138 directly targeted RhoC.
To confirm the regulatory effect of miR-138 on RhoC
in CC cells, the expression levels of RhoC mRNA and protein in the
RBE and QBC939 CC cells transfected with miR-138 mimics (which
successfully enhanced the miR-138 level in CC cells as shown in
Fig. 2B), miR-138 inhibitor or
negative control (NC) miR-138 were detected. The results showed
that the introduction of miR-138 mimics to RBE and QBC939 CC cells
reduced RhoC mRNA (Fig. 2C and D)
and protein (Fig. 2D and E)
expression; however, miR-138 inhibitor increased RhoC mRNA and
protein expression levels. This suggests that miR-138 serves as a
suppressor of RhoC by directly targeting RhoC in CC cells.
Effect of miR-138/RhOC on cell viability
and cell cycle distribution
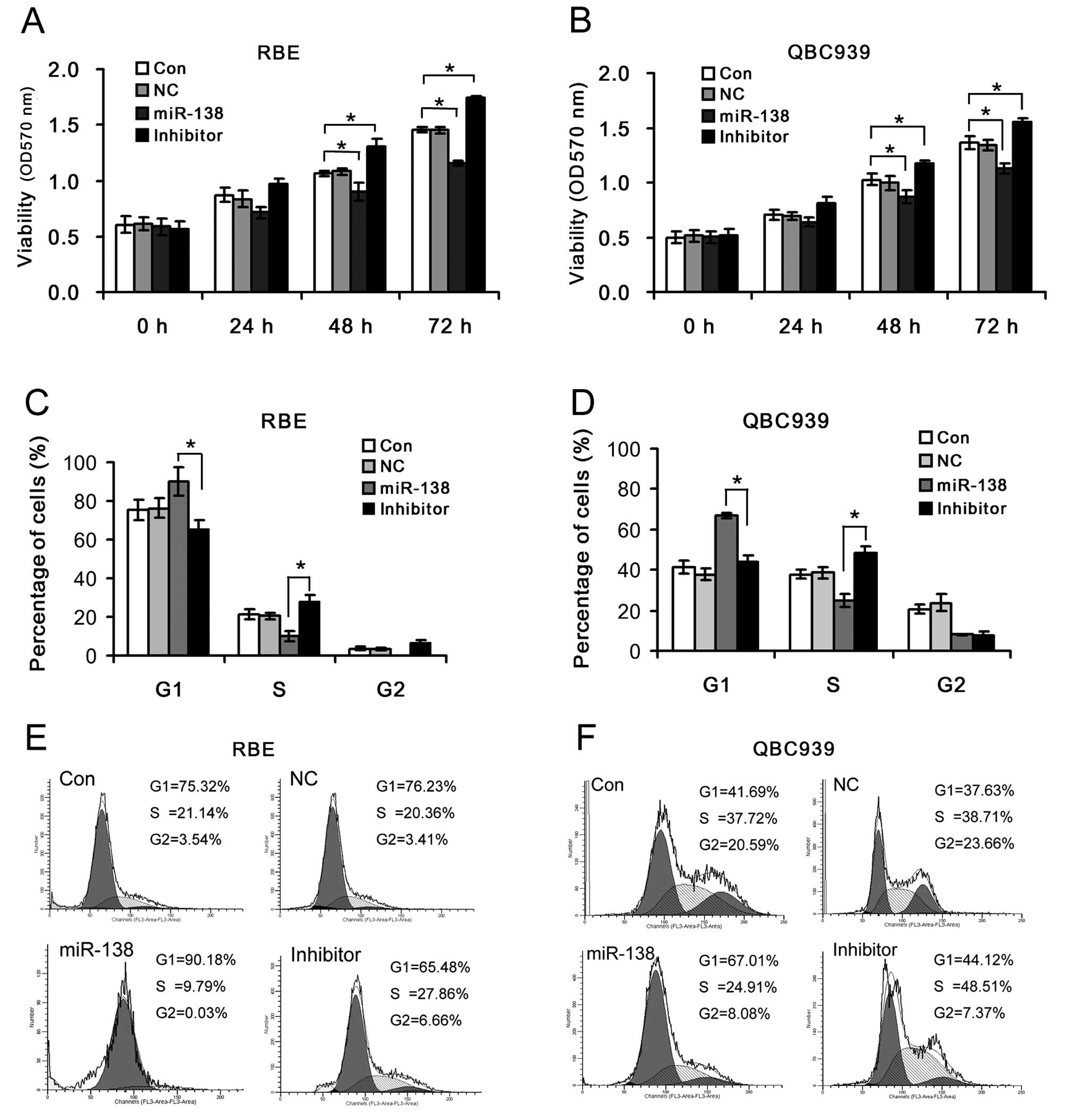
To investigate the effects of miR-138/RhoC on cell
viability and cell cycle distribution in CC, the RBE and QBC939 CC
cells were transfected with miR-138 mimics or miR-138 inhibitor.
Cell viability significantly decreased in the RBE and QBC939 CC
cells transfected with miR-138 mimics, but markedly increased
following transfection with miR-138 inhibitor compared to the
control (Fig. 3C). The percentages
of G1 phase cells in the miR-138 mimics group were significantly
higher than those in the miR-138 inhibitor group. The introduction
of miR-138 mimics enhanced the G1/S ratio in the RBE and QBC939 CC
cells (9.21 and 2.69, respectively); however, the inhibition of
miR-138 reduced the G1/S ratio in the RBE and QBC939 CC cells (2.35
and 0.91, respectively) (Fig. 3D and
E). This suggests that the miR-138-mediated RhoC up- or
downregulation plays an important role in the regulation of CC cell
survival by controlling cell proliferation and G1/S transition.
Effect of miR-138/RhOC on cell migration
and invasion
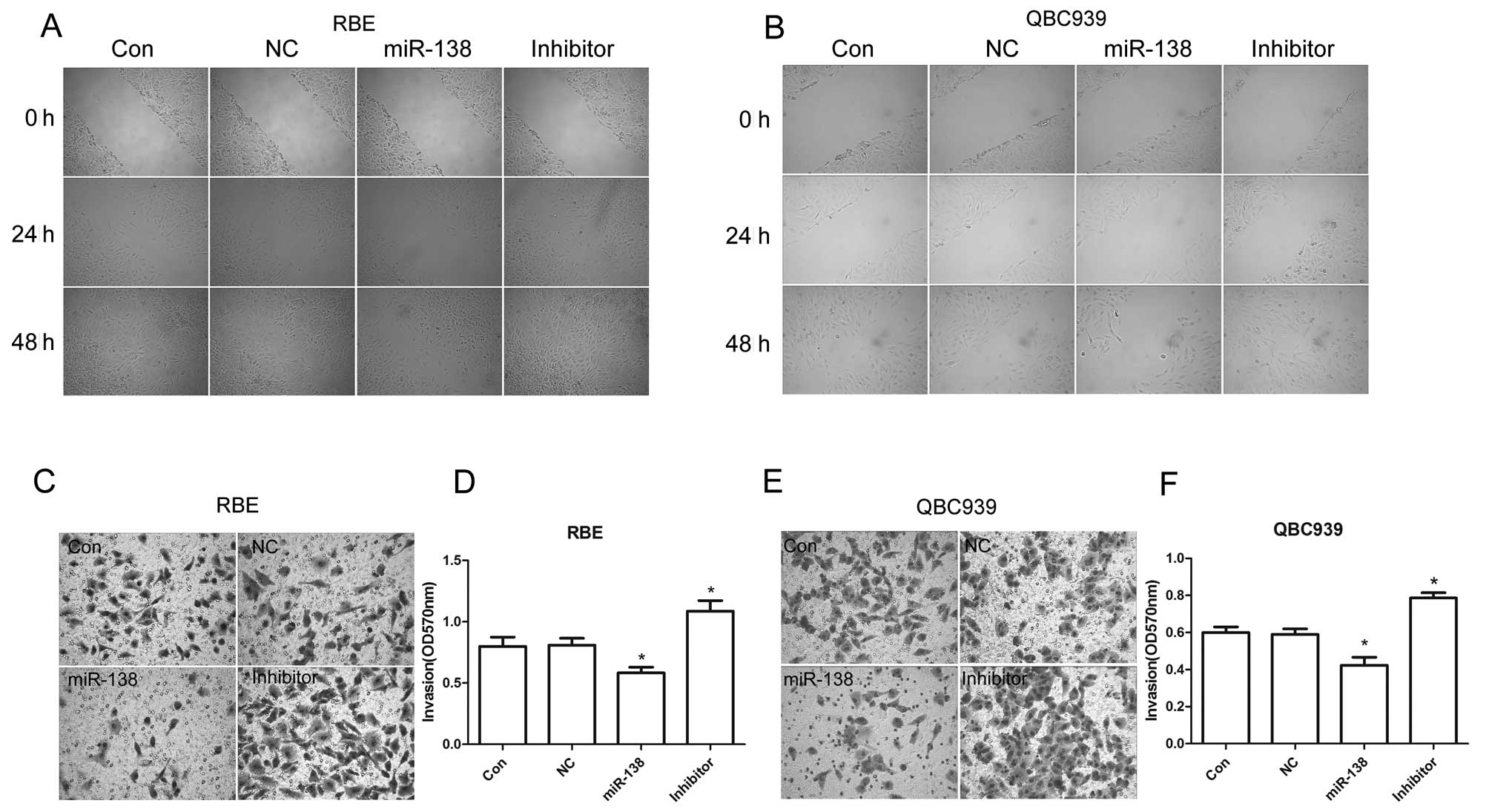
In order to determine the effects of miR-138/RhoC on
CC metastasis, the migration and invasive capability of RBE and
QBC939 CC cells transfected with miR-138 mimics or inhibitor were
examined. We found that compared to the control, introducing
miR-138 mimics to the RBE and QBC939 CC cells significantly
inhibited their migration and invasion. However, migration and
invasion were enhanced with the inhibition of miR-138 in RBE and
QBC939 CC cells (Fig. 4). These
results suggest that the upregulation of RhoC caused by the
downregulation of miR-138 promotes CC cell migration and invasion.
In other words, it contributes to the malignant transformation of
CC.
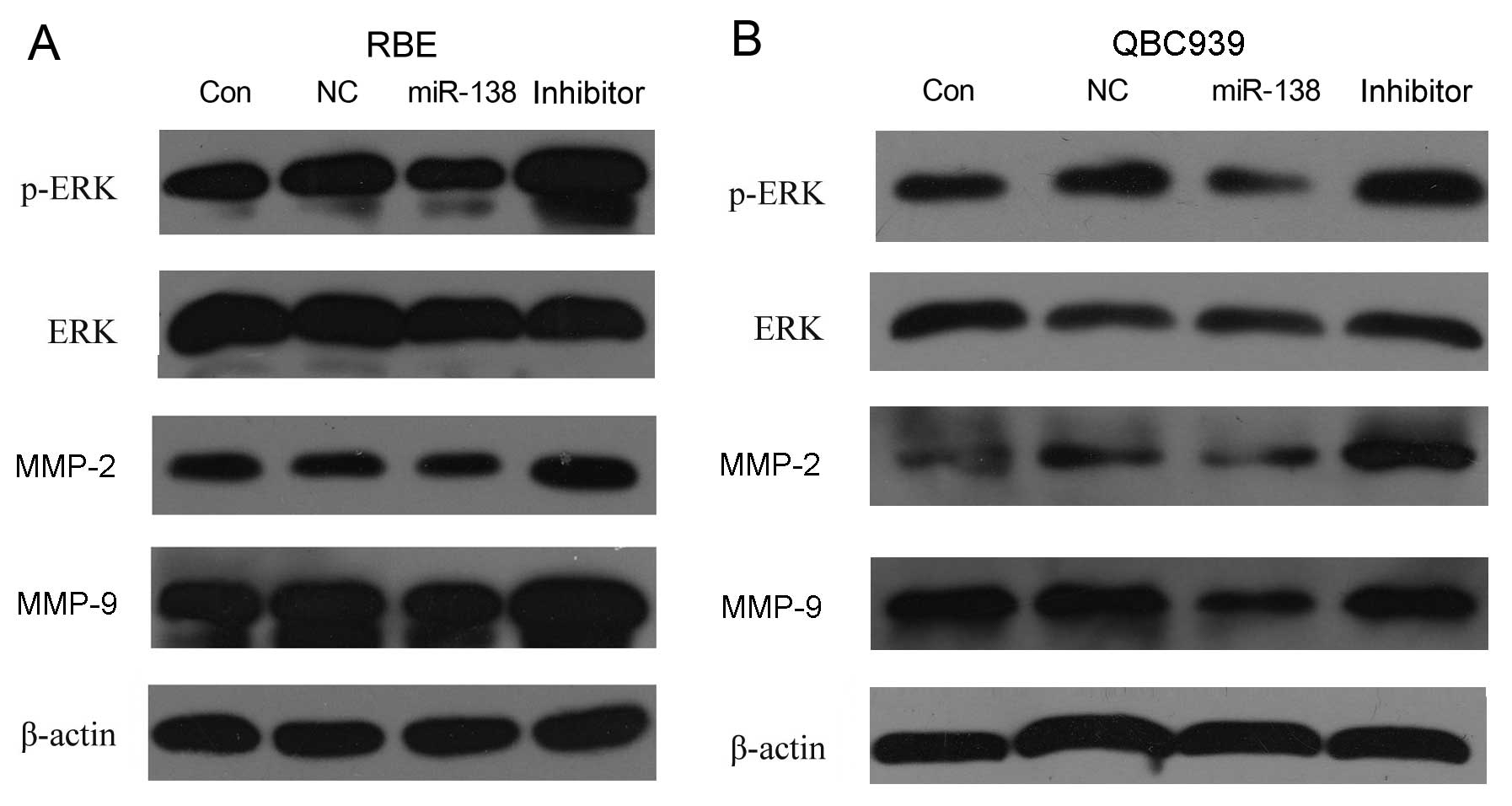
Effect of miR-138/RhoC on ERK, p-ERK,
MMP-2 and MMP-9 expression
To determine which molecules are involved in the
regulation of migration and invasion induced by the dysregulation
of miR-138/RhoC in CC, the protein levels of p-ERK, ERK, MMP-2 and
MMP-9 were analyzed by western blot analysis. The results showed
that introducing miR-138 mimics to the RBE and QBC939 CC cells
decreased p-ERK (without effects on total ERK), MMP-2 and MMP-9
expression. Inversely, the inhibition of miR-138 in RBE and QBC939
CC cells increased p-ERK (without effects on total ERK), MMP-2 and
MMP-9 protein expression (Fig. 5).
This indicates that RhoC upregulation caused by miR-138
downregulation promotes CC cell migration and invasion. The
underlying mechanisms involve the increase in the expression of
p-ERK, MMP-2 and MMP-9.
Discussion
Tumor metastasis is considered the dominant cause of
mortality in cancer patients. Current therapeutic approaches for
highly invasive cholangiocarcinoma are not satisfactory. Therefore,
efforts to elucidate the molecular mechanisms underlying the
metastatic process are of significant clinical importance. Our data
from the comparison of tumor tissues from patients with different
stages of the disease and adjacent non-tumor tissues showed that
the miR-138 expression level gradually decreased with the
enhancement of metastatic potential in CC tissues and the
corresponding RhoC level increased progressively. Therefore,
understanding the functions of miR-138 and RhoC, and their
correlation is important for the clinical diagnosis and treatment
of CC. We then demonstrated the post-transcriptional regulation of
RhoC by miR-138 through the direct binding to its 3′-UTR using dual
luciferase reporter assay. The transfection of RBE and QBC939 CC
cells with miR-138 mimics resulted in a significant increase in the
miR-138 level and a decrease in RhoC mRNA and protein levels. These
data suggest that miR-138 downregulation induces RhoC upregulation
in CC and this process is likely involved in CC progression.
To confirm the functions of miR-138 and RhoC in CC
cells, a series of experiments were performed, demonstrating that
the increased miR-138 expression level suppressed the
proliferation, G1/S transition, migration and invasion of CC cells,
and decreased the expression of RhoC, p-ERK, MMP-2 and MMP-9 in CC
cells. However, the upregulation of RhoC by the inhibition of
miR-138 in CC cells increased the proliferation, G1/S transition,
migration and invasion of the cells, and increased the expression
of p-ERK, MMP-2 and MMP-9. These data suggest that RhoC
upregulation caused by miR-138 downregulation plays an important
role in cell growth regulation, cell cycle transition and
metastasis of CC.
A number of studies have revealed that RhoC plays an
important role in cell proliferation, cycle distribution and tumor
metastasis (23,32). It has been reported that decreasing
the RhoC expression level by transfection with antisense RhoC cDNA
inhibits proliferation and increases the percentage of CC cells in
the G1 phase (32). Our results
also indicated that RhoC downregulation promoted by transfection
with miR-138 mimics suppressed the proliferation and G1/S
transition in CC cells. The inhibition of ERK phosphorylation
induced by RhoC downregulation in RBE and QBC939 CC cells
transfected with miR-138 mimics suppressed cyclin D1 expression,
which plays an important role in the regulation of G1/S transition
and cell proliferation (36,37).
However, whether this process was partly controlled through miR-138
directly targeting cyclin D1 (29)
or cyclin D3 (28), which are 2 of
the the target genes of miR-138, requires further confirmation.
Cell migration and invasion are initial steps in
tumor cell metastasis. Our data indicated that the inhibition of
miR-138 in CC cells or the increase in the RhoC expression level
due to miR-138 downregulation in CC tumor tissues may result in the
enhanced migration and invasion of CC. These effects may be linked
to the increased p-ERK, MMP-2 and MMP-9 expression induced by RhoC
upregulation in RBE and QBC939 CC cells transfected with miR-138
mimics (38,39). Studies have indicated that RhoC
upregulation can promote actin stress fiber and focal adhesion
contact formation and may thus facilitate CC cell migration
(23,40). The upregulation of MMP-2 and MMP-9
can cause the CC cells to break through the surrounding tissue
barrier via enzymatic degradation of the extracellular matrix
(41). Moreover, the activation of
ERK possibly enhances MMP-2 and MMP-9 release from CC cells
(42). In addition, it has been
reported that the upregulation of RhoC and p-ERK expression
promotes VEGF expression, resulting in accelerated angiogenesis and
distant metastasis (43). These
data suggest that RhoC upregulation caused by miR-138
downregulation promotes CC malignant transformation. The mechanisms
underlying these effects involve the activation of ERK and the
upregulation of MMP-2 and MMP-9 expression.
In conclusion, RhoC is highly expressed in CC
tissues and its expression gradually increases with the
downregulation of miR-138. RhoC is a direct target of miR-138 in
CC. The upregulation of miR-138 suppresses the proliferation, G1/S
transition, migration and invasion of RBE and QBC939 CC cells. The
potential mechanisms involved include the inhibition of RhoC
expression, ERK activation, and the downregulation of MMP-2 and
MMP-9 expression. Therefore, it can be concluded that RhoC
upregulation induced by miR-138 downregulation plays an important
role in the malignant progression of CC, and miR-138/RhoC is a
potential target for the clinical diagnosis and treatment of
CC.
References
|
1
|
Shimoda M and Kubota K: Multi-disciplinary
treatment for cholangiocellular carcinoma. World J Gastroenterol.
13:1500–1504. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patel T: Cholangiocarcinoma. Nat Clin
Pract Gastroenterol Hepatol. 3:33–42. 2006. View Article : Google Scholar
|
|
3
|
Reddy SB and Patel T: Current approaches
to the diagnosis and treatment of cholangiocarcinoma. Curr
Gastroenterol Rep. 8:30–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Singh P and Patel T: Advances in the
diagnosis, evaluation and management of cholangiocarcinoma. Curr
Opin Gastroenterol. 22:294–299. 2006. View Article : Google Scholar
|
|
5
|
Kawahigashi Y, Mishima T, Mizuguchi Y, et
al: MicroRNA profiling of human intrahepatic cholangiocarcinoma
cell lines reveals biliary epithelial cell-specific microRNAs. J
Nippon Med Sch. 76:188–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Isomoto H: Epigenetic alterations
associated with cholangiocarcinoma (Review). Oncol Rep. 22:227–232.
2009.PubMed/NCBI
|
|
7
|
Chen L, Yan HX, Yang W, et al: The role of
microRNA expression pattern in human intrahepatic
cholangiocarcinoma. J Hepatol. 50:358–369. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhong XY, Yu JH, Zhang WG, et al:
MicroRNA-421 functions as an oncogenic miRNA in biliary tract
cancer through down-regulating farnesoid X receptor expression.
Gene. 493:44–51. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhang J, Han C and Wu T: MicroRNA-26a
promotes cholangiocarcinoma growth by activating β-catenin.
Gastroenterology. 143:246–256.e248. 2012.PubMed/NCBI
|
|
10
|
Yamanaka S, Campbell NR, An F, et al:
Coordinated effects of microRNA-494 induce G(2)/M arrest in human
cholangiocarcinoma. Cell Cycle. 11:2729–2738. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Razumilava N, Bronk SF, Smoot RL, et al:
miR-25 targets TNF-related apoptosis inducing ligand (TRAIL) death
receptor-4 and promotes apoptosis resistance in cholangiocarcinoma.
Hepatology. 55:465–475. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oishi N, Kumar MR, Roessler S, et al:
Transcriptomic profiling reveals hepatic stem-like gene signatures
and interplay of miR-200c and epithelial-mesenchymal transition in
intrahepatic cholangiocarcinoma. Hepatology. 56:1792–1803. 2012.
View Article : Google Scholar
|
|
13
|
Liu CZ, Liu W, Zheng Y, et al: PTEN and
PDCD4 are bona fide targets of microRNA-21 in human
cholangiocarcinoma. Chin Med Sci J. 27:65–72. 2012.PubMed/NCBI
|
|
14
|
He Q, Cai L, Shuai L, et al: Ars2 is
overexpressed in human cholangiocarcinomas and its depletion
increases PTEN and PDCD4 by decreasing MicroRNA-21. Mol Carcinog.
Dec 28–2011.(Epub ahead of print).
|
|
15
|
Selaru FM, Olaru AV, Kan T, et al:
MicroRNA-21 is overexpressed in human cholangiocarcinoma and
regulates programmed cell death 4 and tissue inhibitor of
metalloproteinase 3. Hepatology. 49:1595–1601. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li B, Han Q, Zhu Y, Yu Y, Wang J and Jiang
X: Down-regulation of miR-214 contributes to intrahepatic
cholangiocarcinoma metastasis by targeting Twist. FEBS J.
279:2393–2398. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen YJ, Luo J, Yang GY, Yang K, Wen SQ
and Zou SQ: Mutual regulation between microRNA-373 and
methyl-CpG-binding domain protein 2 in hilar cholangiocarcinoma.
World J Gastroenterol. 18:3849–3861. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen Y, Luo J, Tian R, Sun H and Zou S:
miR-373 negatively regulates methyl-CpG-binding domain protein 2
(MBD2) in hilar cholangiocarcinoma. Dig Dis Sci. 56:1693–1701.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen Y, Gao W, Luo J, Tian R, Sun H and
Zou S: Methyl-CpG binding protein MBD2 is implicated in
methylation-mediated suppression of miR-373 in hilar
cholangiocarcinoma. Oncol Rep. 25:443–451. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Olaru AV, Ghiaur G, Yamanaka S, et al:
MicroRNA down-regulated in human cholangiocarcinoma control cell
cycle through multiple targets involved in the G1/S checkpoint.
Hepatology. 54:2089–2098. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Meng F, Wehbe-Janek H, Henson R, Smith H
and Patel T: Epigenetic regulation of microRNA-370 by interleukin-6
in malignant human cholangiocytes. Oncogene. 27:378–386. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee ST, Chu K, Im WS, et al: Altered
microRNA regulation in Huntington’s disease models. Exp Neurol.
227:172–179. 2011.
|
|
23
|
Jiang L, Liu X, Kolokythas A, et al:
Downregulation of the Rho GTPase signaling pathway is involved in
the microRNA-138-mediated inhibition of cell migration and invasion
in tongue squamous cell carcinoma. Int J Cancer. 127:505–512. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Chen Z, Yu J, Xia J and Zhou X:
MicroRNA profiling and head and neck cancer. Comp Funct Genomics.
2009:8375142009.PubMed/NCBI
|
|
25
|
Mitomo S, Maesawa C, Ogasawara S, et al:
Downregulation of miR-138 is associated with overexpression of
human telomerase reverse transcriptase protein in human anaplastic
thyroid carcinoma cell lines. Cancer Sci. 99:280–286. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Poliseno L, Haimovic A, Segura MF, et al:
Histology-specific microRNA alterations in melanoma. J Invest
Dermatol. 132:1860–1868. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang L, Dai Y, Liu X, et al:
Identification and experimental validation of G protein alpha
inhibiting activity polypeptide 2 (GNAI2) as a microRNA-138 target
in tongue squamous cell carcinoma. Hum Genet. 129:189–197. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang W, Zhao LJ, Tan YX, Ren H and Qi ZT:
MiR-138 induces cell cycle arrest by targeting cyclin D3 in
hepatocellular carcinoma. Carcinogenesis. 33:1113–1120. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu X, Lv XB, Wang XP, et al: MiR-138
suppressed nasopharyngeal carcinoma growth and tumorigenesis by
targeting the CCND1 oncogene. Cell Cycle. 11:2495–2506. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clark EA, Golub TR, Lander ES and Hynes
RO: Genomic analysis of metastasis reveals an essential role for
RhoC. Nature. 406:532–535. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Iiizumi M, Bandyopadhyay S, Pai SK, et al:
RhoC promotes metastasis via activation of the Pyk2 pathway in
prostate cancer. Cancer Res. 68:7613–7620. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shi Z, Chen ML, He QL and Zeng JH:
Antisense RhoC gene suppresses proliferation and invasion capacity
of human QBC939 cholangiocarcinoma cells. Hepatobiliary Pancreat
Dis Int. 6:516–520. 2007.PubMed/NCBI
|
|
33
|
Arocho A, Chen B, Ladanyi M and Pan Q:
Validation of the 2-DeltaDeltaCt calculation as an alternate method
of data analysis for quantitative PCR of BCR-ABL P210 transcripts.
Diagn Mol Pathol. 15:56–61. 2006.PubMed/NCBI
|
|
34
|
Saadoun S, Papadopoulos MC, Hara-Chikuma M
and Verkman AS: Impairment of angiogenesis and cell migration by
targeted aquaporin-1 gene disruption. Nature. 434:786–792. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Aspenstrom P, Fransson A and Saras J: Rho
GTPases have diverse effects on the organization of the actin
filament system. Biochem J. 377:327–337. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ravenhall C, Guida E, Harris T, Koutsoubos
V and Stewart A: The importance of ERK activity in the regulation
of cyclin D1 levels and DNA synthesis in human cultured airway
smooth muscle. Br J Pharmacol. 131:17–28. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Corona G, Deiana M, Incani A, Vauzour D,
Dessi MA and Spencer JP: Hydroxytyrosol inhibits the proliferation
of human colon adenocarcinoma cells through inhibition of ERK1/2
and cyclin D1. Mol Nutr Food Res. 53:897–903. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ikoma T, Takahashi T, Nagano S, et al: A
definitive role of RhoC in metastasis of orthotopic lung cancer in
mice. Clin Cancer Res. 10:1192–1200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Xue F, Takahara T, Yata Y, et al: Blockade
of Rho/Rho-associated coiled coil-forming kinase signaling can
prevent progression of hepatocellular carcinoma in matrix
metalloproteinase-dependent manner. Hepatol Res. 38:810–817. 2008.
View Article : Google Scholar
|
|
40
|
van Golen KL, Wu ZF, Qiao XT, Bao LW and
Merajver SD: RhoC GTPase, a novel transforming oncogene for human
mammary epithelial cells that partially recapitulates the
inflammatory breast cancer phenotype. Cancer Res. 60:5832–5838.
2000.
|
|
41
|
Blain EJ: Mechanical regulation of matrix
metalloproteinases. Front Biosci. 12:507–527. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ehrenfeld P, Conejeros I, Pavicic MF, et
al: Activation of kinin B1 receptor increases the release of
metalloproteases-2 and -9 from both estrogen-sensitive and
-insensitive breast cancer cells. Cancer Lett. 301:106–118. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ming J, Liu N, Gu Y, Qiu X and Wang EH:
PRL-3 facilitates angiogenesis and metastasis by increasing ERK
phosphorylation and up-regulating the levels and activities of
Rho-A/C in lung cancer. Pathology. 41:118–126. 2009. View Article : Google Scholar : PubMed/NCBI
|