Introduction
Lung cancer is one of the most common types of
malignancies. It is not only the leading cause of cancer-related
deaths, but also the most frequently diagnosed cancer worldwide
(1). Yearly, it accounts for more
than 1.8 million newly diagnosed cases (13% of total) and 1.6
million cancer-related deaths (19.4% of total) (2). Approximately 90% of all lung cancers
are NSCLC, with 25–30% of the NSCLC cases being squamous cell
cancer (SCC) (3). SCC is the second
most common histologic subtype of NSCLC (4). Compared with other histologic types,
SCC exhibits distinct epidemiological, clinicopathological and
molecular characteristics. In particular, SCC lesions are usually
located centrally in segmental, lobar or main bronchi, may show
central cavitation and are associated with increased rates of
hemorrhage compared with other lung cancer types (5). While outcomes have improved with
adenocarcinoma, treatment options and outcomes are still limited in
patients with advanced-stage SCC (4). To date, no agents have been
specifically approved for the treatment of SCC (3).
Surgery, chemotherapy and radiotherapy are major
treatment methods for lung cancer patients (6). Therein, surgery is considered as the
most effective curable method, particularly for early-stage
patients. However, ~70% of lung cancer patients suffer from local
advanced or metastatic disease at the time of diagnosis and are
unsuitable to receive surgical treatment (7). Thus, receiving chemotherapy is
unavoidable for these patients and the efficacy of chemotherapy is
still a matter of concern. The genome DNA is constantly exposed to
various genotoxic insults. The DNA damage response (DDR) is a
complex signal transduction pathway that leads to multiple outcomes
including cell cycle arrest, DNA repair, senescence and apoptosis
(8–10). DNA-damaging agents induce various
types of DNA damage including modification of bases, intra-strand
crosslinks, inter-strand crosslinks, single-strand breaks (SSBs)
and double-strand breaks (DSBs). Each type of DNA damage is
recognized and processed by proteins involved in the DDR. The
response to DNA DSBs and collapsed replication forks is
particularly crucial as these types of damage are difficult to
repair. To respond to DNA damage, ataxia telangiectasia mutated
(ATM) or ataxia telangiectasia and Rad3-related (ATR)
phosphorylates and activates checkpoint kinase 1/2 (Chk1/2), and
then causes cell cycle arrest and DNA repair, otherwise it may lead
to cell apoptosis when DNA damage is too severe.
Arrestins were initially discovered to interact with
G protein-coupled receptors (GPCRs) to mediate their
internalization and desensitization (11,12).
In recent years, more and more novel functions of ARRB1 have been
discovered. Arrestins, including ARRB1, always serve as adaptors,
scaffolds and/or signal transducers, and they connect the activated
receptors with diverse signaling pathways within the cell. ARRB1
plays a vital role in scaffolding and modulating varieties of
intracellular signaling networks such as NF-κB and Mdm2/p53
(13). In this diversity of cell
signaling pathways, ARRB1 may have different effects on the
cellular process. Our former study showed that loss of ARRB1
expression was associated with poor survival of NSCLC particularly
in SCC (14). Recently, it was
reported that ARRB1 may be involved in DNA damage, which is a
common target of chemotherapy and radiotherapy (15,16),
however, whether DDR is involved in this process remains unclear.
We aimed to study the role of ARRB1 in DDR induced by DNA-damaging
agents, and to explore the underlying mechanism of regulation
within it.
Materials and methods
Ethical approval
Written informed consent was obtained from all the
subjects involved and the present study was approved by the
Shandong Provincial Hospital Ethics Committee. All methods used in
the present study were carried out in accordance with the approved
guidelines.
Patients
Fresh frozen tissue samples from 30 patients with
primary lung squamous cancer who had undergone complete surgical
resection were obtained in the present study (Table I). Patients and relevant data were
acquired from the Bio-Bank of Shandong Provincial Hospital from
January 2011 to December 2012.
 | Table I.Characteristics of the patients with
SCC-NSCLC in the present study. |
Table I.
Characteristics of the patients with
SCC-NSCLC in the present study.
| Variables | n | Percentage (%) |
|---|
| All cases | 30 | 100.00 |
| Age at diagnosis
(years) |
|
|
| ≥60 | 21 |
70.00 |
|
<60 | 9 |
30.00 |
| Gender |
|
|
|
Female | 5 |
16.67 |
| Male | 25 |
83.33 |
| Smoking index |
|
|
| ≥400 | 23 |
76.67 |
|
<400 | 7 |
23.33 |
| Tumor
differentiation |
|
|
| Well | 0 |
0.00 |
|
Moderate | 21 |
70.00 |
|
Poor | 9 |
30.00 |
| Pathological
stage |
|
|
|
I+II | 5 |
16.67 |
|
III+IV | 25 |
83.33 |
| Lymph node
status |
|
|
|
Negative | 18 |
60.00 |
|
Positive | 12 |
40.00 |
The Ethics Community of Shandong Provincial Hospital
affiliated to Shandong University permitted the undertaking of the
present study and the informed consent from patients for the use of
clinical data was obtained before surgery.
For the histology and tumor-node-metastasis (TNM)
stage, the classification criteria for lung tumors from the World
Health Organization and International Association for the Study of
Lung Cancer (WHO/IASLC) was applied. For the follow-up, patients
were evaluated every 3 months by thorax CT and abdomen
ultrasonography for the first 2 years after surgery along with
adjuvant treatment, and annually thereafter according to
schedule.
Cell culture and transfection
Two human lung squamous cancer cell lines (H520 and
SK-MES-1) were used in the present study. Both of them were
obtained from the American Type Culture Collection (ATCC; Manassas,
VA, USA). Cells were grown in monolayer cultures in RPMI-1640
medium (HyClone, Logan, UT, USA) containing 10% fetal bovine serum
(Gibco, Carlsbad, CA, USA) in a humidified 37°C atmosphere of 5%
CO2. For both plasmids (pcDNA and ARRB1) and siRNA
transfection, all cells were seeded into 6-well cell culture plates
(diameter of each well was ~35 mm; Corning, Corning, NY, USA). When
the cell density reached ~50–70% confluence the next day,
Lipofectamine 2000 transfection reagent (Invitrogen, Carlsbad, CA,
USA) was applied according to the manufacturers instructions.
MTT and colony formation assays
The
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was used to assess the cytotoxicity of DNA-damaging agents.
Cells were seeded into 96-well culture plates with a density of
3×103 and incubated for 24 h under the conditions
described above for standard cell culture maintenance. Then, the
cells received cisplatin or etoposide treatment at different doses.
After a 48-h treatment, 10 µl MTT reagent was added into each well
and incubated for another 4 h. Finally, 100 µl dimethyl sulfoxide
(DMSO) was added into each of the same wells. Then, the optical
density value of each well was determined using a Bio-Rad model 680
microplate reader at a wavelength of 490 nm.
As for the colony formation assay, cells were plated
into 12-well culture plates with 1,000 cells in each well. On the
following day, the cells received drugs (1.5 µM cisplatin or
etoposide) and were then incubated for 14 days. Subsequently, the
plates were stained with crystal violet, and a colony count was
conducted using ImageJ software.
Western blotting and
co-immunoprecipitation (Co-IP)
Western blotting was performed to detect the
specific protein levels (total or phosphorylated). Cells were lysed
in RIPA buffer supplemented with PhosSTOP (Roche, Mannheim,
Germany) on ice. After BCA protein quantification, loading buffer
was added into the mixture. Then, a standard western blotting
procedure was performed. Nitrocellulose filter membranes with
proteins were covered with primary antibodies overnight. After
washing the membrane 3 times with Tris-buffered saline and Tween-20
(TBST), horseradish peroxidase (HRP)-conjugated anti-mouse,
anti-goat or anti-rabbit antibodies were applied according to the
primary antibodies on the following day. After washing the
membranes 3 times with TBST, the protein levels were detected using
enhanced chemiluminescence (ECL) detection reagent (Advansta Corp.,
Menlo Park, CA, USA).
For Co-IP, cells after treatment were lysed by RIPA
solution for Co-IP on ice for 30 min. Then, BCA protein
quantification was conducted. Each adjusted buffer was added to 5
µl anti-ARRB1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA,
USA) and protein A and G Sepharose beads (Invitrogen) overnight.
The beads were washed 3 times with immune-complex buffer and
resolved by SDS-PAGE. Then 1X sample buffer was added to the
resolved precipitates and the mixture was boiled for 10 min.
Subsequently, western blotting was performed.
Antibodies used in the western blotting are listed
as follows. Anti-GAPDH antibody was purchased from Santa Cruz
Biotechnology. Anti-caspase3, -PARP, -ARRB1 and
anti-phosphorylated-ATR (Ser428), -ATM (Ser1981), -Chk1 (Ser345),
-Chk2 (Thr68), -BRCA1 (Ser1524), -H2AX (Ser139) and -NF-κB p65
(Ser536) antibodies were all purchased from Cell Signaling
Technology (Danvers, MA, USA). Anticancer drugs (cisplatin and
etoposide) were purchased from Sigma-Aldrich (St. Louis, MO,
USA).
Flow cytometry
PE-Annexin V/7-amino-actinomycin D (7-AAD) staining
assay was used to assess apoptosis. Annexin V is a
phospholipid-binding protein with a high affinity for
phosphatidylserine (PS). In normal live cells, PS is located on the
cytoplasmic surface of the cell membrane. When cells undergo
apoptosis, PS is translocated from the inner to the outer leaflet
of the plasma membrane, and becomes available for Annexin V binding
(17). 7-AAD is generally
cell-impermeable for live cells, and undergoes a spectral shift
upon association with DNA. Thus, it has been used to label necrotic
or late apoptotic/dead cells with damaged cell membranes (18). Therefore, combination of PE-Annexin
V and 7-AAD stains can be exploited to distinguish cells undergoing
necrosis and apoptosis to cell death (19). Apoptosis was detected in the present
study, using flow cytometry (FCM). A PE Annexin V apoptosis
detection kit (559763) and a LSRFortessa (both from BD Pharmingen,
Franklin Lakes, NJ, USA) were used for FCM. No adjustments were
made to the instrument configuration and settings.
Comet assay
The comet assay (also called single-cell gel
electrophoresis) is a sensitive and rapid method for DNA
strand-break detection in eukaryotic cells (20). We performed the assay according to
the instructions (Cell Biolabs Inc., San Diego, CA, USA) to
evaluate the DNA damage induced by chemotherapy. Cells were
digested by trypsin and washed twice with cold phosphate-buffered
saline (PBS). Then, OxiSelect Comet Slides were covered with a
mixture of comet agarose and the cells. After lysis, the slides
underwent electrophoresis for 30 min under 1 V/cm at 4°C. Cells
were stained with Vista Green DNA dye and visualized using
epifluorescence microscopy. The percentage of DNA migrated in the
tail (% of tail DNA), the tail moment and the length of DNA
migration (tail length) were analyzed by Comet Assay IV software
V4.3 (Perceptive Instruments, Suffolk, UK).
Animal study
All mouse experimental procedures were performed in
accordance with the Regulations for the Administration of Affairs
Concerning Experimental Animals approved by the State Council of
the People's Republic of China. In addition, the present study was
approved by the Ethics Committee of Shandong Provincial Hospital.
All methods used in the present study were carried out in
accordance with the approved guidelines.
BALB/c-nu mice (female, 5–6 weeks old) were
purchased from the Vital River Laboratory Animal Technology (VRL;
Beijing, China). Mice were bred and maintained under specific
pathogen-free conditions. All animal experimental protocols were
approved by the Ethics Committee of Shandong University. H520 cells
(4×106) were subcutaneously injected into mice. Tumor
growth was monitored and evaluated according to volume (length ×
width × width/2) at the indicated time-points during a 28-day
period after the tumor volume reached 100 mm3. To assess
the effect of ARRB1 on cisplatin in vivo, mice were divided
into groups with matched weight: i) the group treated with 2.5
mg/kg cisplatin (control group); ii) the group treated with 2.5
mg/kg cisplatin after in vivo transfection of ARRB1 (ARRB1+
group). Cisplatin (2.5 mg/kg) dissolved in normal saline was
intraperitoneally injected every third day in both groups. ARRB1
plasmid (1.5 mg/kg) was intratumorally injected 24–48 h before
cisplatin treatment in the ARRB1+ group.
Statistical analysis
All statistical calculations were performed using
SPSS 19.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism 6
(GraphPad Software, Inc., La Jolla, CA, USA). Each experiment was
performed in triplicate, at least 3 independent times. In addition,
data are expressed in the form of count or mean ± standard
deviation, and analyzed by t-test or χ2 test according
to the data characteristics (continuous variables, t-test;
categorical variables, χ2 test). For all calculations,
P-values were all two-sided and <0.05 was considered to indicate
a statistically significant result.
Results
Downregulation of ARRB1 in lung SCC
patients
Characteristics of the patients are shown in
Table I. Compared with
para-carcinoma tissues, a significant downregulation of ARRB1 in
SCC tissues was noted. Of the 30 matched cancer and normal tissues,
the mRNA level of ARRB1 in 16 cancer tissues was decreased in
comparison with this level in the matched para-carcinoma tissues.
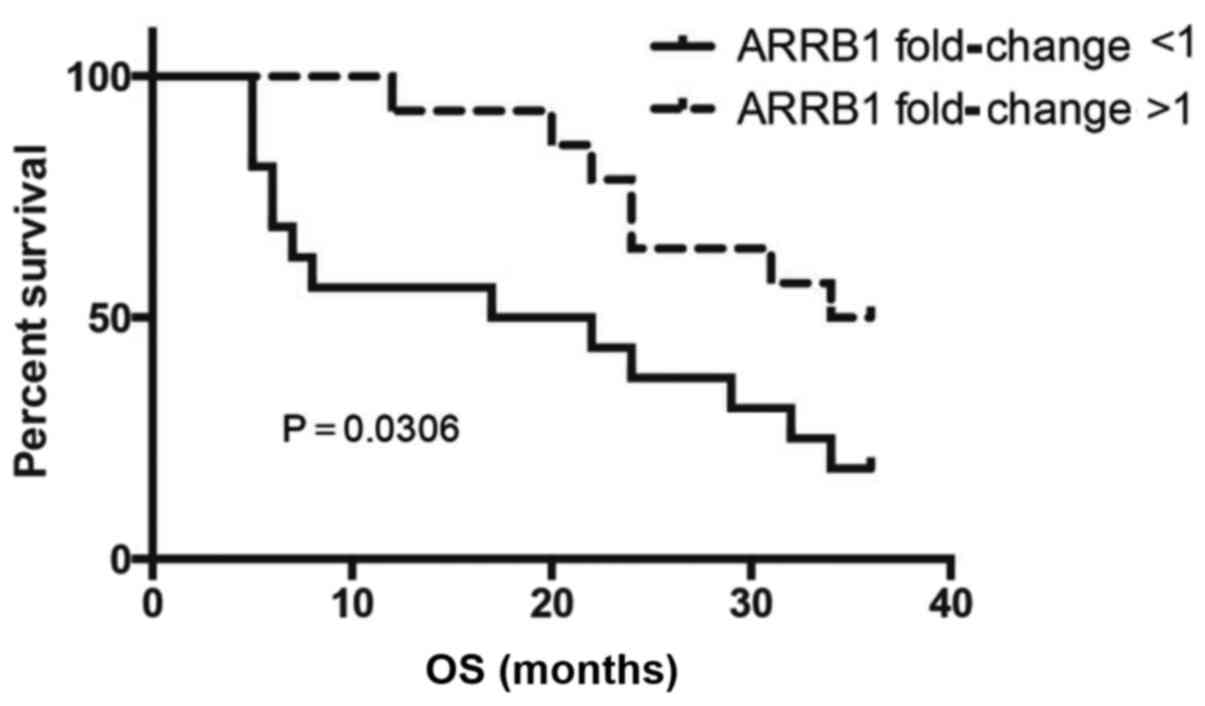
SCC patients with low levels of ARRB1 showed significantly poorer
overall survival (OS) (P=0.0306) when compared with the OS in the
patients with high ARRB1 (Fig.
1).
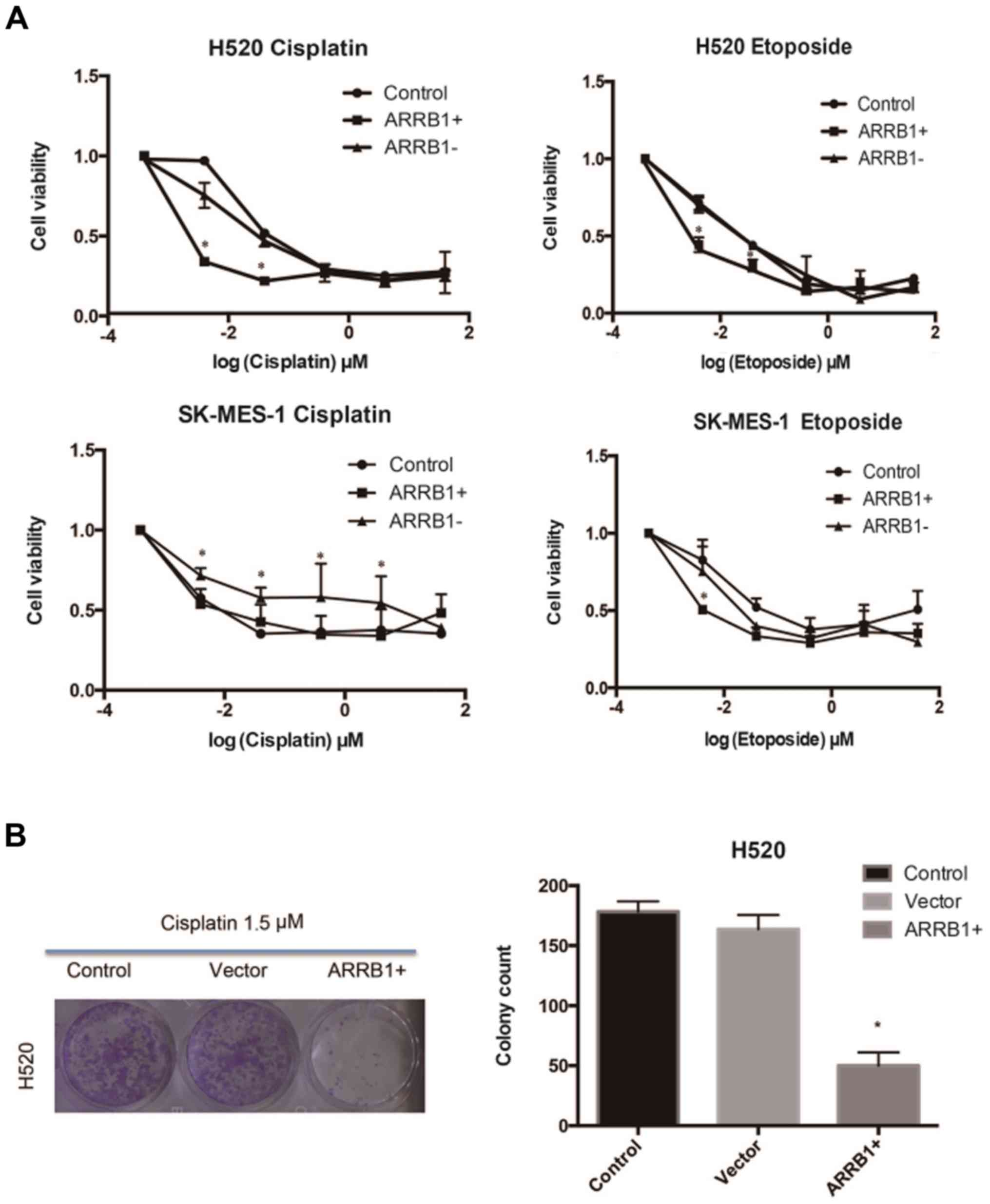
ARRB1 increases the efficacy of
DNA-damaging agents in SCC in vitro
Cisplatin and etoposide were used in H520 and
SK-MES-1 cell lines, respectively, at various concentrations and
the resulting cell viability was assessed using MTT and colony
formation assays (Fig. 2). The data
we obtained suggested that ARRB1 enhanced the sensitivity of tumor
cells to chemotherapy at lower concentrations and that
ARRB1-knockdown increased resistance to chemotherapy.
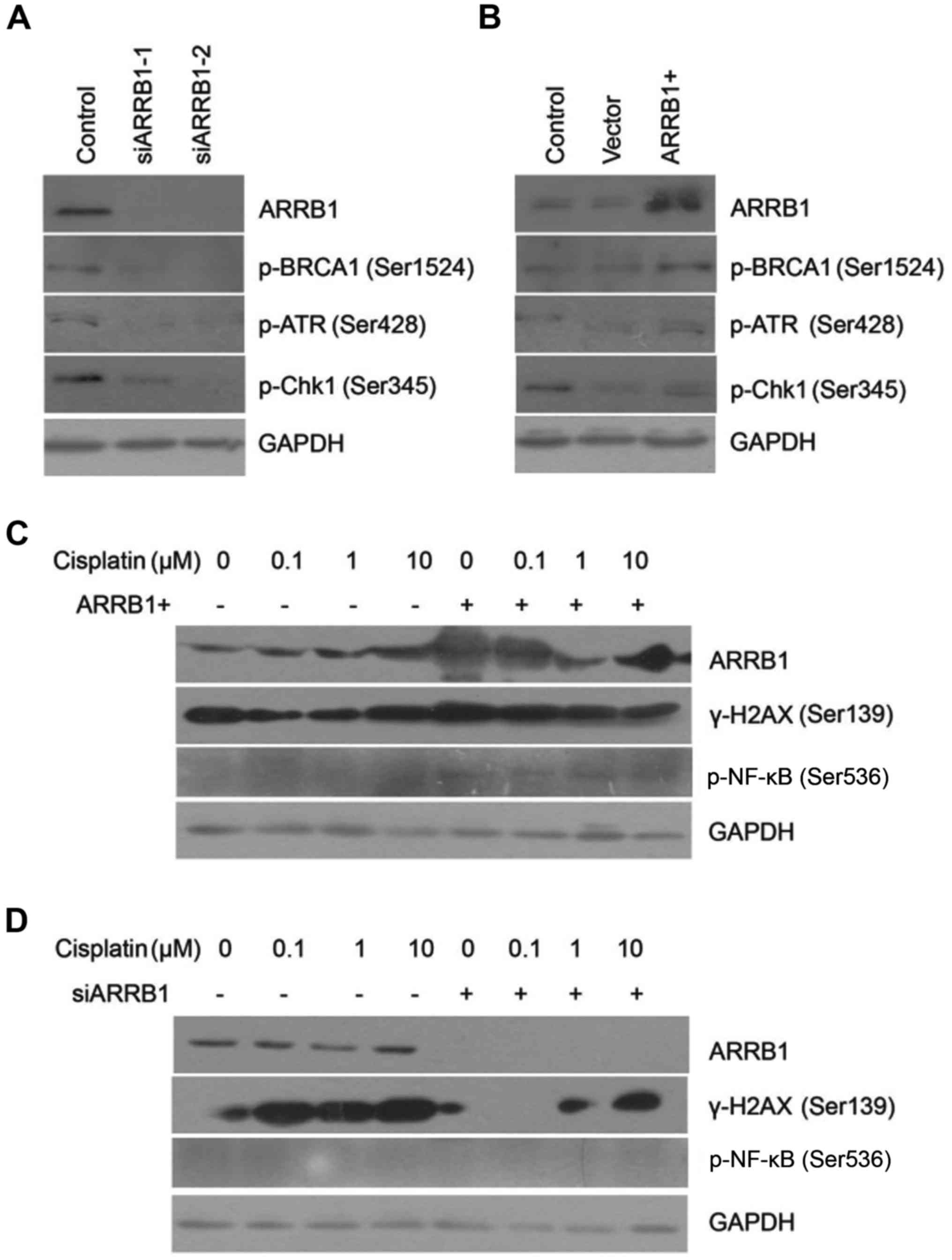
ARRB1 modulates the DDR pathway
activated by DNA-damaging agents
We found that phosphorylated ATR and Chk1 were
downregulated after ARRB1 knockdown. On the contrary, ATR and Chk1
maintained phosphorylation activation with ARRB1 transfection,
which was in accordance with the status of BRCA1 activation
(Fig. 3A and B). Downregulation of
phosphorylated BRCA1, ATR and Chk1 indicated that DDR was
inhibited. However, concurrently, downregulation of phosphorylated
BRCA1 also indicated initiation of DNA repair. We also found that
depletion of ARRB1 inhibited the phosphorylation of H2AX, which is
a biomarker of DSBs. On the contrary, overexpression of ARRB1
increased the phosphorylation of H2AX, which was in accordance with
the status of NF-κB phosphorylation (Fig. 3C and D).
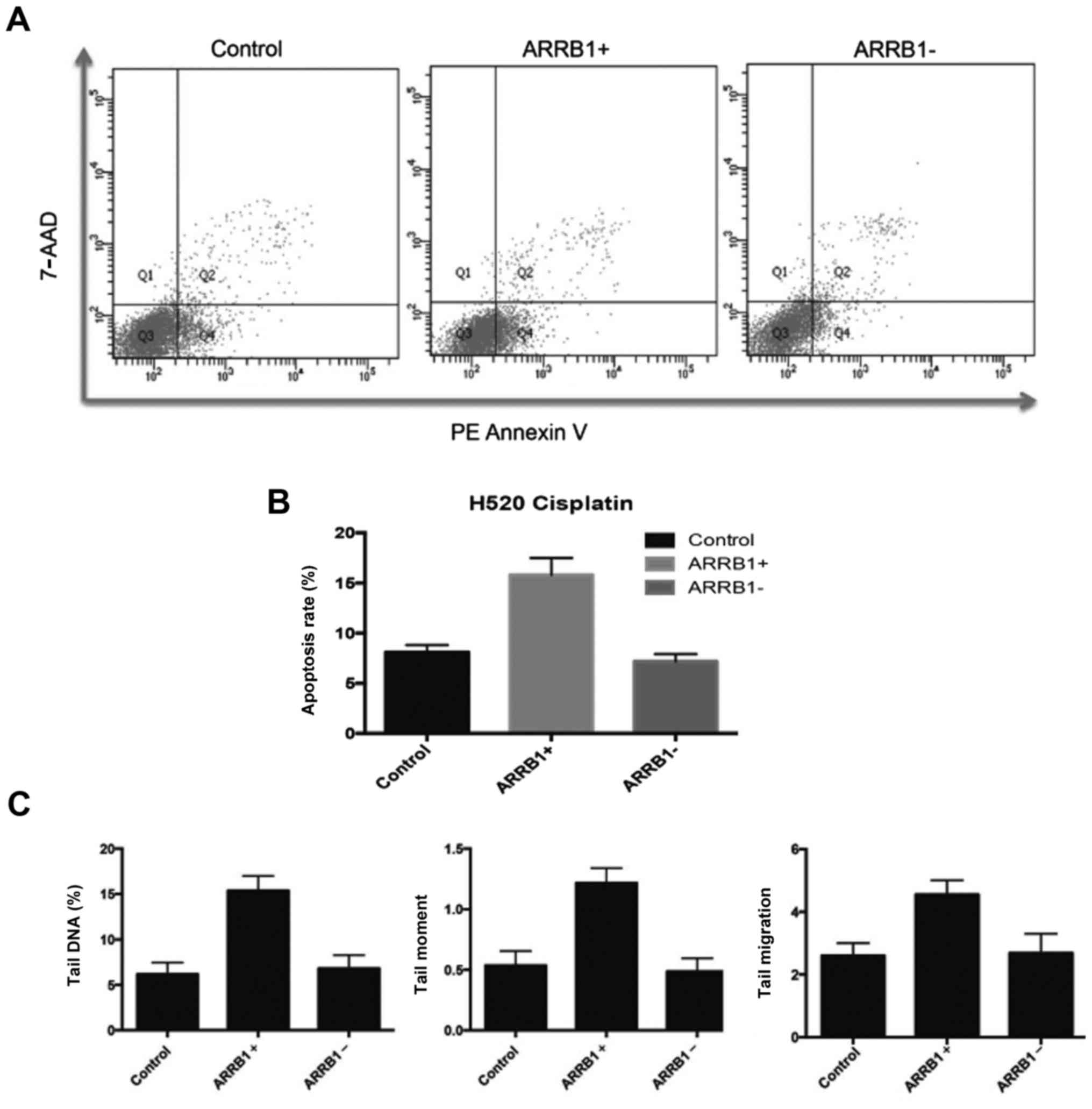
ARRB1 enhances apoptosis and DNA
damage induced by DNA-damaging agents
In order to quantitatively and precisely analyze the
cellular responses and apoptosis, the FCM apoptosis and necrosis
assay (Annexin V and 7-AAD) was performed in the experiments. In
the present study, PE-Annexin V staining was used to detect the
externalization of PS in apoptotic cells, and 7-AAD was applied to
stain necrotic and dead cells, which lose their cell membrane
integrity. Fig. 4A and B, revealed
that there were more apoptotic cells in the ARRB1-overexpressing
cells compared with the control group, and less apoptotic cells in
the ARRB1-knockdown cells. These are in accordance with the DNA
damage results assessed by comet assay (Fig. 4C).
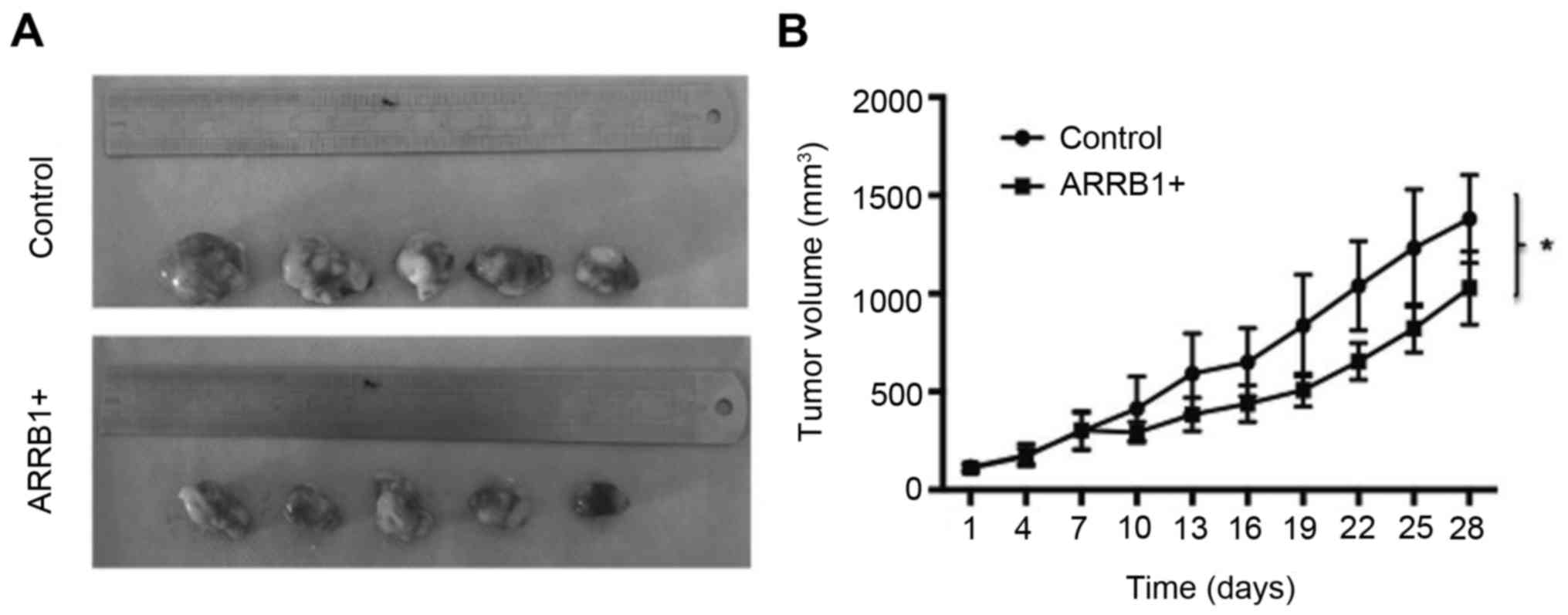
Overexpression of ARRB1 leads to
decreased tumor size in vivo
We examined the effect of ARRB1 overexpression via
treatment with cisplatin on mouse xenograft models of lung cancer
cells (H520). We observed a significant trend of decreased tumor
growth with this treatment (P=0.038) (Fig. 5).
Discussion
In our research, we designed a series of experiments
to explore the possible impacts of ARRB1 expression on chemotherapy
efficacy in NSCLC and relevant mechanisms of regulation of ARRB1 in
DDR induced by DNA-damaging agents. When cells detect DNA damage,
DDR may immediately start. Then, the two key kinases ATR and ATM
are activated. ATR and ATM are vital signaling molecules in the DDR
pathway, which regulate the cell cycle, cell apoptosis and DNA
repair (21). With the ATR-Chk1 and
ATM-Chk2 pathways activated, Chk1 and Chk2 may be phosphorylated.
Then phosphorylated-Chk1 (p-Chk1) phosphorylates the cdc25 family
and other downstream substrates, which induce cell cycle arrest,
DNA repair and other cellular responses. Similarly,
phosphorylated-Chk2 (p-Chk2) phosphorylates p53 and the suppression
of the combination of MDM2 and p53, causes p53 upregulation
(22). Activated p53 enhances the
expression of p21, 14-3-3σ, GADD45 and other signaling molecules,
which ultimately causes cell cycle arrest. Thus, cells may have
plenty of time to complete the DNA repair process. If the DNA
repair process cannot be completed in time, it may lead to cell
apoptosis (23).
Kook et al reported that ARRB1 is cleaved by
caspases, and then binds to truncated BID (tBID) (24), which means that ARRB1 directly
regulates apoptosis. Apart from that, our results revealed that
more ATR and H2AX were phosphorylated compared with the control
group after transfection with the ARRB1 plasmids. As a vital
indicator of DNA DSBs, H2AX is required for checkpoint-mediated
cell cycle arrest and DNA repair in DDR. That is to say, more DNA
damage occurs after ARRB1 levels are increased. This fact also
indicates that ARRB1 may enhance cell apoptosis and promote
chemotherapy efficacy. The potential mechanism may be related with
the interaction between ARRB1 and ATR and H2AX (25). ARRB1 is likely to be involved in the
phosphorylation/activation of H2AX and ATR. In contrast, low
expression of ARRB1 can suppress cell proliferation through
inhibition of cellular pathways such as the NF-κB pathway.
Activation of the NF-κB pathway was reported to be associated with
chemoresistance and anti-apoptosis. Numerous studies have confirmed
that ARRB1 in the nucleus promotes activation of the NF-κB pathway
(26,27). NF-κB is widely used by eukaryotic
cells as a regulator of genes that control cell proliferation and
cell survival. As such, many different types of human tumors have
misregulated NF-κB. A previous study showed that co-expression of
nuclear ARRB1 and NF-κB is associated with cancer progression and
poor prognosis in lung adenocarcinoma (28), which indicates the potential
relationship of ARRB1 and NF-κB.
In conclusion, ARRB1 participates in numerous
cellular pathways and diversely functions under different
circumstances. Our results and data showed that ARRB1 promoted DNA
damage and cell apoptosis and depletion of ARRB1 inhibited DDR
pathway activation. Thus, we surmise that ARRB1 may enhance
chemosensitivity and be beneficial to cancer treatment. However,
various clinical studies have reported that a high level of ARRB1
is related to a poor outcome in cancer treatment (29). Thus, the status of ARRB1 is
associated with different tumor characteristics.
The roles of ARRB1 played in lung cancer treatment
have not been extensively studied, and in particular its clinical
relevance has not been previously addressed. The detailed and
concrete functions of ARRB1 in lung cancer treatment require
further study.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (project 81301728), the Key
Research and Development Program of Shandong Province
(2015GSF118129), the Foundation for Outstanding Young Scientist in
Shandong Province (BS2013YY066), and the Medicine and Health
Science Technology Development Plan of Shandong Province
(2013WS0103).
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2015. CA Cancer J Clin. 65:5–29. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J, Soerjomataram I, Dikshit R, Eser
S, Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: Cancer
incidence and mortality worldwide: sources, methods and major
patterns in GLOBOCAN 2012. Int J Cancer. 136:E359–E386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jin F, Zhu H, Shi F, Kong L and Yu J: A
retrospective analysis of safety and efficacy of weekly
nab-paclitaxel as second-line chemotherapy in elderly patients with
advanced squamous non-small-cell lung carcinoma. Clin Interv Aging.
11:167–173. 2016.PubMed/NCBI
|
|
4
|
Tan HL, Ang YL and Soo RA: Therapeutic
options in advanced squamous cell lung carcinoma. Lung Cancer.
4:75–86. 2015.
|
|
5
|
Koutsoukos K and Mountzios G: Novel
therapies for advanced squamous cell carcinoma of the lung. Future
Oncol. 12:659–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Cufer T and Knez L: Update on systemic
therapy of advanced non-small-cell lung cancer. Expert Rev
Anticancer Ther. 14:1189–1203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rothschild SI: Advanced and metastatic
lung cancer - what is new in the diagnosis and therapy? Praxis.
104:745–750. 2015.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hosoya N and Miyagawa K: Targeting DNA
damage response in cancer therapy. Cancer Sci. 105:370–388. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jackson SP and Bartek J: The DNA-damage
response in human biology and disease. Nature. 461:1071–1078. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ciccia A and Elledge SJ: The DNA damage
response: Making it safe to play with knives. Mol Cell. 40:179–204.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lefkowitz RJ and Whalen EJ:
beta-arrestins: Traffic cops of cell signaling. Curr Opin Cell
Biol. 16:162–168. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lefkowitz RJ and Shenoy SK: Transduction
of receptor signals by beta-arrestins. Science. 308:512–517. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sobolesky PM and Moussa O: The role of
β-arrestins in cancer. Prog Mol Biol Transl Sci. 118:395–411. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ma H, Wang L, Zhang T, Shen H and Du J:
Loss of β-arrestin1 expression predicts unfavorable prognosis for
non-small cell lung cancer patients. Tumour Biol. 37:1341–1347.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hara MR, Kovacs JJ, Whalen EJ, Rajagopal
S, Strachan RT, Grant W, Towers AJ, Williams B, Lam CM, Xiao K, et
al: A stress response pathway regulates DNA damage through
β2-adrenoreceptors and β-arrestin-1. Nature.
477:349–353. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hara MR, Sachs BD, Caron MG and Lefkowitz
RJ: Pharmacological blockade of a β2AR-β-arrestin-1
signaling cascade prevents the accumulation of DNA damage in a
behavioral stress model. Cell Cycle. 12:219–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Engeland M, Nieland LJ, Ramaekers FC,
Schutte B and Reutelingsperger CP: Annexin V-affinity assay: A
review on an apoptosis detection system based on phosphatidylserine
exposure. Cytometry. 31:1–9. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zembruski NC, Stache V, Haefeli WE and
Weiss J: 7-Aminoactinomycin D for apoptosis staining in flow
cytometry. Anal Biochem. 429:79–81. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patra B, Peng CC, Liao WH, Lee CH and Tung
YC: Drug testing and flow cytometry analysis on a large number of
uniform sized tumor spheroids using a microfluidic device. Sci Rep.
6:210612016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Fairbairn DW, Olive PL and O'Neill KL: The
comet assay: A comprehensive review. Mutat Res. 339:37–59. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maréchal A and Zou L: DNA damage sensing
by the ATM and ATR kinases. Cold Spring Harb Perspect Biol.
5:a0127162013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cheng Q and Chen J: Mechanism of p53
stabilization by ATM after DNA damage. Cell Cycle. 9:472–478. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhou BB and Elledge SJ: The DNA damage
response: Putting checkpoints in perspective. Nature. 408:433–439.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kook S, Zhan X, Cleghorn WM, Benovic JL,
Gurevich VV and Gurevich EV: Caspase-cleaved arrestin-2 and BID
cooperatively facilitate cytochromec release and cell death. Cell
Death Differ. 21:172–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao K, McClatchy DB, Shukla AK, Zhao Y,
Chen M, Shenoy SK, Yates JR III and Lefkowitz RJ: Functional
specialization of β-arrestin interactions revealed by proteomic
analysis. Proc Natl Acad Sci USA. 104:12011–12016. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bernal-Mizrachi L, Lovly CM and Ratner L:
The role of NF-κB-1 and NF-κB-2-mediated resistance to apoptosis in
lymphomas. Proc Natl Acad Sci USA. 103:9220–9225. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cianfrocca R, Tocci P, Semprucci E,
Spinella F, Di Castro V, Bagnato A and Rosanò L: β-Arrestin 1 is
required for endothelin-1-induced NF-κB activation in ovarian
cancer cells. Life Sci. 118:179–184. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu J, Wang L, Zhang T, Shen H, Dong W, Ni
Y and Du J: Co-expression of β-arrestin1 and NF-кB is associated
with cancer progression and poor prognosis in lung adenocarcinoma.
Tumour Biol. 36:6551–6558. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lundgren K, Tobin NP, Lehn S, Stål O,
Rydén L, Jirström K and Landberg G: Stromal expression of
β-arrestin-1 predicts clinical outcome and tamoxifen response in
breast cancer. J Mol Diagn. 13:340–351. 2011. View Article : Google Scholar : PubMed/NCBI
|