Introduction
Lung cancer is one of the most common types of
cancer and the leading cause of cancer-related deaths worldwide
(1,2). Lung cancer is classified into two
groups. One is small cell lung cancer and the other is non-small
cell lung cancer (NSCLC). NSCLC includes squamous cell carcinoma,
adenocarcinoma and large cell carcinoma, which account for 80–85%
of all lung cancer cases (1).
Although there has been a significant development in the therapies
of NSCLC for decades, the 5-year survival rate is still less than
15% (3). Therefore, new therapeutic
targets are required to improve survival and quality of life.
Death-associated kinase 3 (DAPK3) is a member of the
DAPK family. The DAPK family also includes DAPK1 and DAPK2. These
proteins contain a similar N-terminal kinase domain and play a role
in the induction of cell death (4).
Moreover, DAPK3 was found to mediate DAPK1-induced apoptosis in HEK
293T cells (5), and to promote
starvation-induced autophagy, an alternate type of programmed cell
death, through the regulation of Atg9-mediated autophagosome
formation (6). Furthermore, the
expression of DAPK3 was found to be decreased in several squamous
cell carcinomas (7,8). In view of this evidence, DAPK3 has
been regarded as a tumor suppressor.
In contrast, a recent study showed that DAPK3
promotes cancer cell proliferation rather than the induction of
apoptosis in various cancer cell lines. In prostate cancer cells,
DAPK3 promoted cell proliferation through the activation of the
androgen receptor (9). In gastric
cancer cells, overexpression of DAPK3 propagated cell
proliferation, migration and invasion via activation of Akt and
NF-κB signals and promotion of stemness (8). Furthermore, knockdown of DAPK3
prevented cell proliferation in colon cancer cells through the
inhibition of Wnt/β-catenin signals (10). These studies suggest the possibility
of DAPK3 as a novel therapeutic target of cancer.
In lung cancer tissues, the DAPK3 gene was
heterozygously mutated at a frequency of 3.2% (11). Nevertheless, it remains to be
clarified how the wild-type DAPK3 gene controls NSCLC pathogenesis
through a signaling pathway. We therefore examined whether
knockdown of the DAPK3 gene affects NSCLC progression via cellular
signaling. To the very best of our knowledge, in the present study
we demonstrated for the first time that the DAPK3 gene regulates
cell proliferation, migration and invasion through ERK/c-Myc
signaling in A549 cells. It was also established that the DAPK3
gene plays a role in the tumor growth of A549 cells in
vivo.
Materials and methods
Materials
Antibody sources were as follows: phospho-ERK,
phospho-Akt, total-ERK, total-JNK, total-Akt, total-c-Myc (Cell
Signaling Technology, Inc., Danvers, MA, USA); phospho-c-Myc (Bio
Academia, Inc., Osaka, Japan); phospho-JNK (Promega, Madison, WI,
USA); total-VCP (GeneTex, Inc., Irvine, CA, USA); total-cyclin D1
(Bioss, Inc., Woburn, MA, USA). Total-DAPK3 was provided by Dr
Tachibana (Osaka City University, Japan).
Cell culture
A549 (human lung adenocarcinoma) cells were kindly
provided by Dr B. Shimizu (Yamaguchi University, Ube, Japan). A549
cells were cultured in Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% fetal bovine serum (FBS; Invitrogen,
Carlsbad, CA, USA).
Short hairpin (sh)RNA and lentivirus
production
Human DAPK3 small hairpin RNA (shRNA) and shNon
target (shNT) expressing cells were produced as previously
described (12). Briefly, shRNA
complementary DNA strands (shNT, 5′-CAACAAGATGAGAGCACCA-3′;
shDAPK3, 5′-GACGGACGTGGTCCTCATC-3′) were annealed and ligated into
MluI/ClaI sites of the pLV-mC vector (Takara, Tokyo,
Japan). To produce lentiviruses, 1 µg of pLV-mC, 0.77 µg of a
packaging plasmid (psPAX2) and 0.43 µg of a protein-coated plasmid
expressing vesicular stomatitis virus G protein (pMD2.G) were
transfected into Lenti-X 293T cells using 2.5 µl of PEI Max
(Polysciences, Inc., Warrington, PA, USA) dissolved in 333 µl of
Opti-MEM (Invitrogen). After 48 h, viral supernatants were
collected and filtered. A549 cells were treated with them for 8
h.
Cell proliferation analysis
Cells (1×104) were seeded on a 12-well
culture plate. Cell proliferation was examined by cell counting
using Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) as
previously described (13,14). The absorbance of the medium at 450
nm was read on a standard plate reader (Beckman Coulter DU-800;
Beckman Coulter, Inc., Brea, CA, USA).
Flow cytometry
After A549 cells expressing DAPK3 shRNA or shNT were
seeded at a density of 2×105 cells on a 6-well plate and
incubated for 24 h, they were trypsinized and collected into a 1.5
ml tube. After the cells were fixed with 70% ethanol for 20 min at
−20̊C, they were washed with HEPES-buffered saline (HBS) and
treated with RNase Staining Solution (200 µg/ml; Cell Signaling)
for 30 min. Next, they were washed with HBS and stained with
propidium iodide (50 µg/ml; Cell Signaling) for 30 min at room
temperature in the dark. The cell cycle data were obtained using BD
Accuri™ C6 (BD Biosciences, San Jose, CA, USA). In addition, the
cell population data were analyzed using BD Accuri software.
Western blotting
Western blotting was performed as previously
described (14). Protein lysates
were obtained by homogenizing cells with Triton-based lysis buffer
[50 mM Tris-HCl (pH 8.0), 5 mM EDTA, 5 mM EGTA, 1% Triton X-100, 1
mM Na3VO4, 20 mM sodium pyrophosphate and
Roche Complete protease inhibitor mixture]. Loading proteins (20–40
µg) were separated by SDS-PAGE (10%) and transferred to a
nitrocellulose membrane (Wako, Osaka, Japan). After being blocked
with 3% bovine serum albumin or 0.5% skim milk, the membranes were
incubated with primary antibodies at 4̊C overnight. Then, the
membranes were treated with secondary antibodies (1:10,000
dilution, 1 h) and ECL Pro (PerkinElmer, Freiburg, Germany). The
results were visualized using LAS-3000 (FujiFilm, Tokyo, Japan) and
quantified using ImageJ densitometric analysis software (National
Institutes of Health, Bethesda, MD, USA).
Boyden chamber and invasion
assays
The Boyden chamber assay was performed as previously
described (12,15). After membranes were coated with 2%
gelatin (for the Boyden chamber assay) or 2% Matrigel (for the
invasion assay), A549 cells expressing DAPK3 shRNA or shNT were
seeded at a density of 1×104 cells in the upper chamber.
To migrate the cells, 600 µl of medium with 10% FBS was added to
the lower chamber. The membranes to which the cells migrated or
invaded were fixed with methanol for 15 min and stained with Giemsa
solution. The numbers of migrated and invaded cells through the
membranes were counted under a light microscope and averaged.
Colony formation assay
The colony formation assay was performed as
previously described (12). Cells
(1×103) were seeded on a 60-mm dish and cultured for 1
week. After the cells were fixed with 99.5% ethanol, colonies were
stained with Giemsa. The number of surviving colonies was
counted.
Soft-agar colony formation assay
For the soft-agar colony formation assay,
2×103 cells were seeded in 1.5 ml top agar (DMEM
containing 10% FBS, 2.8% NaHCO3, 1X
antibiotic/antimicotic and 0.36% agarose) on a 2.5 ml bottom layer
of solidified bottom agar (DMEM containing 10% FBS, 2.8%
NaHCO3, 1X antibiotic/antimicotic and 0.75% agarose) in
a 6-well culture plate and cultured for 3 weeks. Colonies were
stained with crystal violet solution and the number of colonies was
counted.
Mouse xenograft assay
NOD.CB17 mouse Prkdcscid/J mice
were obtained from Japan SLC (Hamamatsu, Japan). All studies
involving mice were conducted according to the Guide to Animal Use
and Care of the Yamaguchi University and approved by the Ethics
Committee. After A549 cells expressing shDAPK3 or shNT were
subcutaneously injected into the flanks of NOD/SCID mice (5–6
weeks, 1×106 cells each), the tumor volumes were
measured every week for 6 weeks and estimated using the following
formula: Tumor volume = 0.5 × (length × width2).
Subsequently, the mice were euthanized, the tumor tissues were
isolated and the tumor weights were assessed.
Statistical analysis
Data are shown as the means ± SEM. Statistical
evaluations were performed using Student's t-test between two
groups. Values of P<0.05 were considered statistically
significant.
Results
Effects of DAPK3 gene knockdown on
proliferation of A549 cells
Since several studies have shown that the DAPK3 gene
is mutated in several types of cancer cells (11), we investigated whether DAPK3 is
mutated in A549 cells. No DAPK3 mutation was observed in the A549
cells (data not shown). To clarify the role of the wild-type DAPK3
gene in NSCLC, we generated DAPK3 shRNA-expressing A549 cells. We
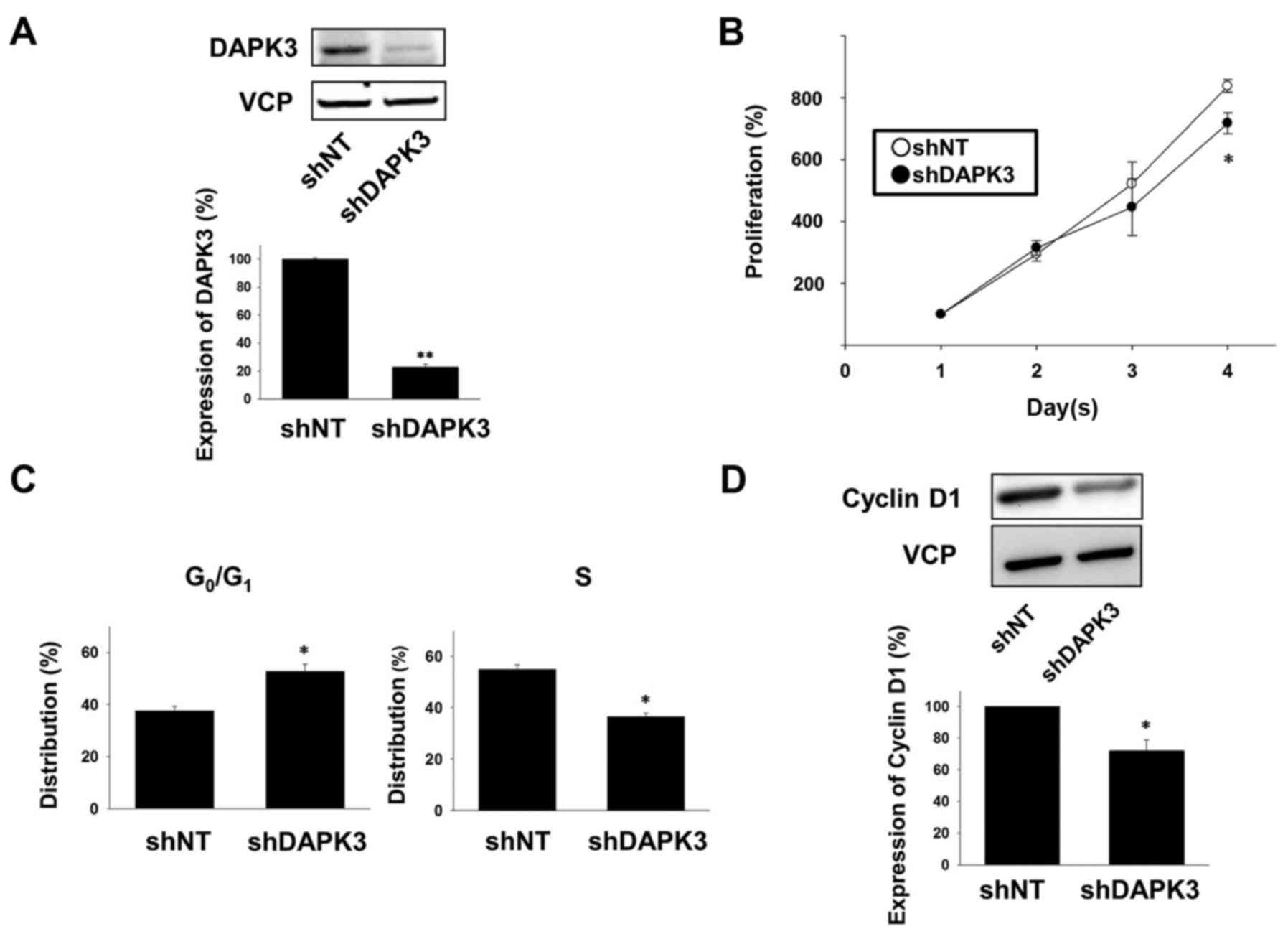
confirmed that the expression of the DAPK3 protein was
significantly decreased in the shDAPK3-expressing A549 cells
compared with the shNT-expressing cells (Fig. 1A). In order to examine the effects
of DAPK3 gene knockdown on A549 cell proliferation, we used a cell
counting assay. The cell proliferation at day 4 was significantly
lower in the DAPK3-knocked down cells compared with the
proliferation of the shNT-expressing cells (Fig. 1B).
Effects of DAPK3 gene knockdown on
cell cycle arrest in A549 cells
We next performed cell cycle analysis by flow
cytometry. In the DAPK3-knockdown A549 cells, the number of cells
in the G0/G1 phase was significantly
increased and the number of cells in the S phase was significantly
decreased (Fig. 1C). We further
examined the effects of DAPK3 gene knockdown on the expression of a
cell cycle regulatory protein, cyclin D1, which regulates
progression of the G1 phase (16). Expression of cyclin D1 was
significantly decreased in the DAPK3-knockdown A549 cells (Fig. 1D).
Effects of DAPK3 gene knockdown on
proliferation-related signals in A549 cells
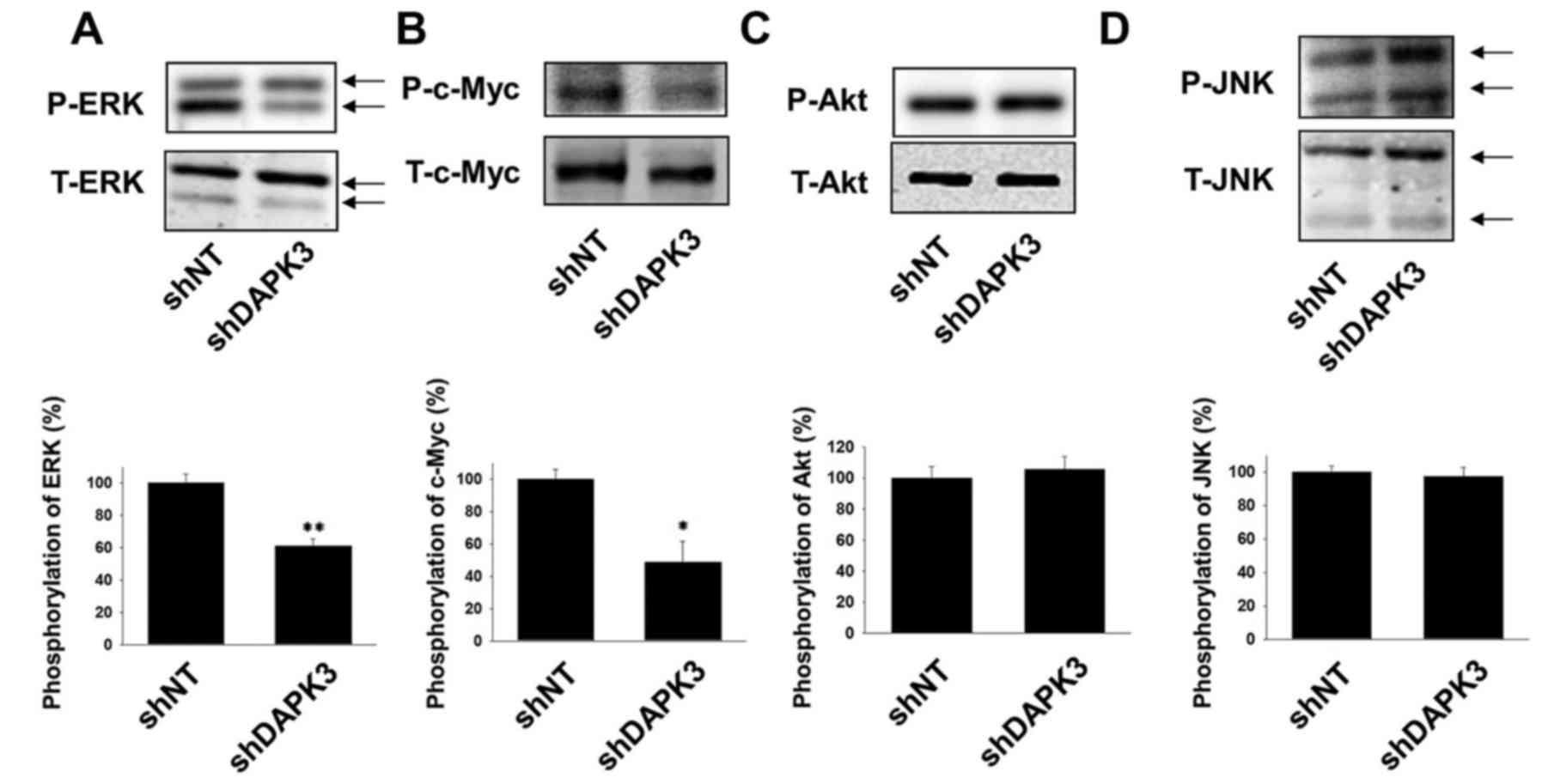
We next examined whether DAPK3 mediates the
proliferation-related signals in A549 cells. Phosphorylation of ERK
and c-Myc was significantly inhibited by DAPK3 gene knockdown
(Fig. 2A and B). In contrast, DAPK3
gene knockdown had no effect on the phosphorylation of Akt and JNK
(Fig. 2C and D).
Effects of DAPK3 gene knockdown on
migration, invasion and colony formation in A549 cells
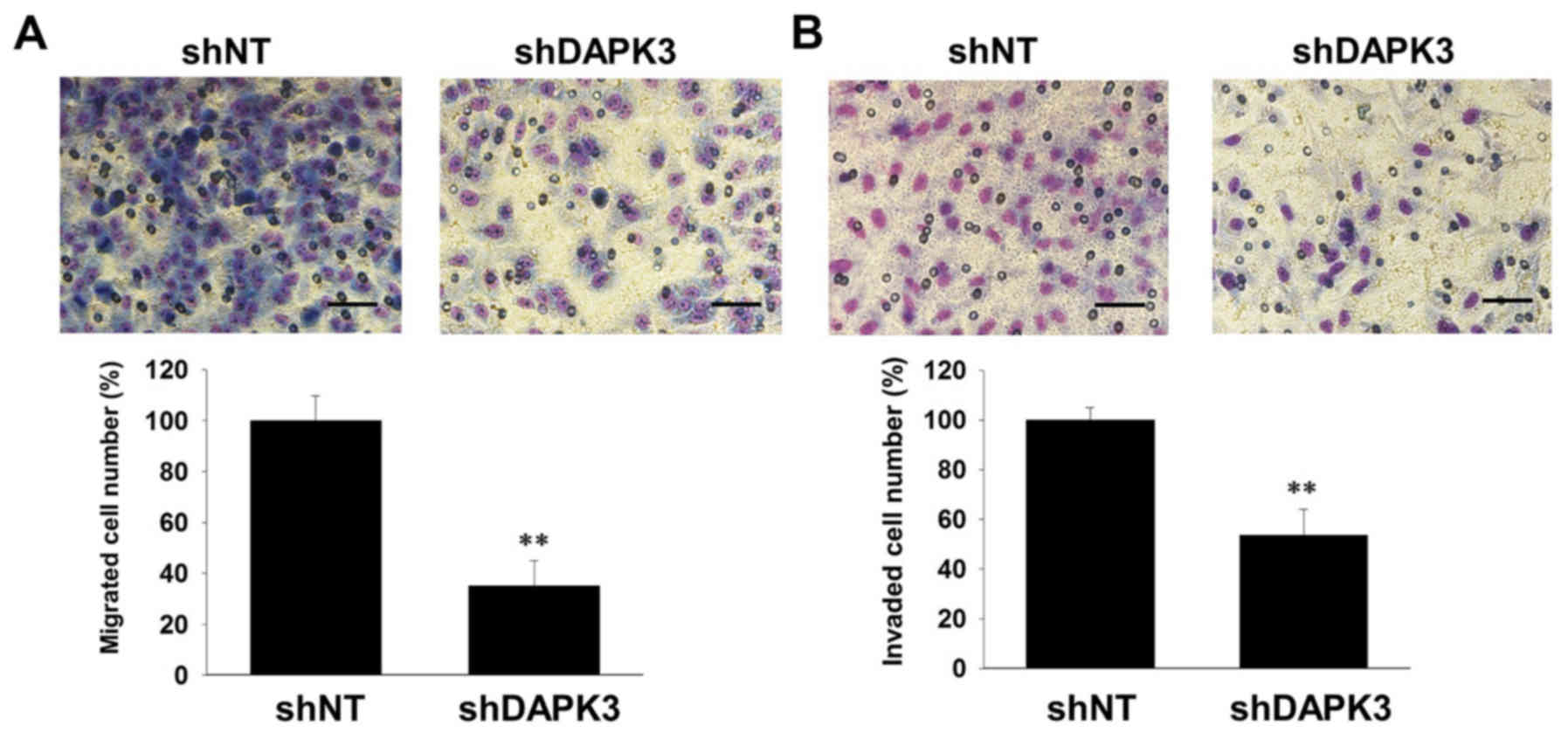
Cell migration and invasion are essential for tumor
metastasis (17). We thus examined
the effects of DAPK3 gene knockdown on the migration and invasion
of A549 cells. DAPK3 gene knockdown significantly inhibited cell
migration (Fig. 3A) and invasion
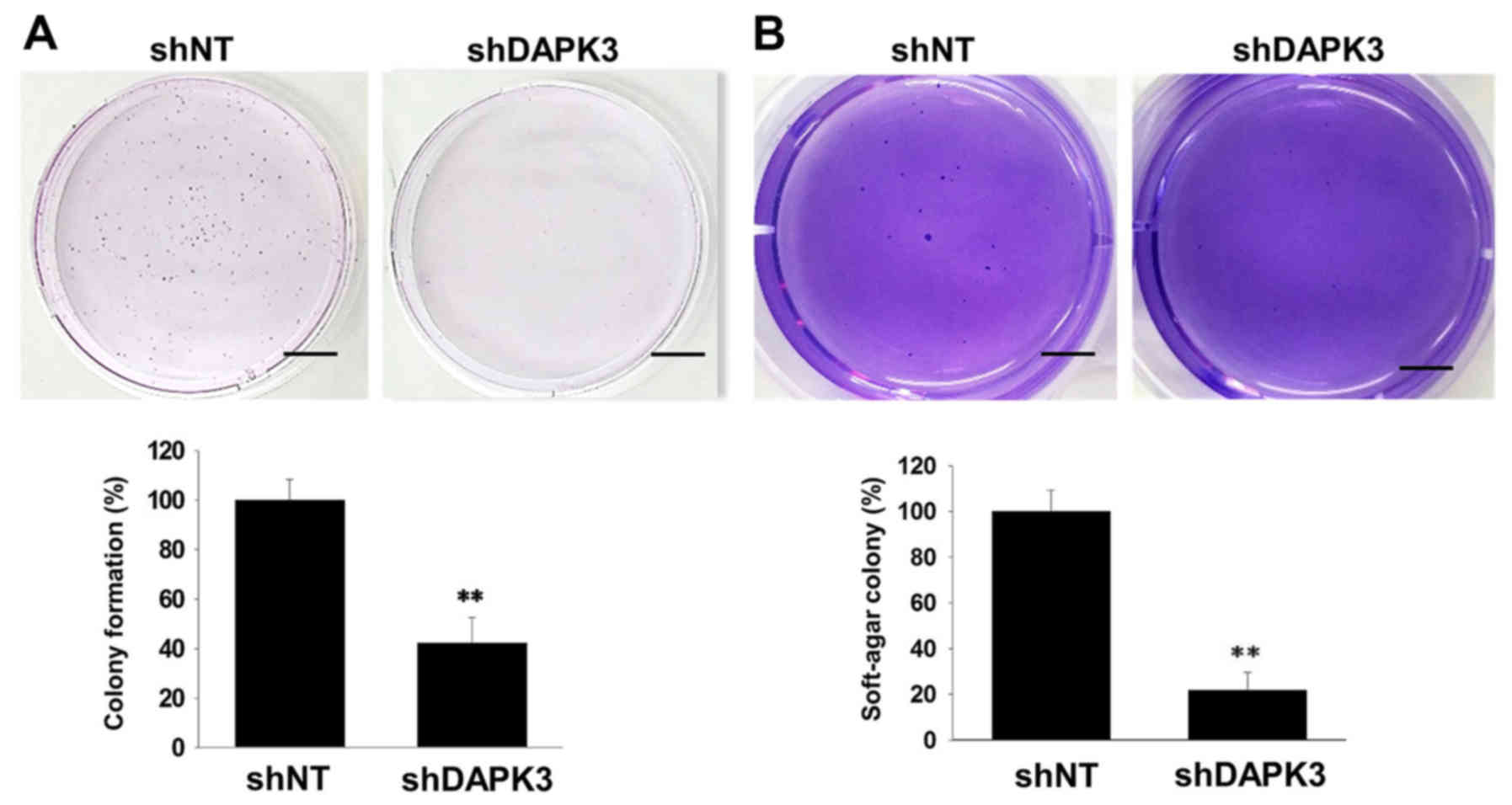
(Fig. 3B). Since cell colonization
is also an important step in tumor metastasis (18), we examined the effects of DAPK3 gene
knockdown on colony formation in A549 cells. Both
anchorage-dependent colony formation (Fig. 4A) and anchorage-independent colony
formation in soft-agar (Fig. 4B)
were decreased by DAPK3 gene knockdown.
Effects of DAPK3 gene knockdown on
tumor growth in a xenograft mouse model
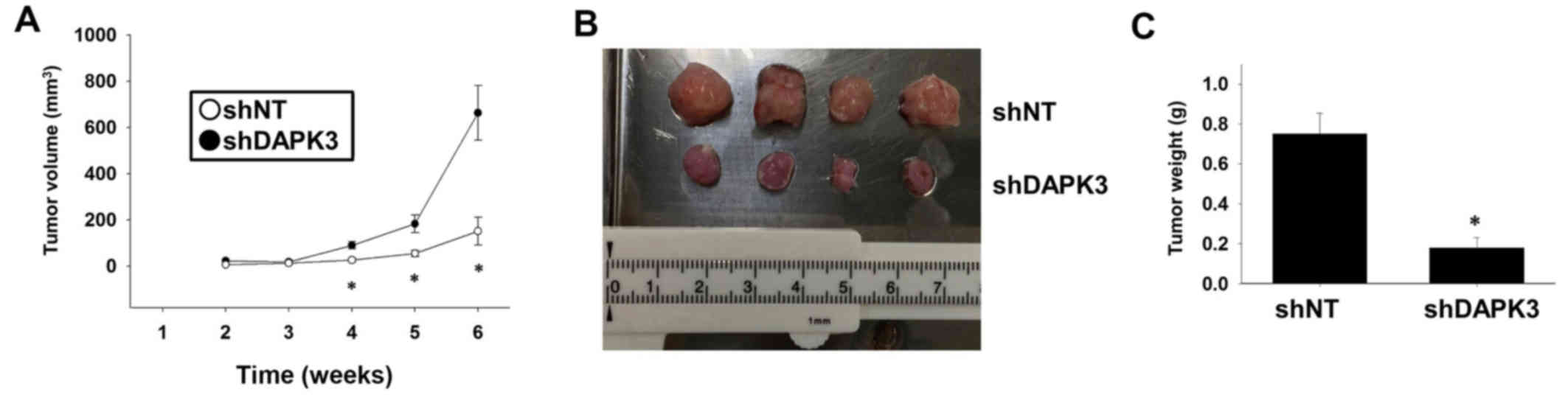
We finally examined whether DAPK3 gene knockdown
inhibits tumor growth in vivo. Injection of DAPK3
shRNA-expressing A549 cells into immunodeficient mice significantly
slowed down tumor growth (Fig. 5A and
B) and decreased tumor weight (Fig.
5C).
Discussion
In the present study, we examined whether DAPK3
mediates the tumor progression of A549 cells. The major findings of
the present study are that the knockdown of the DAPK3 gene
inhibited proliferation, cell cycle and activation of ERK and c-Myc
(Figs. 1 and 2). It was also found that the knockdown of
the DAPK3 gene inhibited migration and invasion (Fig. 3A and B). In addition, we showed that
the knockdown of the DAPK3 gene inhibited colony formation
(Fig. 4A and B). It was also shown
that the knockdown of the DAPK3 gene slowed down tumor growth in a
mouse xenograft model (Fig. 5).
Collectively, our results indicate that DAPK3 mediates the tumor
progression of A549 cells via cell proliferation, migration,
invasion and colony formation through the activation of ERK/c-Myc
signals.
In the present study, we showed that DAPK3 gene
knockdown inhibited cell proliferation in A549 cells (Fig. 1). Since several studies have shown
that DAPK3 has a pro-apoptotic effect on cancer cells (19), DAPK3 has been regarded as a tumor
suppressor in various tumors. However, accumulating evidence has
revealed that DAPK3 alternatively promotes cancer cell
proliferation in some cancer cell lines. In human colon carcinoma
cells, knockdown of the DAPK3 gene inhibited proliferation through
the inhibition of Wnt/β-catenin signaling (10). In gastric cancer cell lines,
overexpression of DAPK3 promoted proliferation through the
upregulation of Akt and NF-κB signaling (8). The results of the present study concur
with these studies. In the same study, the expression of DAPK3 in
primary gastric tumor tissues was lower compared with non-tumor
tissues. However, the expression of DAPK3 increased in metastatic
tissues compared with primary tumors. These clinical data suggest
that DAPK3 may change into a tumor promoter from a tumor suppressor
during the course of gastric tumor progression. Therefore, we
speculate that DAPK3 also acquires a role as a metastatic promoter
in the progression of NSCLC. Further clinical study on the
correlation of DAPK3 expression and lung cancer metastasis may be
required.
In NSCLC, KRAS mutations were observed at a
frequency of ~30% (20). KRAS
mutations were also identified in A549 cells which are used in the
present study (21). Mutated KRAS
increases its activity and promotes cell proliferation through the
interaction with Raf, which causes activation of MEK/ERK signaling
(22). In the present study, we
demonstrated that DAPK3 gene knockdown inhibited ERK activity in
A549 cells (Fig. 2). Nevertheless,
it was unclear how DAPK3 controls ERK activity. In several cell
lines, DAPK3 is regulated by DAPK1 (23) and a recent study showed that Raf
interacts with DAPK1 in mitochondria, whose interaction increases
reactive oxygen species (ROS) production (24). In addition, it was reported that
inhibition of ROS prevented ERK signaling in A549 cells (25). In the previous study, we showed that
DAPK3 promoted TNF-α-induced activation of JNK, p38 and Akt through
ROS generation (26). Considering
these data, DAPK3 may mediate the interaction between Raf and
DAPK1, which promotes ROS production and ERK activity in
KRAS-mutated NSCLC cells.
Metastasis is responsible for the low 5-year
survival rate of NSCLC (27).
Migrated cancer cells gradually invade into blood or lymphatic
vessels, which extravasate into distant tissue. In the previous
study, we demonstrated that DAPK3 mediates platelet-derived growth
factor BB-induced migration of vascular smooth muscle cells through
p38/HSP27 signals (15). Another
study showed that DAPK3 promoted migration and invasion of gastric
cancer cell lines via activation of Akt and NF-κB signals (8). In the present study, we found that
DAPK3 gene knockdown inhibited migration and invasion of A549 cells
(Fig. 3A and B). In addition, DAPK3
gene knockdown inhibited phosphorylation of ERK (Fig. 2A) but not p38 and NF-κB (data not
shown) in A549 cells. Since ERK signaling regulates not only cell
proliferation, but also migration and invasion of A549 cells
(25), we hypothesize that DAPK3
mediates migration and invasion through ERK activity in the A549
cells. These data imply that DAPK3 plays a significant role in the
metastasis of NSCLC.
Cancer stem cells have self-renewal potential and
are able to form new tumors. A recent study showed that
overexpression of DAPK3 increased colony formation of gastric
cancer cells through the upregulation of stemness-related gene
expression (8). In the present
study, we showed that knockdown of the DAPK3 gene inhibited fossil
formation and soft-agar colony formation (Fig. 4A and B). Although we did not
investigate whether DAPK3 gene knockdown affects the expression of
stemness-related genes in A549 cells, these results imply that
DAPK3 regulates stemness of lung cancer cells, which leads to the
formation of new tumors.
Although a previous study showed that overexpression
of mutated DAPK3 promotes proliferation of lung cancer cells
(11), it has not been determined
whether DAPK3 mediates tumor growth in vivo. To the very
best of our knowledge, in the present study we demonstrated for the
first time that tumor growth of A549 cells was slowed down by DAPK3
gene knockdown in a mouse xenograft model (Fig. 5). While investigating whether DAPK3
is mutated in A549 cells, we observed no DAPK3 mutation (data not
shown). Therefore, we speculated that wild-type DAPK3 functions as
an oncogene in the course of tumor growth in NSCLC. Further
clinical investigations may be needed to confirm this theory.
In summary, to the very best of our knowledge, in
the present study we demonstrated for the first time that DAPK3
controls proliferation, migration and invasion of A549 cells. It
was also suggested that DAPK3 may be responsible for tumor growth
and metastasis. Further studies on DAPK3 may contribute to the
development of new therapies and knockdown of the DAPK3 gene may
become a new strategy used in non-small lung cancer.
Acknowledgements
We thank Dr Tachibana and Dr Shimizu for kindly
providing the DAPK3 antibody and A549 cells, respectively.
References
|
1
|
Jemal A, Siegel R, Ward E, Hao Y, Xu J and
Thun MJ: Cancer statistics, 2009. CA Cancer J Clin. 59:225–249.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Torre LA, Bray F, Siegel RL, Ferlay J,
Lortet-Tieulent J and Jemal A: Global cancer statistics, 2012. CA
Cancer J Clin. 65:87–108. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bialik S and Kimchi A: The
death-associated protein kinases: Structure, function, and beyond.
Annu Rev Biochem. 75:189–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shani G, Marash L, Gozuacik D, Bialik S,
Teitelbaum L, Shohat G and Kimchi A: Death-associated protein
kinase phosphorylates ZIP kinase, forming a unique kinase hierarchy
to activate its cell death functions. Mol Cell Biol. 24:8611–8626.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tang HW, Wang YB, Wang SL, Wu MH, Lin SY
and Chen GC: Atg1-mediated myosin II activation regulates
autophagosome formation during starvation-induced autophagy. EMBO
J. 30:636–651. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mallipeddi R, Wessagowit V, South AP,
Robson AM, Orchard GE, Eady RA and McGrath JA: Reduced expression
of insulin-like growth factor-binding protein-3 (IGFBP-3) in
Squamous cell carcinoma complicating recessive dystrophic
epidermolysis bullosa. J Invest Dermatol. 122:1302–1309. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li J, Deng Z, Wang Z, Wang D, Zhang L, Su
Q, Lai Y, Li B, Luo Z, Chen X, et al: Zipper-interacting protein
kinase promotes epithelial-mesenchymal transition, invasion and
metastasis through AKT and NF-kB signaling and is associated with
metastasis and poor prognosis in gastric cancer patients.
Oncotarget. 6:8323–8338. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leister P, Felten A, Chasan AI and
Scheidtmann KH: ZIP kinase plays a crucial role in androgen
receptor-mediated transcription. Oncogene. 27:3292–3300. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Togi S, Ikeda O, Kamitani S, Nakasuji M,
Sekine Y, Muromoto R, Nanbo A, Oritani K, Kawai T, Akira S, et al:
Zipper-interacting protein kinase (ZIPK) modulates canonical
Wnt/beta-catenin signaling through interaction with Nemo-like
kinase and T-cell factor 4 (NLK/TCF4). J Biol Chem.
286:19170–19177. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Brognard J, Zhang YW, Puto LA and Hunter
T: Cancer-associated loss-of-function mutations implicate DAPK3 as
a tumor-suppressing kinase. Cancer Res. 71:3152–3161. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Enjoji S, Yabe R, Fujiwara N, Tsuji S,
Vitek MP, Mizuno T, Nakagawa T, Usui T, Ohama T and Sato K: The
therapeutic effects of SET/I2PP2A inhibitors on canine melanoma. J
Vet Med Sci. 77:1451–1456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Usui T, Nijima R, Sakatsume T, Otani K,
Kameshima S, Okada M and Yamawaki H: Eukaryotic elongation factor 2
kinase controls proliferation and migration of vascular smooth
muscle cells. Acta Physiol. 213:472–480. 2015. View Article : Google Scholar
|
|
14
|
Yabe R, Miura A, Usui T, Mudrak I, Ogris
E, Ohama T and Sato K: Protein phosphatase methyl-esterase PME-1
protects protein phosphatase 2A from ubiquitin/proteasome
degradation. PLoS One. 10:e01452262015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Usui T, Sakatsume T, Nijima R, Otani K,
Kazama K, Morita T, Kameshima S, Okada M and Yamawaki H:
Death-associated protein kinase 3 mediates vascular structural
remodelling via stimulating smooth muscle cell proliferation and
migration. Clin Sci. 127:539–548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hunter T and Pines J: Cyclins and cancer.
II: Cyclin D and CDK inhibitors come of age. Cell. 79:573–582.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bravo-Cordero JJ, Hodgson L and Condeelis
J: Directed cell invasion and migration during metastasis. Curr
Opin Cell Biol. 24:277–283. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Scheel C and Weinberg RA: Cancer stem
cells and epithelial-mesenchymal transition: Concepts and molecular
links. Semin Cancer Biol. 22:396–403. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kawai T, Matsumoto M, Takeda K, Sanjo H
and Akira S: ZIP kinase, a novel serine/threonine kinase which
mediates apoptosis. Mol Cell Biol. 18:1642–1651. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guin S, Ru Y, Wynes MW, Mishra R, Lu X,
Owens C, Barn AE, Vasu VT, Hirsch FR, Kern JA, et al: Contributions
of KRAS and RAL in non-small-cell lung cancer growth and
progression. J Thorac Oncol. 8:1492–1501. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oneyama C, Ikeda J, Okuzaki D, Suzuki K,
Kanou T, Shintani Y, Morii E, Okumura M, Aozasa K and Okada M:
MicroRNA-mediated downregulation of mTOR/FGFR3 controls tumor
growth induced by Src-related oncogenic pathways. Oncogene.
30:3489–3501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
An S, Yang Y, Ward R, Liu Y, Guo XX and Xu
TR: A-Raf: A new star of the family of raf kinases. Crit Rev
Biochem Mol Biol. 50:520–531. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Endo A, Surks HK, Mochizuki S, Mochizuki N
and Mendelsohn ME: Identification and characterization of
zipper-interacting protein kinase as the unique vascular smooth
muscle myosin phosphatase-associated kinase. J Biol Chem.
279:42055–42061. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tsai YT, Chuang MJ, Tang SH, Wu ST, Chen
YC, Sun GH, Hsiao PW, Huang SM, Lee HJ, Yu CP, et al: Novel cancer
therapeutics with allosteric modulation of the mitochondrial
C-Raf-DAPK complex by Raf inhibitor combination therapy. Cancer
Res. 75:3568–3582. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ku MJ, Kim JH, Lee J, Cho JY, Chun T and
Lee SY: Maclurin suppresses migration and invasion of human
non-small-cell lung cancer cells via anti-oxidative activity and
inhibition of the Src/FAK-ERK-β-catenin pathway. Mol Cell Biochem.
402:243–252. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Usui T, Okada M, Hara Y and Yamawaki H:
Death-associated protein kinase 3 mediates vascular inflammation
and development of hypertension in spontaneously hypertensive rats.
Hypertension. 60:1031–1039. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marcus AI and Zhou W: LKB1 regulated
pathways in lung cancer invasion and metastasis. J Thorac Oncol.
5:1883–1886. 2010. View Article : Google Scholar : PubMed/NCBI
|