Introduction
Prostate cancer (PCa) is the most common cancer type in males and the second-leading cause of cancer-related mortality in the United States, with an estimated 248,530 new cases and 34,130 cancer-related deaths in 2018 (1). Between 20 and 40% of clinically localized PCa patients could have a recurrence following the initial therapy (radiotherapy or prostatectomy) (2) and will then have to receive androgen deprivation therapy (ADT). Unfortunately, castration-resistant PCa (CRPC) would develop in all patients following 12–18 months of ADT, with disease progression (3). In pathology, androgen receptor (AR) gene amplification was identified in 30 to 50% of CRPC patients, resulting in the overexpression of the AR protein (4,5). Clinical research has revealed that AR amplification is clearly related to poor prognosis, with lower overall survival and progression-free survival (6,7).
In 2012, Muller et al (8) reported the passenger genomic alteration in multiple solid cancers. They revealed the co-deletion of genes with certain key tumor suppressive functions located in the same regions of chromosomes, such as TP53. Conversely, it was suggested that there could also exist a co-amplification of genes with certain key tumor driver genes. Furthermore, these co-amplified genes could participate in the bio-behavior of cancer cells. Thus, The Cancer Genome Atlas (TCGA) database was searched and the amplification of AR copies was identified in the majority of PCa tumors. In addition, the gene oligophrenin 1 (OPHN1) was identified, which is located in Xq12, the same region of the AR gene, and is amplified in most PCa cases, associated with the AR gene (9).
OPHN1 is considered a Rho-GTPase-activating protein (Rho-GAP) involved in the regulation of the G-protein cycle. The aberrant expression of this gene, including nonsense, frameshift, missense variants, and chromosomal deletions, is responsible for X-linked mental retardation, which is associated with cerebellar hypoplasia and distinctive facial features (9–11). In addition, the positive expression of OPHN1 has been identified in PCa by Goto et al, and the overexpression of OPHN1 has been revealed to be related to the high Gleason score and poor prognosis of PCa (12). Du et al reported that OPHN1 is related to the prognosis of CRPC (13). Therefore, it was hypothesized that OPHN1 is amplified in PCa and that the overexpression of OPHN1 could contribute to PCa progression. In the present study, the potential effects of OPHN1 on the pathology of CRPC development were investigated. The results revealed that the expression of OPHN1 could enhance PCa cell resistance to ADT. Additionally, OPHN1 could promote PCa cell proliferation, migration, and resistance to apoptosis.
Materials and methods
Genomic data and analysis
TCGA is a landmark cancer genomics program that provides genomic, epigenomic, transcriptomic, and proteomic data spanning 33 types of cancer, including PCa (14,15). TCGA database was employed to evaluate the expression of both AR and OPHN1 in PCa, and 4 datasets of Pca were recruited: A dataset of metastatic prostate adenocarcinoma (MCTP; 61 samples) (16), another dataset of metastatic prostate adenocarcinoma (SU2C/PCF Dream Team; 444 samples) (17), a dataset of metastatic castration-sensitive PCa (MSK; 424 samples, CRPC) (18), and a dataset of the metastatic PCa project (provisional; 75 samples). However, in these four datasets, there were 965 cases that provided the genomic data of PCa tumors, which were involved for analysis in the present study. Clinical information (data including age, stage, therapy, and clinical variables) was downloaded from TCGA database. All copy number variation (CNV) data of genes (AR and OPHN1) of PCa were obtained and analyzed from TCGA Data Portal: cBioPortal (https://www.cbioportal.org/). Normally, a copy number of more than 1 of each gene was considered amplification.
Cell cultures
The LNCaP (CRL-1740), 22RV1 (CRL-2505), PC3 (CRL-1435) and 293T (CRL-3216) cell lines were purchased from the American Type Culture Collection (ATCC), and maintained in RPMI-1640 medium (ATCC), or Dulbecco's modified Eagle's medium (DMEM; HyClone; Cytiva). This was supplemented with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.), penicillin (100 U/ml) and streptomycin (10 mg/ml; both from Invitrogen; Thermo Fisher Scientific Inc.) and maintained at 37°C and 5% CO2. FBS was treated with dextran-coated charcoal (final concentration 0.25%; Sigma-Aldrich; Merck KGaA) to remove any hormonal effects. Bicalutamide (AR antagonist) was purchased from Sigma-Aldrich; Merck KGaA (product no. B9061). Cells were treated with 1 µM bicalutamide in DMSO (19,20).
Northern blot analysis
Following treatment of PCa cells with bicalutamide, total RNA was extracted using an RNeasy kit (Qiagen, Inc.) for northern blot analysis. Equal amounts of the sample (10 µg) were loaded and separated on 1% agarose-formaldehyde gels by electrophoresis and then transferred to the membranes. The probes were labeled with P32-OPHN1 by random labeling and hybridized overnight at 42°C for 16 h. The internal control used was an 18S rRNA probe. The blots were visualized using Kodak XAR film. The probe of OPHN1 was 5′-TCTTAGGCGGATGCAGTCAA-3′, the probe of AR was 5′-TTGGAGCATCTGAGTCCAGG-3′, and the probe of 18S was 5′-TCGGAACTGAGGCCATGATT-3′, to generate DNA against the target gene by PCR.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from PCa cells using an RNeasy kit (Qiagen, Inc.), following the manufacturer's protocol, and was then reverse-transcribed into cDNA using SuperScript™ III Reverse Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.). qPCR (SYBR™ Green; cat. no. 4309155; Thermo Fisher Scientific, Inc.) was subsequently performed using an ABI PRISM 7500 Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.), according to the manufacturer's protocol. The cycling conditions for the reaction were as follows: An initial hold for 10 min at 95°C; then 40 cycles of 15 sec at 95°C denaturation, 30 sec at an annealing temperature of 60°C and a 30-sec extension at 72°C. The following primer pairs were used for the qPCR: GAPDH forward, 5′-AATGGACAACTGGTCGTGGAC−3′ and reverse, 5′-CCCTCCAGGGGATCTGTTTG-3′; AR forward, 5′-CCAGGGACCATGTTTTGCC-3′ and reverse, 5′-CGAAGACGACAAGATGGACAA−3′; and OPHN1 forward, 5′-TGGAGACACTCTGACTGATGAT-3′ and reverse, 5′-TACCTCGTTGAGCAATTCAGC−3′. Gene expression levels were quantified using the 2−ΔΔCq method and normalized to the internal reference gene GAPDH (21). Each experiment was performed in triplicate.
Mouse xenograft models and treatments
All animal care procedures and experiments were conducted in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines (22) and approved by Ethics Committee of Animal Experiments of the Hebei General Hospital (approval no. 20200147), Shijiazhuang, China). The experiments were conducted by the Department of Urology of Hebei General Hospital. BALB/c NU/NU nude mice (male; 8 weeks old; body weight 20–30 g; 40 mice in total) were purchased from Hebei Medical University and housed in the Experimental Animal Facility (five mice per cage) of the hospital under standard laboratory conditions (18-23°C; 40–60% humidity; 12-h light/dark cycle) with unlimited access to food and water. Subcutaneous injections of 1×106 cells (regular LNCaP or LNCaP-transfected with OPHN1 recombinant lentiviral vectors) suspended in 100 µl Matrigel (BD Biosciences) were administered in the flank of each mouse (at 8 weeks old). Every two days, each tumor volume was measured with a dial caliper, while the bodyweights of the mice were measured. The tumor volumes were determined by the following formula: length × width2 ×0.5.
The mice (20/group) were randomly assigned into the following treatment groups: i) Mice injected with regular LNCaP cells transfected with empty vectors; and ii) mice injected with LNCaP cells transfected with OPHN1 recombinant lentiviral vectors. Subsequently, the tumor volumes of both groups were monitored until they exceeded 500 mm3. Thereafter, the mice were castrated by surgery (anesthetized using 87.5 mg/kg ketamine and 12.5 mg/kg xylazine by intraperitoneal injection), and the tumor volumes continued to be monitored. At the end of the experiment, the mice were euthanized via CO2 inhalation (50% of the chamber volume/min), and the tumor tissues were collected and weighed. In addition, the maximum tumor diameters observed were 12.43×9.68 mm during the experiments.
Cell viability assay
The PCa cells (LNCaP, 22RV1, and PC3) were seeded in 96-well plates (5,000 cells/well). The LNCaP and 22RV1 cells were maintained with/without bicalutamide at a concentration of 1 µM for 72 h at 37°C, while the PC3 cells were maintained under regular conditions for 72 h at 37°C. Subsequently, cell viability was determined via an MTT assay (Beyotime Institute of Biotechnology), following the manufacturer's instructions. The formazan crystals were dissolved in DMSO and the absorbance was measured at 440 nm with a multimode plate reader.
Cell apoptosis assay and caspase-3/8 activity assay
The LNCaP and 22RV1 cells received different pre-treatments with: i) ADT by bicalutamide (1 µM); ii) transfection of OPHN1 recombinant lentiviral vectors; iii) ADT + transfection of OPHN1 recombinant lentiviral vectors; or iv) ADT + transfection of OPHN1 siRNA for 72 h, while the PC3 cells received pre-treatments with transfection of OPHN1 siRNA for 72 h. Subsequently, the cells were collected and resuspended in 500 µl binding buffer. A total of 5 µl Annexin V-FITC (Beyotime Institute of Biotechnology) was added into the cell suspension and incubated for 10 min at room temperature in the dark. Next, 10 µl of propidium iodide (PI; Beyotime Institute of Biotechnology) was added to the cell suspension, which was then incubated for 15 min on ice or at room temperature in the dark. Following incubation, the cell suspensions were loaded onto a FACScan flow cytometer (BD Biosciences) to evaluate cell apoptosis. The number of early apoptotic (Annexin V-positive) and late apoptotic (Annexin V- and PI-positive) cells indicates the total percentage of gated cells. The results were analyzed using FlowJo software (version 10.6; BD Biosciences) to determine the apoptotic rate. The Caspase-3/8 activity was assayed using the Caspase-3 Assay kit and the Caspase-8 Assay kit (cat. no. C1168S and cat. no. C1152, respectively; Beyotime Institute of Biotechnology) according to the manufacturer's instructions. The total protein lysates were collected from cells using a cell lysis buffer (cat. no. P0013; Beyotime Institute of Biotechnology). Then, a reaction buffer (85 µl), combined with Leu-Glu-His-Asp-p-nitroanilide (5 µl; cat. no. P9728; Beyotime Institute of Biotechnology), was added to each sample and incubated at 37°C for 2 h. The absorbance was measured using a multiplate reader at 450 nm.
Cancer invasion assay
The LNCaP, 22RV1 and PC3 cells received different pre-treatments (transfection of OPHN1 recombinant lentiviral vectors or transfection of OPHN1 siRNA). A 24-well Transwell chamber plate (pore size 5.0 µm; Invitrogen; Thermo Fisher Scientific, Inc.) was used to evaluate the invasion ability of cancer cells. The upper chamber was precoated with 100 µg Matrigel for 30 min at 37°C, and then the cells (200 µl) were added to the upper chamber at a concentration of 1×105 cells/ml, while a 500-µl RPMI-1640 culture medium containing 10% FBS was added into the lower chamber. The cells (LNCaP, 22RV1 and PC3) were maintained in the chamber system for 24 h at 37°C. Subsequently, the filter was collected, fixed (2.5% glutaraldehyde, 10 min, room temperature), and stained with a 0.1% crystal violet for 20 min at room temperature. The filter was observed under a light microscope at a magnification of ×100, and the number of cells that passed through the filter in five random fields was counted as the invasion ability.
Western blotting
The cell pellets were collected, and a radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich; Merck KGaA) containing protease inhibitors was used to extract proteins from the cell pellets. The protein levels were determined by bicinchoninic acid (BCA) method. Equal amounts of protein were loaded on 8–10% sodium dodecyl sulfate (SDS)-polyacrylamide gels at 20 µg/lane and were then separated for electrophoresis. Thereafter, the protein on the gels was transferred to nitrocellulose membranes. Next, following washing of the membranes using Tris-Cl-buffered saline (TBST, 0.1% Tween-20) and blocking with 5% BSA for 1 h at room temperature, the membranes were incubated overnight with primary antibodies at 4°C. Subsequently, the membranes were washed and incubated with secondary antibodies: Horseradish peroxidase (HRP)-conjugated anti-mouse (1:5,000; cat. no. sc-2357; Santa Cruz Biotechnology, Inc.) or anti-rabbit (1:5,000; product no. 7074s, Cell Signaling Technology, Inc.) at room temperature for 1 h. The membranes were then stained using an enhanced chemiluminescence (ECL) detection kit (Pierce; Thermo Fisher Scientific, Inc.). The signals were detected via a chemiluminescence detection system (Bio-Rad Laboratories, Inc.). The following primary antibodies were used: rabbit polyclonal OPHN1 (1:3,000; product code ab229655; Abcam) and mouse monoclonal GAPDH (1:3,000; cat. no. sc-32233; Santa Cruz Biotechnology, Inc.). GAPDH was used as the loading control. The protein expression levels were quantified with ImageJ software (version 1.8.0; National Institutes of Health).
Plasmid construction and transfection
Human OPHN1 cDNA (Origene Technologies, Inc.) was sequenced and subcloned into the PGL3-basic vector (OriGene Technologies, Inc.) with GFP in accordance with the manufacturer's protocol. The 293T cells were then transfected with a 4 µg lentiviral vector (2nd generation, lentiviral plasmid: packaging vector: envelope at 4:3:1) and pCMV-OPHN1-plasmid constructs using a Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) transfection reagent for 48 h in accordance with the manufacturer's protocol. Subsequently, 12-h post-transfection, the medium was replaced with DMEM supplemented with 5% FCS and incubated at 5% CO2 for 48 h prior to the collection of viral supernatant. The conditioned medium was centrifuged at 1,500 rpm for 5 min at 4°C and then passed through a filter pore (pore size, 0.45 µm; EMD Millipore). The OPHN1 recombinant lentiviral vectors were harvested from the cell supernatant for further experiments. Subsequently, lentiviral vectors were then added with a multiplicity of infection (MOI) of 30. The target cells were incubated at 37°C with the vectors, and were used for further experiments after 72 h. Western blotting was performed to verify the interference efficiency.
siRNA transfection
Cells (LNCaP, 22RV1 and PC3) were seeded (1×105 cells/well) into 12-well culture plates and transfected with 40 nM OPHN1 siRNA (5′-GAGCUCACACAGGAUUUCCUCCCAU-3′; MyBioSource, Inc.) (12) or scrambled negative control siRNA (5′-UUCUUCGAACGUGUCACGUTT−3′; Santa Cruz Biotechnology, Inc.) using Lipofectamine 3000 (Thermo Fisher Scientific, Inc.) for 24 h at 37°C. Following 24 h of transfection, the transfection efficiency in each type of PCa cell was validated by western blotting, and then the cells were used for subsequent experiments.
Statistical analysis
The data were expressed as the mean ± standard error of the mean (SEM). One-way analysis of variance (ANOVA), repeated measures of two-way ANOVA, followed by Tukey's post hoc test or Student's t-test was used for statistical analysis in the R environment (version 3.6; r-project.org). P<0.05 was considered to indicate a statistically significant difference.
Results
Amplification of OPHN1 in PCa tumors
TCGA database was searched. A total of 4 PCa datasets were included, with a total of 965 cases for reanalysis. The details of the clinicopathological characterization of PCa cohorts used in the present study is presented in Table SI. AR amplifications were identified in 26.53% (256 cases) of the selected cases. Both the AR and OPHN1 genes are located in the same region on chromosome X, and OPHN1 amplifications were identified in 18.96% (183 cases) of the selected cases. The co-amplification of AR with OPHN1 was revealed in 18.13% of the selected samples, with a significant positive association (P<0.001), which may be a phenomenon in PCa due to the amplification of a segment of chromosome X.
OPHN1 promotes resistance to ADT in PCa cells
A previous study revealed that the treatment of bicalutamide could create an androgen deprivation condition, induce the amplification of AR, and result in CRPC development (23). Therefore, in the present study, to investigate whether ADT could lead to the amplification of OPHN1, bicalutamide was used to treat LNCaP and 22RV1 cells (both are androgen-sensitive PCa cell lines) for 4 weeks. The data revealed that bicalutamide treatment induced AR amplification as well as a copy of OPHN1 as revealed by Northern blotting and RT-qPCR (Fig. 1).
 |
Figure 1.
Expression levels of AR and OPHN1 in bicalutamide-treated PCa cells. Both LNCaP and 22RV1 cells were treated with 1 µM bicalutamide for one week, while the same passages of cells treated with DMSO were considered as controls. Subsequently, the cells were collected and total RNA was extracted for analysis. (A) Northern blot analysis of the expression of AR and OPHN1 in LNCaP and 22RV1 cells, as well as the data, revealed that treatment with bicalutamide clearly increased both AR and OPHN1 level compared with that in parental cells. (B) Relative expression levels of AR and OPHN1 mRNA as determined by reverse transcription-quantitative PCR. The OPHN1 mRNA level was ~10 times higher in LNCaP cells and ~14 times higher in 22RV1 cells than in controls (parental LNCaP and 22RV1). Data are presented as the mean ± SEM of three replicates (*P<0.05 vs. the control, determined by Student's t-test). OPHN1, oligophrenin 1; AR, androgen receptor; PCa, prostate cancer.
|
To investigate the mechanism(s) by which OPHN1 regulates ADT resistance, the expression of OPHN1 in PCa cell lines (LNCaP, 22RV1, and PC3) was overexpressed or knocked down (Fig. 2A). Subsequently, the LNCaP and 22RV1 cells were cultured in an androgen-depleted medium containing bicalutamide to simulate ADT. Bicalutamide treatment led to the cell death of regular LNCaP and 22RV1. By contrast, less cell death was observed in cells with enhanced OPHN1 expression. In addition, our results indicated that the expression of OPHN1 could promote the cell viability rate in androgen-dependent cell lines LNCaP and 22RV1 with or without the pressure of bicalutamide (Fig. 2B). Therefore, the data may indicate that OPHN1 promoted PCa cell survival under androgen-depleted conditions. More interestingly, following overexpression of OPHN1 by transfection in PC3 cells, an androgen-insensitive cell line, it was revealed that overexpression of OPHN1 could also promote the viability of PC3 cells. Conversely, blocking the expression of OPHN1 clearly inhibited the viability of LNCaP, 22RV1 and PC3 cells (Fig. 2B).
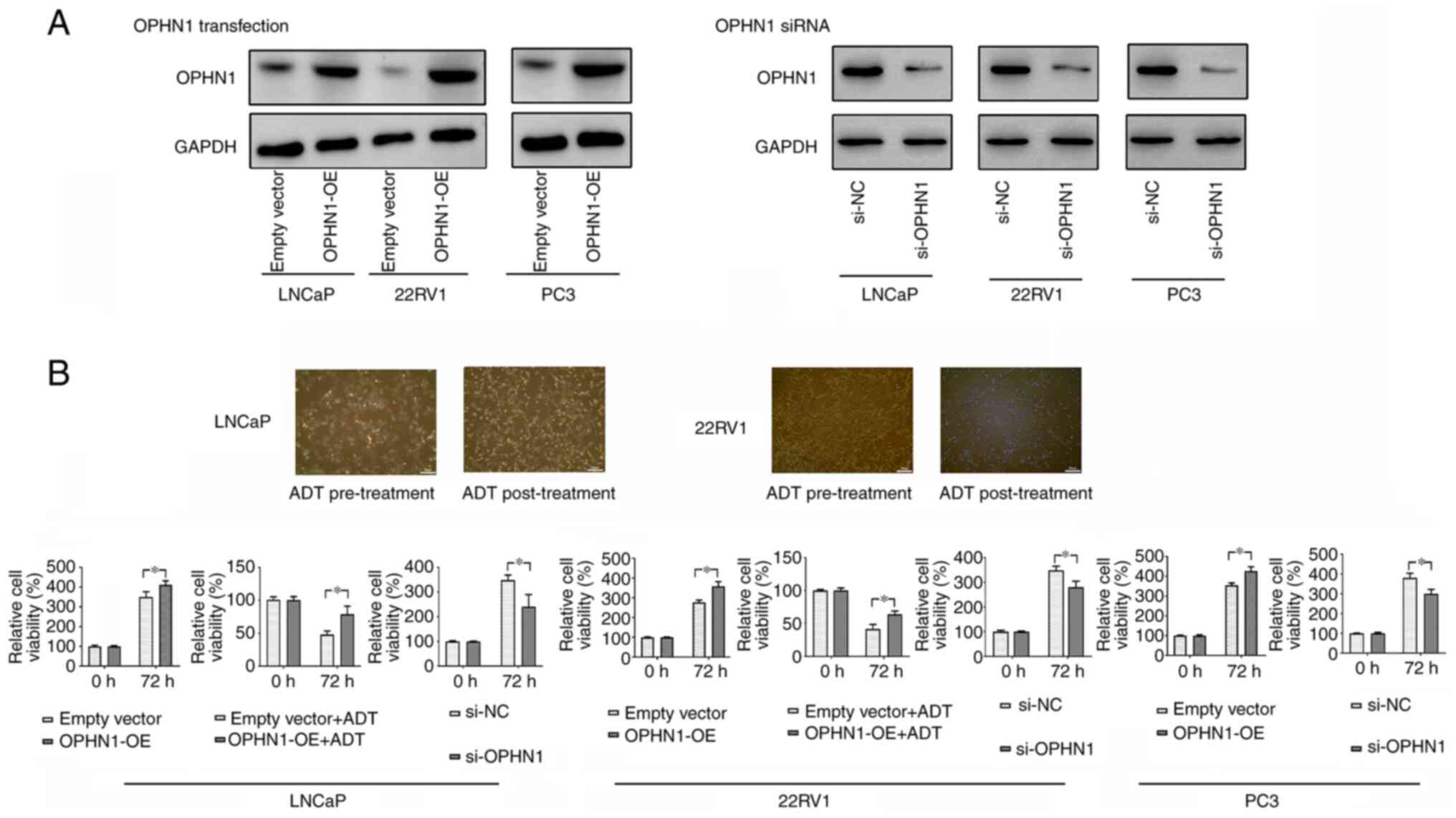 |
Figure 2.
Expression of OPHN1 promotes PCa cell resistance to ADT. (A) The transfection of OPHN1 recombinant lentiviral vectors increased OPHN1 expression (OPHN1 over expression, OPHN1-OE) in the LNCaP, 22RV1, and PC3 cells, while the knockdown of OPHN1 by siRNA (si-OPHN1) inhibited OPHN1 expression in the LNCaP, 22RV1, and PC3 cells (representative images are shown), as determined by western blotting. (B) Effects of OPHN1 on the cell viability of PCa cells. The expression of OPHN1 was overexpressed or knocked down in LNCaP, 22RV1, and PC3 cells, which were then cultured in vitro under various conditions for 72 h. Thereafter, cell viability rates (%) were determined using an MTT assay. The treatment groups were compared with the controls. The data revealed that the enhanced expression of OPHN1 promoted the viability of LNCaP, 22RV1 and PC3 cells, while blocking the expression of OPHN1 by siRNA decreased the viability of LNCaP, 22RV1 and PC3 cells. In addition, under ADT (bicalutamide 1 µM) conditions, the overexpression of OPHN1 contributed to the survival of cells of LNCaP and 22RV1. Data are presented as the mean ± SEM of three replicates. *P<0.05 vs. the control, by Student's t-test. OPHN1, oligophrenin 1; PCa, prostate cancer; ADT, androgen deprivation therapy; si-NC, siRNA negative control.
|
Expression of OPHN1 promotes PCa cell invasion and inhibits cell apoptosis
Using Transwell and apoptotic assays, the effect of OPHN1 on PCa cell invasion and apoptosis was investigated. The Transwell invasion assay indicated that the invasion ability of PCa cells was significantly increased via the overexpression of OPHN1 compared with that of the control group. However, the knockdown of the expression of OPHN1 in PCa cells significantly inhibited cell invasion ability in all three (LNCaP, 22RV1 and PC3) PCa cell lines (Fig. 3). Consistently, the apoptotic assay revealed that the expression of OPHN1 facilitated LNCaP and 22RV1 cell resistance to bicalutamide, with a lower apoptotic rate (Fig. 4A) and caspase-3/8 activities (Fig. 4B), while the knockdown of the expression of OPHN1 increased the apoptotic rate and caspase-3/8 activities of LNCaP and 22RV1 under the pressure of bicalutamide. Additionally, in PC3 cells, knockdown of the expression of OPHN1 increased the apoptosis rate and caspase-3/8 activities (Fig. 4). Therefore, it was concluded that the upregulation of OPHN1 promoted the invasion and inhibited the apoptosis of PCa cells in vitro.
 |
Figure 3.
Expression of OPHN1 promotes the invasion capacity of PCa. The expression of OPHN1 was enhanced by the transfection of recombinant lentiviral vectors (OPHN1-OE) or blocked by the transfection of siRNA (si-OPHN1). Subsequently, the cells (LNCaP, 22RV1 and PC3) were loaded into the Transwell chambers to evaluate the invasion ability of cells. The number of cells that passed through the filter in five random fields was counted as the invasion ability. The data revealed that the overexpression of OPHN1 significantly promoted the invasion capacity of the LNCaP, 22RV1 and PC3 cells, with more cells passing through the filters, whereas knockdown OPHN1 inhibited the invasion capacity of LNCaP, 22RV1 and PC3 cells by decreasing the cell numbers that passed through the filters. Scale bar, 50 µm. The results are expressed as the mean ± SEM for three replicate determinations. *P<0.05 and **P<0.01, by Student's t-test. OPHN1, oligophrenin 1; PCa, prostate cancer; si-NC, siRNA negative control.
|
 |
Figure 4.
Expression of OPHN1 prevents cell apoptosis in vitro. The expression of OPHN1 in PCa cells (LNCaP, 22RV1 and PC3) was overexpressed by the transfection of recombinant lentiviral vectors (OPHN1-OE) or blocked by the transfection of siRNA (si-OPHN1). Then the cells were cultured in vitro under various conditions for 72 h. (A) Different types of PCa cells (LNCaP and 22RV1) with OPHN1 overexpression or knockdown were treated with ADT for 72 h, and the PC3 cells with the OPHN1 knockdown by siRNA continued to be cultured for 72 h. Following treatment, all the PCa cells were collected, stained with Annexin V and PI, and analyzed by FACS. The number of early apoptotic (Annexin V-positive) and late apoptotic (Annexin V- and PI-positive) cells indicates the total percentage of gated cells. Representative images and relative quantifications are shown. Compared with the cells without ADT, both the LNCaP and 22RV1 cells under ADT conditions had a higher percentage of apoptosis. However, overexpression of OPHN1 alleviated the pro-apoptotic effect of ADT in both LNCaP and 22RV1 cells. Under ADT conditions, both LNCaP and 22RV1 cells with OPHN1 overexpression displayed a lower percentage of apoptosis than the LNCaP and 22RV1 cells transfected with empty vectors. In addition, under ADT conditions, the knockdown of the expression of OPHN1 in LNCaP and 22RV1 cells increased the apoptotic rates compared with the cells that were transfected with negative control RNA. In addition, in PC3 cells, the knockdown of the expression of OPHN1 promoted apoptosis and displayed a higher percentage of apoptosis than cells transfected with negative control RNA. (B) The data on the activities of caspase-3 and caspase-8 were consistent with the apoptotic assay of flow cytometry. ADT promoted the activities of both caspase-3 and caspase-8 in both LNCaP and 22RV1 cells compared with the cells without ADT, which could be alleviated by the overexpression of OPHN1. Furthermore, under ADT conditions, blocking the expression of OPHN1 increased caspase-3 and caspase-8 activities in LNCaP and 22RV1 cells. Blocking the expression of OPHN1 in PC3 cells promoted the activities of both caspase-3 and caspase-8. All the experiments were performed in triplicate, and the data are presented as the mean ± SEM. *P<0.05 and, by one-way ANOVA followed by Tukey's post hoc test, or by Student's t-test. OPHN1, oligophrenin 1; PCa, prostate cancer; ADT, androgen deprivation therapy; si-NC, siRNA negative control.
|
Expression of OPHN1 promotes castration resistance in vivo
The effects of OPHN1 expression on tumor growth were examined. Xenografts were created by injecting overexpressed OPHN1 in LNCaP cells in mice, and the data revealed that OPHN1-overexpressing tumors had a growth rate higher than that of the empty vector controls. In addition, mice were castrated when the tumor size exceeded 500 cm3, and our data demonstrated that the expression of OPHN1 was more resistant to castration with slower tumor size shrinkage (Fig. 5 and representative tumor image in Fig. S1). Therefore, our data demonstrated that OPHN1 overexpression conferred resistance to androgen deprivation in vivo.
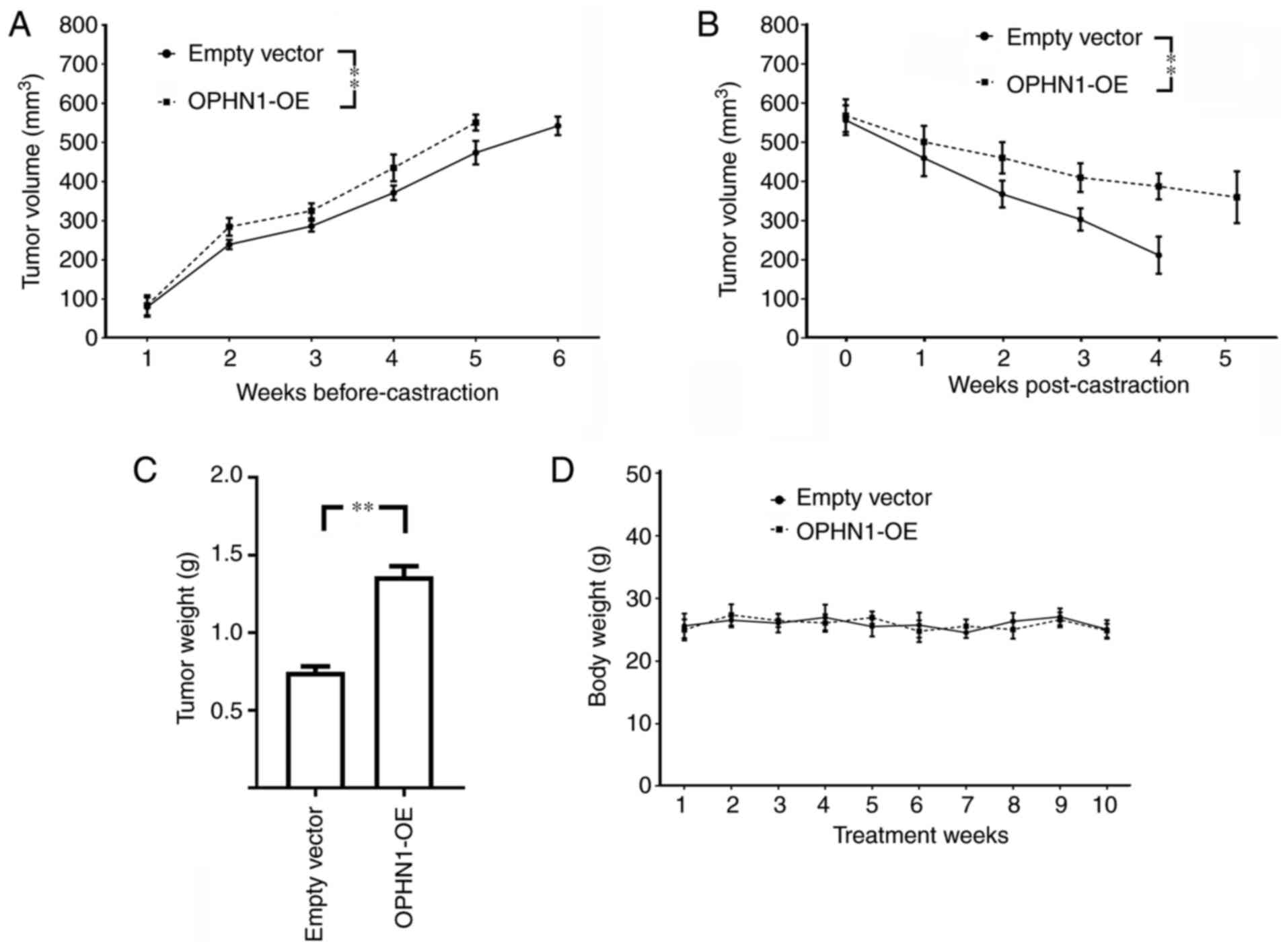 |
Figure 5.
Effects of OPHN1 on tumor xenograft growth in mice. LNCaP (1×106 cells) transfected with recombinant lentiviral vectors (OPHN1 overexpression, OPHN1-OE) were injected into mice subcutaneously (20 mice/group), and then the tumor growth was monitored, while the LNCaP cells transfected with empty vectors were injected as controls. (A) The tumor volumes of both groups were monitored until they exceeded 500 mm3, as revealed with growth curves for xenografts in each group. The data revealed that the tumors of OPHN1-OE LNCaP cells had a higher rate of tumor growth (F=14.81; P<0.001), which exceeded 500 mm3 at ~5 weeks, whereas the tumors in the control group exceeded 500 mm3 at ~6 weeks. Thereafter, the mice were castrated by surgery, and the tumor volumes continued to be monitored. (B) The data revealed that the mouse xenografts of OPHN1-OE LNCaP exhibited a significantly lower decrease rate in tumor volume than the control group (F=35.34; P<0.001). (C) In addition, the mouse xenografts of OPHN1-OE LNCaP exhibited a significantly higher tumor weight than the control group. (D) There was no difference in body weight between the two groups of mice (F=0.519; P=0.861). The mouse xenograft experiments were performed as described in the Materials and methods section. The data are presented as the mean ± SEM. **P<0.01, by two-way ANOVA followed by Tukey's post hoc test, or by Student's t-test. OPHN1, oligophrenin 1.
|
Discussion
In the present study, our results revealed that ADT could induce the amplification of OPHN1, which could then contribute to the survival of PCa cells in an ADT environment. Additionally, our results revealed that the expression of OPHN1 could promote the viability and invasion of PCa cells, which could lead to PCa progression.
Gene amplification is normally considered an outcome of an increase in the copy number of a restricted region of a chromosome arm, and this amplified region is called an ‘amplicon.’ Furthermore, gene amplification, particularly oncogenes, is frequently observed in various solid cancers through amplicons and is expected to contribute to tumor evolution by altering gene expression, which is likely to reflect the numerous routes taken by individual tumors to escape normal protective mechanisms (24). In fact, previous studies (25–27) have demonstrated that multiple genes co-amplify with oncogenes in amplicons and cancers. Additionally, these co-amplification genes, particularly genes located in the same region of the chromosome with the oncogenes, may contribute to cancer progression. For example, in the 17q12-q21 amplicon, ERBB2 amplification was observed in gastric (28), breast (25), and esophageal cancers (28). ERBB2 is important in tumorigenesis as an oncogene by promoting cancer cell proliferation and survival signaling pathways. Growth factor receptor-bound protein 7 (GRB7) (29), retinoic acid receptor alpha (RAR) (30), and steroidogenic acute regulatory protein (StAR)-related lipid transfer domain containing 3 (STARD3) (31) are all frequently observed as co-amplified genes with ERBB2. GRB7 can bind with receptor tyrosine kinase to mediate downstream signal transduction, and the activation of GRB7 may induce progression in multiple types of cancers. In addition, RAR and STARD3 may both contribute to cancer progression (32). However, the mechanism of the increase in copy number in an amplicon remains unknown. At present, there have been some hypotheses concerning the potential mechanism of an amplicon, including double rolling-circle replication, extra replication and recombination, replication fork stalling and template switching, and the breakage-fusion-bridge cycle. Nevertheless, more research is clearly required to explore the biochemical mechanism of amplicons, particularly in carcinogenesis (33).
TCGA is a landmark cancer genomics platform that provides genomic, epigenomic, transcriptomic, and proteomic data spanning 33 types of cancer, including prostate cancer. Therefore, TCGA database was accessed, multiple datasets of PCa were downloaded and reanalysis was performed. In our study, by analyzing the gene CNV in PCa in TCGA, the increased CNV of AR was identified in 26.53% of all these cases. Thus, it was suggested that AR could play a key role in PCa pathology. In fact, AR is one of the most investigated genes in PCa research. Increased AR gene copy numbers were observed in ~80% of CRPCs, 30% of which had high levels of amplification (34). The role of AR in CRPC pathology has been demonstrated for numerous years. The majority of studies suggest that AR remains active through multiple pathways in CRPC despite systemic castration (35–37). For example, various AR mutations are observed in CRPC, and these mutations could lead to decreased specificity of AR-ligand interaction, and allow AR activation by alternative steroidal molecules, such as, corticosteroids, and progesterone (38,39). AR splice variants are another phenomenon observed in CRPC. Researchers have disclosed that one of the most studied variants, AR-V7 may activate the AR pathway with a ligand-independent manner, in response to ADT, and then contribute to castration resistance (40). In addition, AR amplification in CRPC led to higher levels of AR protein, which then contributed to maintain the activation of the AR signaling pathway at lower levels of androgens (41). In the present study, it was revealed that amplification of OPHN1 may provide an additional mechanistic explanation for the development of CRPC. An increase of OPHN1 CNV was identified in 18.96% of all the involved cases of the present study. In addition, it was demonstrated that the gene OPHN1 was co-amplified with AR, and the increased CNV of OPHN1 and AR were positively associated. Thus, it was hypothesized that a fragment of the chromosome X is amplified in PCa, particularly in the PCa under ADT, which then results in increased CNV of both AR and OPHN1 since they are located in the same region of chromosome X. In fact, CNV amplification is one of the major causes of gene overexpression in cancer, and the increased CNV of OPHN1 could result in the overexpression of OPHN1 in PCa tumors, as demonstrated in the in vitro experiments of the present study. Additionally, Visakorpi et al suggested that ADT could result in a gain of chromosome X, which in turn increases the AR gene expression level (4). Zhang et al revealed that ADT leads to X-chromosome polysomy (42). OPHN1 is also located in chromosome X, which could be amplified.
Our results revealed that the expression of OPHN1 may promote cancer survival and invasion. Thus, the expression of OPHN1 could promote PCa progression, which is consistent with certain other studies on PCa (12,43). In addition, in gastric cancer, Dicken et al revealed that the expression of OPHN1 was related to lymphovascular invasion (44). Furthermore, it is known that OPHN1 normally regulates the Rho-GAP pathway in cells, including RhoA, Cdc42, and Rac1. RhoA is involved in PCa tumorigenesis and progression, and Chen et al demonstrated that the activation of RhoA and Rac1 promoted PCa migration and invasion (45). Another previous study revealed that a high expression of RhoA had a poor prognosis for PCa, with poor tumor differentiation and higher prostate-specific antigen (PSA) relapse (46). Additionally, it was previously revealed that by activating the Rho-Gap pathway, CDC42, could facilitate cancer cell resistance to the inhibitor of the PI3K/Akt/mTOR pathway (47). The activation of CDC42 could also promote cell motility, proliferation, and resistance to apoptosis, all of which would result in PCa invasion and metastasis (48). Conversely, blocking Rac1/Cdc42 inhibited PCa tumor growth in mice models by downregulating the PI3K/Akt/mTOR and PAK signaling pathway in PCa cells (49). In addition, the activation of Rac1 could directly contribute to CRPC development activation, and Rac1 is closely related to androgen-independent cell proliferation (50). Both Lyons et al (51) and Chen et al (52) revealed that androgen deprivation increased the expression of Rac1. Rac1 induced AR-dependent gene expression (51), while blocking Rac1 enhanced the efficacy of enzalutamide in enzalutamide-resistant xenograft tumors (52). In addition, a previous study has revealed that blocking Rac1 or CDC42 may inhibit tumor growth and progression in PCa (49). Zins et al indicated that blocking Rac1/Cdc42 GTPase by small molecule inhibitor could suppress growth of primary human PCa xenografts in mice (49). Therefore, in addition to the AR pathway, the OPHN1-Rho-GAP pathway may contribute to CRPC pathology (51). Additionally, the present study had certain limitations. The increased expression of OPHN1 at the protein level needs to be demonstrated in the human PCa tumors. Further investigations are required to determine whether blocking OPHN1 could inhibit tumor growth in a mouse model, although it was observed that expression of OPHN1 promoted PCa progression, and blocking OPHN1 expression inhibited viability and invasion of PCa cells in vitro. Additionally, further research is required to comprehend the potential pathway regulated by OPHN1 in PCa.
In summary, in prostate cancer, ADT induced the amplification of OPHN1, which contributed to the development of CRPC. The overexpression of OPHN1 facilitated PCa survival under ADT by contributing to PCa viability, invasion, and progression. Therefore, targeting OPHN1 could be used to reverse endocrine therapy resistance in CRPC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
JL and YZ designed and performed experiments, analyzed the data and wrote the manuscript. SL and FS analyzed the data and performed the experiments. GW and DW performed the experiments. TY and SG contributed to study design. JL and YZ confirm the authenticity of all the raw data. All authors read and approved the final manuscript.
Ethics approval and consent to participate
All animal care procedures and experiments were conducted in accordance with the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines and approved by Ethics Committee of Animal Experiments of the Hebei General Hospital (Shijiazhuang, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
|
1
|
Global Burden of Disease Cancer Collaboration, ; Fitzmaurice C, Allen C, Barber RM, Barregard L, Bhutta ZA, Brenner H, Dicker DJ, Chimed-Orchir O, Dandona R, et al: Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 32 cancer groups, 1990 to 2015: A systematic analysis for the global burden of disease study. JAMA Oncol. 3:524–548. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Roehl KA, Han M, Ramos CG, Antenor JA and Catalona WJ: Cancer progression and survival rates following anatomical radical retropubic prostatectomy in 3,478 consecutive patients: Long-term results. J Urol. 172:910–914. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang SS: Treatment options for hormone-refractory prostate cancer. Rev Urol. 9 (Suppl 2):S13–S18. 2007.PubMed/NCBI
|
|
4
|
Visakorpi T, Hyytinen E, Koivisto P, Tanner M, Keinänen R, Palmberg C, Palotie A, Tammela T, Isola J and Kallioniemi OP: In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat Genet. 9:401–406. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
LaTulippe E, Satagopan J, Smith A, Scher H, Scardino P, Reuter V and Gerald WL: Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 62:4499–4506. 2002.PubMed/NCBI
|
|
6
|
Jernberg E, Bergh A and Wikstrom P: Clinical relevance of androgen receptor alterations in prostate cancer. Endocr Connect. 6:R146–R161. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Merson S, Yang ZH, Brewer D, Olmos D, Eichholz A, McCarthy F, Fisher G, Kovacs G, Berney DM, Foster CS, et al: Focal amplification of the androgen receptor gene in hormone-naive human prostate cancer. Br J Cancer. 110:1655–1662. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al: Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 488:337–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bergmann C, Zerres K, Senderek J, Rudnik-Schoneborn S, Eggermann T, Häusler M, Mull M and Ramaekers VT: Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain. 126:1537–1544. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Al-Owain M, Kaya N, Al-Zaidan H, Al-Hashmi N, Al-Bakheet A, Al-Muhaizea M, Chedrawi A, Basran RK and Milunsky A: Novel intragenic deletion in OPHN1 in a family causing XLMR with cerebellar hypoplasia and distinctive facial appearance. Clin Genet. 79:363–370. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Busa T, Caietta E, Chabrol B, Girard N, Philip N and Missirian C: Large in-frame intragenic deletion of OPHN1 in a male patient with a normal intelligence quotient score. Clin Dysmorphol. 26:47–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Goto K, Oue N, Hayashi T, Shinmei S, Sakamoto N, Sentani K, Teishima J, Matsubara A and Yasui W: Oligophrenin-1 is associated with cell adhesion and migration in prostate cancer. Pathobiology. 81:190–198. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du M, Tian Y, Tan W, Wang L, Wang L, Kilari D, Huang CC, Wang L and Kohli M: Plasma cell-free DNA-based predictors of response to abiraterone acetate/prednisone and prognostic factors in metastatic castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 23:705–713. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al: The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2:401–404. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al: Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 6:pl12013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al: The mutational landscape of lethal castration-resistant prostate cancer. Nature. 487:239–243. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Abida W, Cyrta J, Heller G, Prandi D, Armenia J, Coleman I, Cieslik M, Benelli M, Robinson D, Van Allen EM, et al: Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA. 116:11428–11436. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Stopsack KH, Nandakumar S, Wibmer AG, Haywood S, Weg ES, Barnett ES, Kim CJ, Carbone EA, Vasselman SE, Nguyen B, et al: Oncogenic genomic alterations, clinical phenotypes, and outcomes in metastatic castration-sensitive prostate cancer. Clin Cancer Res. 26:3230–3238. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hodgson MC, Astapova I, Hollenberg AN and Balk SP: Activity of androgen receptor antagonist bicalutamide in prostate cancer cells is independent of NCoR and SMRT corepressors. Cancer Res. 67:8388–8395. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hara T, Miyazaki J, Araki H, Yamaoka M, Kanzaki N, Kusaka M and Miyamoto M: Novel mutations of androgen receptor: A possible mechanism of bicalutamide withdrawal syndrome. Cancer Res. 63:149–153. 2003.PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kilkenny C, Browne W, Cuthill IC, Emerson M and Altman DG; NC3Rs Reporting Guidelines Working Group, : Animal research: Reporting in vivo experiments: The ARRIVE guidelines. Br J Pharmacol. 160:1577–1579. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kawata H, Ishikura N, Watanabe M, Nishimoto A, Tsunenari T and Aoki Y: Prolonged treatment with bicalutamide induces androgen receptor overexpression and androgen hypersensitivity. Prostate. 70:745–754. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Albertson DG: Gene amplification in cancer. Trends Genet. 22:447–455. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Monni O, Barlund M, Mousses S, Kononen J, Sauter G, Heiskanen M, Paavola P, Avela K, Chen Y, Bittner ML and Kallioniemi A: Comprehensive copy number and gene expression profiling of the 17q23 amplicon in human breast cancer. Proc Natl Acad Sci USA. 98:5711–5716. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Grasso C, Butler T, Rhodes K, Quist M, Neff TL, Moore S, Tomlins SA, Reinig E, Beadling C, Andersen M and Corless CL: Assessing copy number alterations in targeted, amplicon-based next-generation sequencing data. J Mol Diagn. 17:53–63. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boeva V, Popova T, Lienard M, Toffoli S, Kamal M, Le Tourneau C, Gentien D, Servant N, Gestraud P, Rio Frio T, et al: Multi-factor data normalization enables the detection of copy number aberrations in amplicon sequencing data. Bioinformatics. 30:3443–3450. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Press MF, Ellis CE, Gagnon RC, Grob TJ, Buyse M, Villalobos I, Liang Z, Wu S, Bang YJ, Qin SK, et al: HER2 Status in advanced or metastatic gastric, esophageal, or gastroesophageal adenocarcinoma for entry to the TRIO-013/LOGiC trial of lapatinib. Mol Cancer Ther. 16:228–238. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bivin WW, Yergiyev O, Bunker ML, Silverman JF and Krishnamurti U: GRB7 expression and correlation with HER2 amplification in invasive breast carcinoma. Appl Immunohistochem Mol Morphol. 25:553–558. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Doi A, Ishikawa K, Shibata N, Ito E, Fujimoto J, Yamamoto M, Shiga H, Mochizuki H, Kawamura Y, Goshima N, et al: Enhanced expression of retinoic acid receptor alpha (RARA) induces epithelial-to-mesenchymal transition and disruption of mammary acinar structures. Mol Oncol. 9:355–364. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Katoh M and Katoh M: MGC9753 gene, located within PPP1R1B-STARD3-ERBB2-GRB7 amplicon on human chromosome 17q12, encodes the seven-transmembrane receptor with extracellular six-cystein domain. Int J Oncol. 22:1369–1374. 2003.PubMed/NCBI
|
|
32
|
Glynn RW, Miller N and Kerin MJ: 17q12-21-the pursuit of targeted therapy in breast cancer. Cancer Treat Rev. 36:224–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsui A, Ihara T, Suda H, Mikami H and Semba K: Gene amplification: Mechanisms and involvement in cancer. Biomol Concepts. 4:567–582. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Waltering KK, Urbanucci A and Visakorpi T: Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol. 360:38–43. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Karantanos T, Corn PG and Thompson TC: Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene. 32:5501–5511. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Feng Q and He B: Androgen receptor signaling in the development of castration-resistant prostate cancer. Front Oncol. 9:8582019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chandrasekar T, Yang JC, Gao AC and Evans CP: Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl Androl Urol. 4:365–380. 2015.PubMed/NCBI
|
|
38
|
Zhao XY, Malloy PJ, Krishnan AV, Swami S, Navone NM, Peehl DM and Feldman D: Glucocorticoids can promote androgen-independent growth of prostate cancer cells through a mutated androgen receptor. Nat Med. 6:703–706. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Buttigliero C, Tucci M, Bertaglia V, Vignani F, Bironzo P, Di Maio M and Scagliotti GV: Understanding and overcoming the mechanisms of primary and acquired resistance to abiraterone and enzalutamide in castration resistant prostate cancer. Cancer Treat Rev. 41:884–892. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K and Sawyers CL: Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proc Natl Acad Sci USA. 107:16759–16765. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Edwards J, Krishna NS, Grigor KM and Bartlett JM: Androgen receptor gene amplification and protein expression in hormone refractory prostate cancer. Br J Cancer. 89:552–556. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang X, Hong SZ, Lin EJ, Wang DY, Li ZJ and Chen LI: Amplification and protein expression of androgen receptor gene in prostate cancer cells: Fluorescence in situ hybridization analysis. Oncol Lett. 9:2617–2622. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Du M, Huang CC, Tan W, Kohli M and Wang L: Multiplex digital PCR to detect amplifications of specific androgen receptor loci in cell-free DNA for prognosis of metastatic castration-resistant prostate cancer. Cancers (Basel). 12:21392020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dicken BJ, Graham K, Hamilton SM, Andrews S, Lai R, Listgarten J, Jhangri GS, Saunders LD, Damaraju S and Cass C: Lymphovascular invasion is associated with poor survival in gastric cancer: An application of gene-expression and tissue array techniques. Ann Surg. 243:64–73. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Chen X, Cheng H, Pan T, Liu Y, Su Y, Ren C, Huang D, Zha X and Liang C: mTOR regulate EMT through RhoA and Rac1 pathway in prostate cancer. Mol Carcinog. 54:1086–1095. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen W, Delongchamps NB, Mao K, Beuvon F, Peyromaure M, Liu Z and Dinh-Xuan AT: High RhoA expression at the tumor front in clinically localized prostate cancer and association with poor tumor differentiation. Oncol Lett. 11:1375–1381. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu J, Wang L, Zhang Y, Li S, Sun F, Wang G, Yang T, Wei D, Guo L and Xiao H: Induction of entosis in prostate cancer cells by nintedanib and its therapeutic implications. Oncol Lett. 17:3151–3162. 2019.PubMed/NCBI
|
|
48
|
Ahmat Amin MKB, Shimizu A, Zankov DP, Sato A, Kurita S, Ito M, Maeda T, Yoshida T, Sakaue T, Higashiyama S, et al: Epithelial membrane protein 1 promotes tumor metastasis by enhancing cell migration via copine-III and Rac1. Oncogene. 37:5416–5434. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zins K, Lucas T, Reichl P, Abraham D and Aharinejad S: A Rac1/Cdc42 GTPase-specific small molecule inhibitor suppresses growth of primary human prostate cancer xenografts and prolongs survival in mice. PLoS One. 8:e749242013. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Kobayashi T, Inoue T, Shimizu Y, Terada N, Maeno A, Kajita Y, Yamasaki T, Kamba T, Toda Y, Mikami Y, et al: Activation of Rac1 is closely related to androgen-independent cell proliferation of prostate cancer cells both in vitro and in vivo. Mol Endocrinol. 24:722–734. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA and Burnstein KL: Ligand-independent activation of androgen receptors by Rho GTPase signaling in prostate cancer. Mol Endocrinol. 22:597–608. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chen X, Yin L, Qiao G, Li Y, Li B, Bai Y and Feng F: Inhibition of Rac1 reverses enzalutamide resistance in castration-resistant prostate cancer. Oncol Lett. 20:2997–3005. 2020. View Article : Google Scholar : PubMed/NCBI
|














