Introduction
Cancer remains a public health threat worldwide,
with the International Agency for Research on Cancer reporting a
staggering 19.3 million new cases and nearly 10 million deaths in
2020 (1). Despite significant
advances in its treatment, the battle against cancer is hampered by
the significant side effects of traditional therapies and the
inevitable emergence of resistance associated with such treatments.
Therefore, the identification of more effective and
better-tolerated anticancer therapies is critical to meet clinical
requirements. As first described by Dixon et al (2) in 2012, ferroptosis is a distinct form
of cell death characterized by the accumulation of LPOs (3,4).
Recently, ferroptosis has garnered attention owing to its
involvement in numerous diseases, ranging from neurodegeneration to
lethal malignancies, such as liver, breast, and lung cancers
(5–8). Over the past two decades,
comprehensive studies have investigated the intricacies of
ferroptosis in diverse cancer types, elucidating its pivotal role
in governing cancer progression via diverse signaling cascades.
Cancer cells that exhibit resistance to conventional therapies or
possess a high propensity for metastasis are more susceptible to
ferroptosis (9–11). Therefore, the regulation of
ferroptosis and its target proteins represents a novel and
promising strategy for cancer treatment (12,13).
The various signaling pathways involved in ferroptosis have been
extensively summarized those studies. Ferroptosis primarily occurs
through the L-cystine/glutamate antiporter (system Xc-)/glutathione
(GSH)/GSH peroxidase 4 (GPX4) pathway, the GTP cyclohydroxylase-1
(GCH1)/tetrahydrobiopterin (BH4) pathway, the dihydroorotate
dehydrogenase (DHODH) pathway, the O-acyltransferase 1/2
(MBOAT1/2)-monounsaturated fatty acid (MUFA) pathway, the nuclear
factor-erythroid 2-related factor 2 (Nrf2) pathways, the mevalonate
pathway and the apoptosis-inducing factor mitochondria-associated 2
(AIFM2, also known as FSP1 and referred to as FSP1 throughout)
pathway-mainly the FSP1-coenzyme Q10 (CoQ10)-NAD(P)H, FSP1-vitamin
K (VK)-NAD(P)H and FSP1-endosomal sorting complex, required for
transport (ESCRT)-III pathways (Fig.
1). Among these systems, the Xc-/GSH/GPX4 pathway has garnered
significant attention as a crucial regulatory mechanism of
ferroptosis, sparking extensive research on its ability to induce
ferroptosis in tumor cells. However, the clinical application of
inhibitors targeting this system has been hindered by their poor
selectivity, toxicity and the presence of FSP1, which runs parallel
to GSH-GPX4. Furthermore, certain cancer cells exhibit resistance
to ferroptosis induction (14–16).
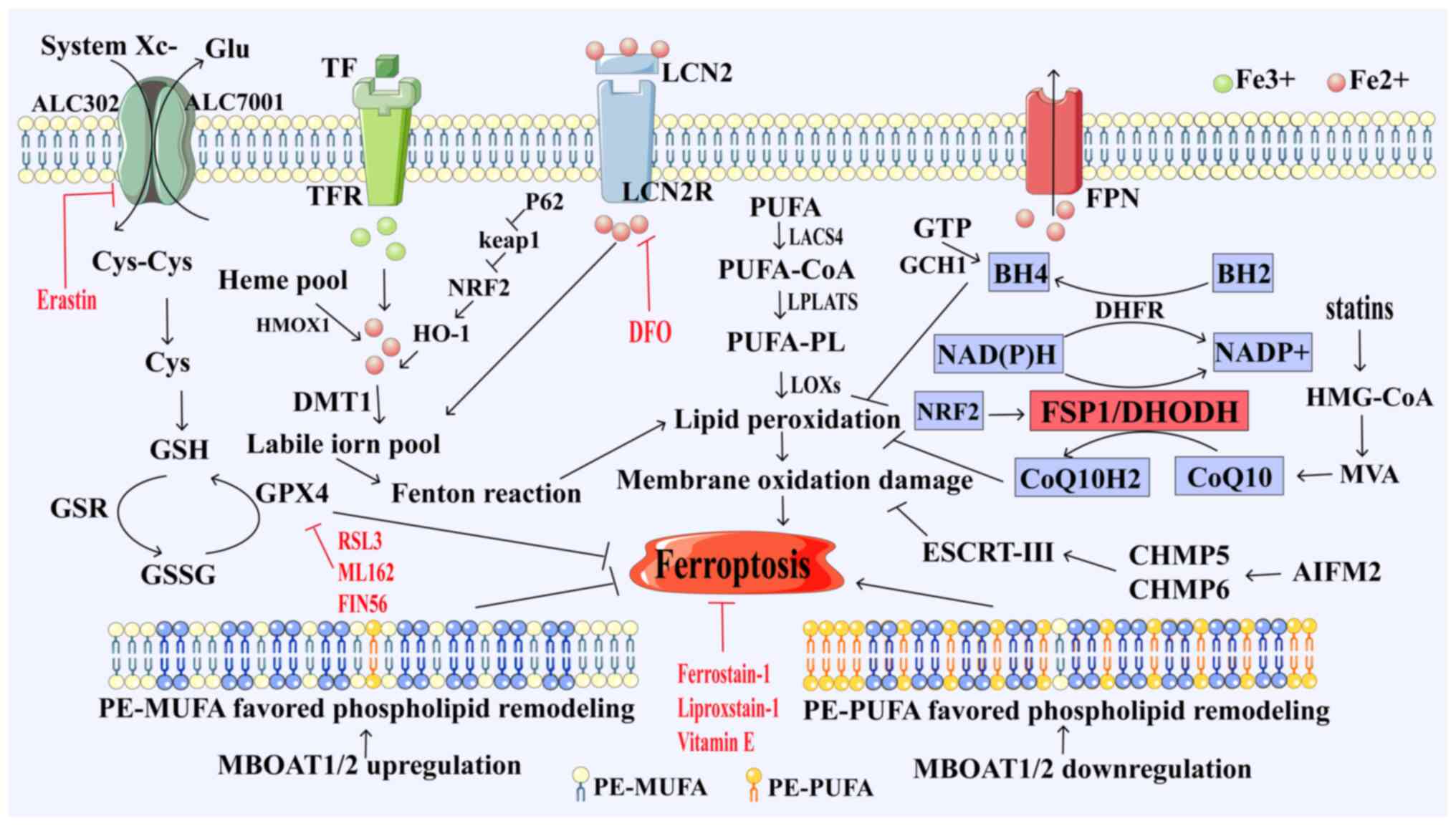 | Figure 1.Ferroptosis pathway. System Xc uptake
of cystine from extracellular sources, conversion of cystine to
cysteine for GSH biosynthesis, GPX4 catalyzes the conversion of GSH
to GSSG while reducing peroxidative unsaturated fat, protecting
lipid bilayer cells from oxidative damage and inhibiting
ferroptosis; long-chain LACS4 reduces arachidonic acid and
adrenaline. PUFA is activated as CoA, oxidizes to perform lipid
peroxidation and induces ferroptosis, while TFR1 transports
Fe3+ into cells and, under the action of DMT1, converts
Fe3+ to Fe2+, ferroptosis is induced by the
Fenton reaction. FSP1 inhibits ferroptosis by reducing ubiquitin to
pantothenol through the NAD(P)H pathway. DHODH reduces ubiquitin to
pantothenol, a process that produces lipophilic antioxidants;
blocking the accumulation of lipid peroxidation inhibits
ferroptosis. GCH1 promotes the synthesis of BH4, which exerts its
antioxidant capacity to protect cells from lipid peroxidation and
induce ferroptosis. MBOAT1 and MBOAT2 inhibit ferroptosis by
reprogramming membrane phospholipids. GSH, glutathione; GSSG, GSH
oxide; GPX4, GSH peroxidase 4; GSR, GSH reductase; FSP1,
ferroptosis inhibitory protein 1; CoQ10, ubiquinone; CoQ10H2,
ubiquinol; DHODH, dihydroorotate dehydrogenase; GTP,
phosphohydrolases; GCH1, cyclohydrolase-1; BH2, dihydrobiopterin;
BH4, tetrahydrobiopterin; LACS4, long-chain acyl-CoA synthetase
family member 4; LOX, lipoxygenase; LPCATs,
lyso-phosphatidylcholine acyltransferase; PUFA, polyunsaturated
fatty acids; CoA, coenzyme A; PUFA-PL, polyunsaturated fatty
acids-polyunsaturated; HMG-CoA, 3-hydroxyl-3-methylglutaryl CoA;
DHFR, dihydrofolate reductase; HO-1, heme oxygenase 1; Nrf2,
nuclear factor erythroid 2-related factor 2; p62, tumour suppressor
gene Tp62; TFR, transferrin receptor; DMT1, divalent metal
transporter 1; Keap1, Kelch-like ECH associated protein 1; LOX,
lipoxygenase; MBOAT1/2, o-acyltransferase 1/2; AIFM2,
apoptosis-inducing factor mitochondria-associated 2; ESCRT,
endosomal sorting complex, required for transport; CHAM5/6, charged
multivesicular proteins 5 and 6; MVA, mevalonate; HMOX1, heme
oxygenase 1; Glu, glutamic acid; PE-PUFA, unsaturated fat
phospholipids; LCN2R, lipocalin-2 receptors; FPN, ferroportin. |
FSP1, a crucial modulator of ferroptosis, has
emerged as a key player in ferroptosis resistance, operating
independently of the GSH-GPX4 pathway. This protein primarily
blocks ferroptosis in tumor cells by regulating the oxidation of
NADPH, thereby hampering the efficacy of cancer treatment
strategies (17). Furthermore, FSP1
is highly expressed in various cancer cells and its expression has
been linked to poor prognosis (18,19).
Therefore, targeting FSP1 to inhibit its activity, thus promoting
ferroptosis, may be a promising approach for cancer therapy,
particularly in the case of difficult-to-treat tumors. This article
provides a comprehensive review of the ferroptosis pathway, the
mechanism by which FSP1 modulates ferroptosis and the potential of
FSP1-related molecular inhibitors in cancer treatment.
Molecular mechanism and pathway of
ferroptosis
Ferroptosis is a non-apoptotic form of
iron-dependent cell death. Key hallmarks of ferroptosis include
elevated iron levels, lipid peroxidation and disruption of the
mitochondrial architecture (20–22).
Ferroptosis has been associated with various factors, including
iron metabolism level, GPX4, GCH1/BH4, DHODH, FSP1 and MBOAT1/2
(Fig. 1). There is a significant
association between intracellular iron metabolism and ferroptosis.
Excessive iron loading can lead to ferroptosis. Transferrin
receptor 1, a marker of ferroptosis, has a pivotal role in
facilitating the transport of Fe3+ into cells (23). Within the cellular milieu, divalent
metal transporter 1 converts Fe3+ into Fe2+,
which is subsequently stored in ferritin. However, when
intracellular levels of labile Fe2+ become excessively
high, it reacts with hydroxyl radicals via the Fenton reaction,
triggering a surge in reactive oxygen species (ROS) production and
the accumulation of lipid peroxidation. These processes ultimately
induce ferroptosis (24).
Furthermore, a phospholipase present in the cytoplasm, namely
platelet-activating factor (PAF)-acetylhydrolase (II), specifically
inhibits short-chain fatty acid oxidation, interfering with the
cell's redox capacity and blocking the aggregation of oxidized
phospholipids such as PAF; this leads to membrane rupture,
inhibiting cell ferroptosis (25).
Acetyl-coenzyme A (CoA) of the mevalonate pathway inhibits
ferroptosis by triggering the NADPH-FSP1-CoQ10 pathway via the
regulation of CoQ10 synthesis. Furthermore, various types of fatty
acids have different degrees of regulatory roles in ferroptosis.
Among these, polyunsaturated fatty acids (PUFAs) are particularly
prone to peroxidation during ferroptosis. This process involves the
esterification of membrane-forming phospholipids with the
assistance of acyl-CoA synthetase long-chain family member 4, which
is a fatty acid-activating enzyme found in the endoplasmic
reticulum and outer mitochondrial membrane. In addition,
lysophosphatidylcholine acyltransferase (LPLAT) contributes to the
destruction of the lipid bilayer structure, leading to enhanced
membrane permeability and ultimately triggering ferroptosis
(26,27). Conversely, the inhibition of lipid
peroxidation has a pivotal role in preventing ferroptosis by
suppressing the formation of LPOs and free radicals. This involves
crucial pathways discussed in the following paragraphs.
System Xc-/GSH/GPX4 pathway
The system Xc-, GPX4 and GSH (system Xc-/GSH/GPX4)
pathways have been identified as crucial players in ferroptosis
(28,29) (Fig.
1). Among these pathways, GPX4 stands out as a principal
regulator of ferroptosis (30,31).
Inhibition GPX4 activity results in elevated intracellular lipid
peroxide (LPO) levels, which leads to ferroptosis (32–34).
GSH is a primary antioxidant that suppresses ferroptosis and acts
as a crucial cofactor for GPX4 (35). It reduces the iron concentration by
converting the peroxidised lipids accumulated during ferroptosis
into non-toxic isoalcohols (36,37).
In addition, GPX4 has a critical role in the repair of lipid
peroxides. In the presence of GSH, GPX4 reduces the accumulation of
lipid peroxidation. However, upon inactivation of GPX4 or depletion
of GSH, lipid peroxidation in the cell induces ferroptosis
(38,39).
GCH1/BH4 pathway
GCH1 and BH4 have a vital role in the process of
ferroptosis (40) (Fig. 1). BH4, which is derived from its
precursor GTP via three enzymatic reactions catalyzed by GCH1,
6-pyruboyl tetrahydrobiopterin synthase and sepiapterin reductase,
exhibits robust antioxidant activity both in vivo and in
vitro, directly protecting cells from lipid peroxidation. Among
these enzymes, GCH1 serves as the rate-limiting step, thus
determining the cellular resistance to ferroptosis to a certain
extent. Inhibition of GCH1 leads to a dysfunctional state of BH4
biosynthesis, resulting in ROS accumulation and ferroptosis
(41). Conversely, overexpression
of GCH1 has been shown to selectively enhance BH4 biosynthesis,
decrease ROS production and thereby inhibit ferroptosis (42).
Furthermore, BH4 pairs with dihydrobiopterin (BH2)
to establish a redox cycle that reduces endogenous oxygen radicals
and suppresses ferroptosis (43).
Dihydrofolate reductase (DHFR) controls the regeneration of BH2 to
BH4, in which NAD(P)H is the principal cofactor that supplements
BH4 but not BH2. Increased BH4 levels trigger cellular lipid
remodeling (42), which effectively
halts the utilization of phospholipids containing two
polyunsaturated fat acyl tails, thereby stalling ferroptosis
(44). In addition, BH4 enhances
the biosynthesis of CoQ10 by influencing the synthesis of its
precursor, 4-OH-benzoate. This connection bridges the GCH1-BH4-DHFR
pathway and the FSP1-CoQ10 axis, whose collaboration impedes the
progression of ferroptosis.
DHODH pathway
The DHODH is another pathway involved in cellular
resistance to ferroptosis (45)
(Fig. 1). DHODH is a
flavin-dependent protein, present in the inner mitochondrial
membrane, that converts DHO to orotate, reducing ubiquitin to
pantothenol and producing lipophilic antioxidants, which inhibits
LPO accumulation and ferroptosis (46). DHODH also exhibits a remarkable
synergistic interaction with mitochondrial GPX4, working in concert
to mitigate lipid peroxidation and forestall ferroptosis within the
mitochondrial inner membrane (47,48).
DHODH holds a pivotal position in the biosynthetic pathway of
pyrimidine nucleotides, playing a crucial role in the production of
DNA and RNA (49), Inhibition of
DHODH disrupts this pathway, leading to abnormalities in, or
cessation of, pyrimidine nucleotide synthesis. This, in turn,
results in a decrease in pyrimidine nucleotide availability,
halting the purine-pyrimidine pairing process and ultimately
impeding RNA synthesis (50). Given
RNA is a cofactor of GSH, a decrease in RNA causes an increase in
GSH levels. GPX4 restores LPO levels and the reduction of LPO
accumulation inhibits ferroptosis (51,52).
MBOAT1/2-MUFA pathway
Liang et al (53) discovered that membrane-bound MBOAT1
and MBOAT2 are novel inhibitors of sex hormone-dependent
ferroptosis. As LPLATs, they selectively introduce MUFAs to
lyso-phosphatidylethanolamine (lyso-PE), elevating the PE-MUFA
content and concurrently decreasing the levels of PE-PUFAs.
PE-PUFAs, being the preferred substrate for LPO, exert inhibitory
effects on cell ferroptosis through unsaturated fatty acid membrane
phospholipids (53,54). MBOAT1 and MBOAT2 are regulated by
estrogen receptor and androgen receptor, respectively. This
cellular defense mechanism against ferroptosis operates
independently of GPX4 and FSP1 (Fig.
1).
FSP1 mediated pathway
FSP1 possesses a myristylation sequence at its
N-terminal region and mutations within this sequence are implicated
in the induction of ferroptosis. CoQ10, alternatively known as
ubiquinone, is an essential component of lipid membranes. It
contributes to the production of ATP in the mitochondria and exists
in its reduced form. FSP1 inhibits ferroptosis by mediating the
NAD(P)H pathway and reducing NAD(P)H-dependent ubiquinone (oxidized
form of CoQ10) to ubiquinol (reduced form of CoQ10) (Fig. 1). FSP1 acts as a GSH-independent
ferroptosis suppressor and is compatible with GPX4 to inhibit lipid
peroxidation (55).
All of the abovementioned antioxidant pathways
inhibit the occurrence of ferroptosis to varying degrees (56,57).
FSP1 holds a prominent position, exhibiting profound ferroptosis
suppression. The mechanism by which FSP1 inhibits ferroptosis is
described in detail in the next chapter.
FSP1 and ferroptosis
AIF, a group of flavoproteins, possess the ability
to initiate caspase-independent apoptosis. The following isozymes
of AIF have been reported in humans: AIFM1, AIFM2 and AIFM3. Among
them, AIFM1-the most abundant isozyme-is initially translated and
then transported to the mitochondrial membrane (58). It folds on the mitochondrial
membrane and attains its functional conformation through the
assistance of flavin adenine dinucleotide (FAD). AIFM2 (also known
as FSP1) lacks a mitochondrial targeting sequence, preventing its
entry into mitochondria. Instead, it adheres to the outer
mitochondrial membrane and possesses an N-terminal myristylation
motif. FSP1 is a member of the nicotinamide adenine dinucleotide
II-H (NADH): The quinone oxidoreductase (NDH-2) family (59), named after NDH-1, which is complex I
of the respiratory chain. In fact, NDH-2 is thought to be a branch
of the traditional mitochondrial respiratory system, whose
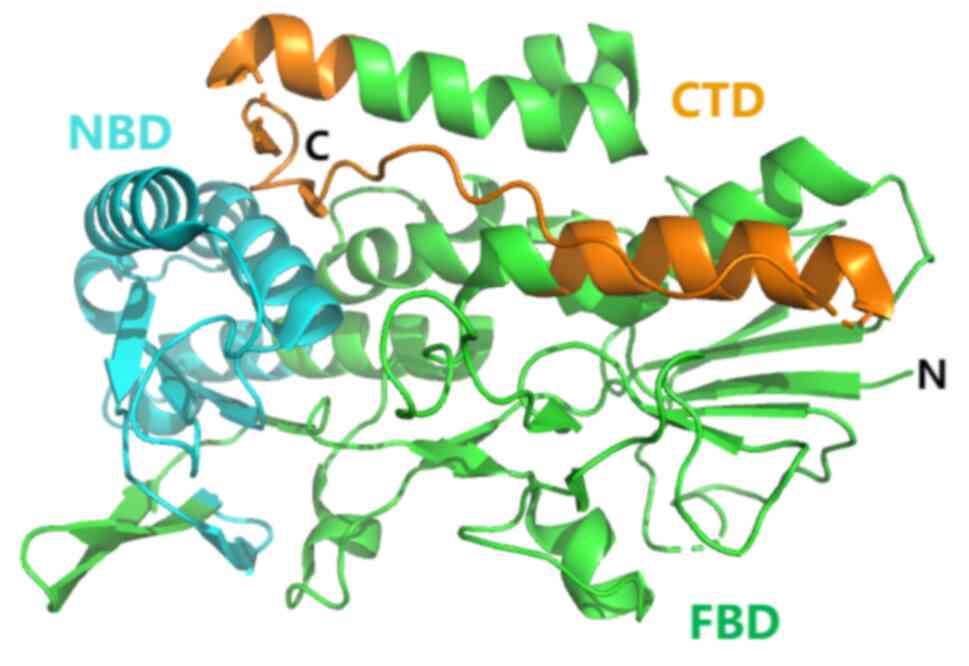
structure includes an N-terminal hydrophobic membrane domain (aa
1–27), NADH oxidoreductase domain (AA 81–285) and FAD domain (aa
286–308) (60) (Fig. 2). FSP1 is an important cellular
anti-ferroptotic factor that has a role in regulating iron
metabolism and protecting cells from iron-dependent death. The
antioxidant pathway mediated by FSP1 is parallel to the GSH-GPX4
pathway.
FSP1 exhibits oxidoreductase activity linked to
ferroptosis, encompassing NADP/NADPH-dependent CoQ10 oxidoreductase
and VK reductase activities (61).
The reductase activity is influenced by its cofactors and
substrates [such as NAD(P)H, FAD, CoQ10 and VK]. These cofactors
and substrates can reduce CoQ10 or VK to their respective forms,
including pantothenol and VK hydroquinone (VKH2). In addition, FSP1
mediates the production of compounds that function as antioxidants,
trapping free radicals and suppressing LPO accumulation in the cell
membrane, thus inhibiting ferroptosis in tumor cells (62). FSP1 inhibitors therefore have
potential therapeutic value in cancer treatment, either as
monotherapeutic agents or in combination with other ferroptosis
inducers, for refractory tumor management (19,63).
Therefore, FSP1 may represent a promising target for cancer therapy
(64).
FSP1 mediates FSP1-CoQ10-NAD(P)H and
FSP1-VK-NAD(P)H to inhibit ferroptosis
Zhang et al (65) demonstrated that FSP1 does not bind
NADH to reduce LPO levels. Instead, it specifically binds NADPH to
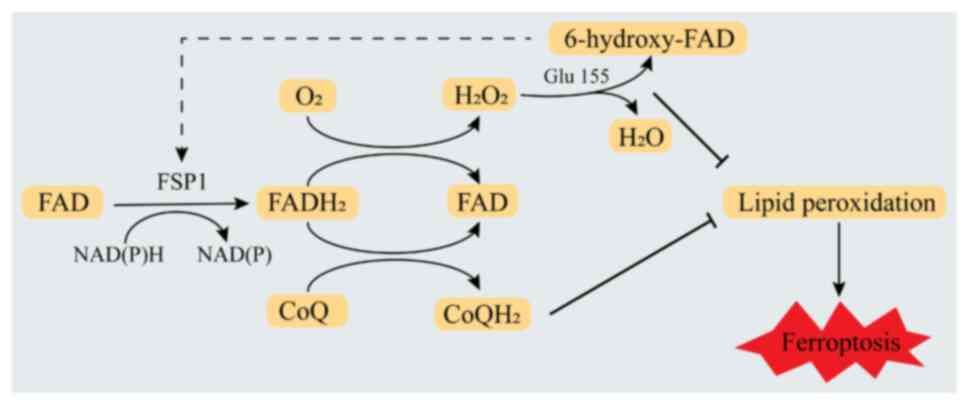
inhibit ferroptosis. Lv et al (66) proposed that when FSP1 binds FAD in
the presence of NAD(P)H and substrate (CoQ10 or oxygen), it first
accepts two electrons from NAD(P)H to form FADH2 and
then transfers two electrons to CoQ10 to form reduced CoQ10 or
transfers electrons to oxygen to form H2O2.
These two pathways cooperate to promote the oxidation of NAD(P)H.
Furthermore, under the condition that the NAD(P)H redox cycle is
widespread, the FAD bound by FSP1 in the presence of produced
hydrogen peroxide undergoes hydroxylation to form 6-hydroxy-FAD.
This reaction is catalyzed by Glu155 (Glu156 in human FSP1) within
a specific FAD hydroxylation pocket. Finally, 6-hydroxy-FAD serves
as a cofactor and potent antioxidant for FSP1, further enhancing
its catalytic activity. These pathways work together to mediate the
inhibition of ferroptosis by FSP1 (Fig.
3). These results indicate that FSP1 is an NADPH-selective
enzyme. Of note, the anti-ferroptotic activity of FSP1 is strictly
dependent on its affinity for NADPH. Its ferroptosis monitoring
system uses NADPH as an electron transport system to inhibit
ferroptosis through the oxidation of NADPH.
Guo et al (67) observed that FSP1 inhibits
ferroptosis by generating an antioxidant form of CoQ10, thereby
enhancing tumor cell resistance to ferroptosis; as a result, tumor
cells contain a lower concentration of the oxidized form of CoQ10
(68,69). However, the addition of exogenous
CoQ10 was not found to alleviate ferroptosis in FSP1-deficient
cells, indicating that exogenous CoQ10 does not directly affect
ferroptosis but rather regulates it indirectly through endogenous
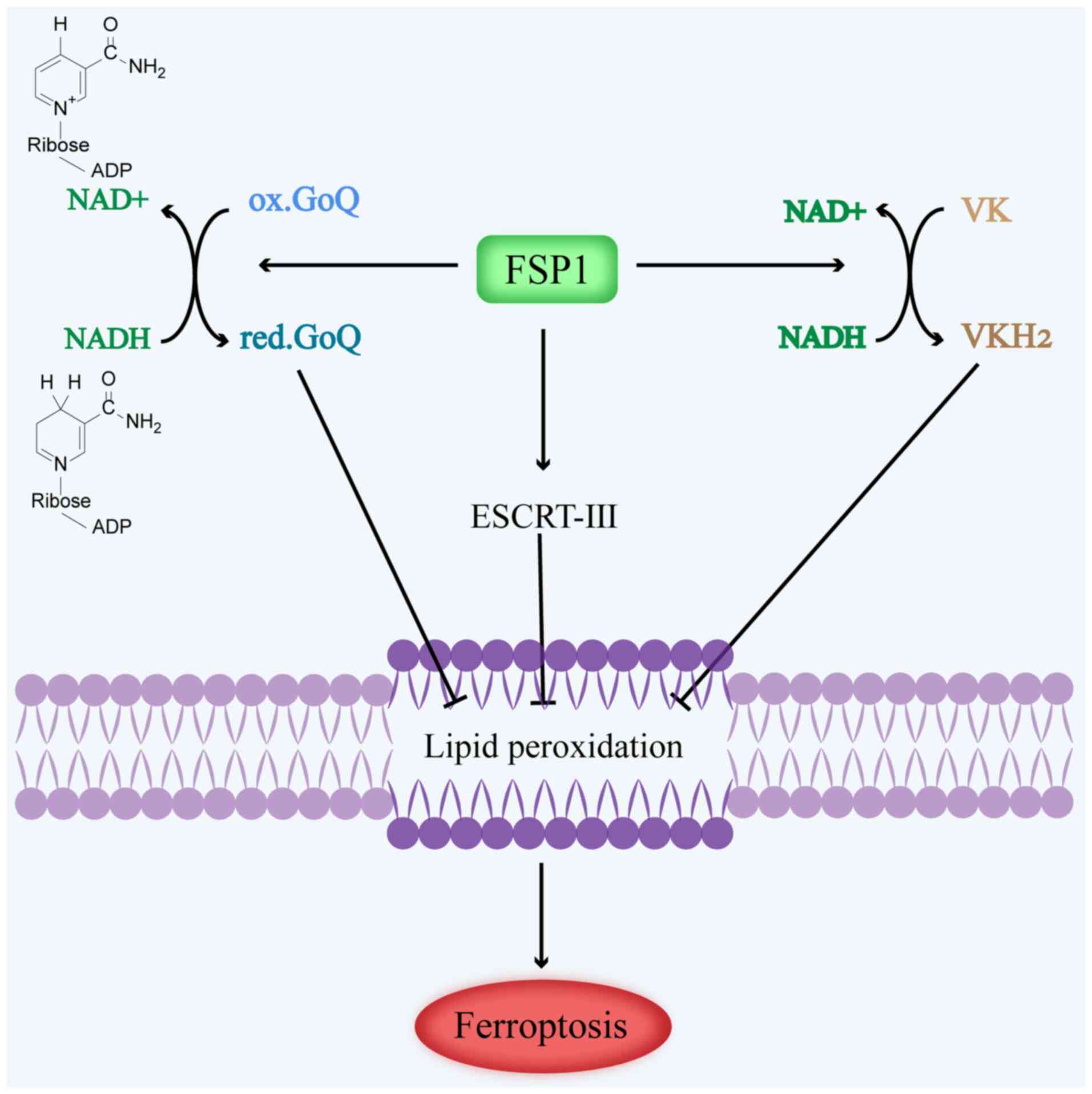
CoQ10. In summary, FSP1 offsets the loss of GPX4 by mediating the
conversion of NAD(P)H-dependent ubiquinone to ubiquinol; in turn,
pantothenol acts as a lipophilic antioxidant to prevent the
accumulation of lipid peroxidation (70), which inhibits ferroptosis in tumor
cells (Fig. 4).
VK comprises a group of lipophilic molecules
characterized by their 2-methyl-1,4-naphthoquinone structure and
polyisoprene side chains, which exhibit variable lengths and
hydrophobicity. VK exists in two forms in nature: The first, called
phylloquinone (also known as VK1), is found in photosynthetic
organisms, such as green plants, cyanobacteria and algae; the
other, namely menadione (also known as VK2), is found in animals
and bacteria. VK is a redox-active naphthoquinone (71,72),
which can be converted into its corresponding hydroquinone VKH2 and
is found mainly in the mature VK cycle (61,73).
Recently, Mishima et al (74) found that FSP1 reduces VK in the
manner of ubiquitin ketone. VKH2 exhibits remarkable antioxidant
properties, effectively trapping free radicals and inhibiting lipid
peroxidation, particularly that of phosphorus lipids (74). When recombinant human FSP1, NAD(P)H
and VK were cultured in vitro, a notable consumption of
NAD(P)H coincided with the generation of VK-hydroquinone. These
findings suggest that FSP1 serves as a reductase for VK, consuming
NAD(P)H and generating VKH2. This process not only blocks lipid
peroxidation but also inhibits ferroptosis (17,75).
In summary, FSP1-mediated VK reduction protects cells from harmful
lipid peroxidation and ferroptosis during the atypical VK cycle
(Fig. 4).
ESCRT-III-dependent membrane repair
ESCRT-III-dependent membrane repair is another
mechanism underlying FSP1-mediated ferroptosis resistance. A
crucial aspect of cell death involves damage to the plasma
membrane. ESCRT-III, a membrane repair-dependent endosomal sorting
complex critical for transportation, has a pivotal role in membrane
deformation and fission, exerting a regulatory function that
inhibits cancer cell ferroptosis (76). Studies have demonstrated that FSP1
can potentiate cell membrane repair and inhibit ferroptosis through
the ESCRT-III-dependent membrane repair pathway, independently of
CoQ10 (77). The presence of FSP1
has been shown to inhibit the occurrence of ferroptosis in tumor
cells after the induction of ferroptosis using a ferroptosis
inducer. Of note, FSP1 knockdown in cells resulted in
blockage of the RAS-selective lethal 3 (RSL3)-induced expression of
charged multivesicular protein (CHMP)5 and CHMP6 at the plasma
membrane. However, overexpression of CHMP5, a crucial subunit of
the ESCRT-III-dependent membrane repair pathway, was shown to
reverse ferroptosis induced by ferroptosis inducers and exert
protective effects against cell death in FSP1 knockdown
cells (78). These results suggest
that FSP1 mediates ferroptosis through the FSP1-ESCRT-III dependent
membrane repair pathway (Fig.
4).
Regulators control FSP1 levels to induce
ferroptosis
Nrf2 serves as a crucial regulator of cellular
antioxidant responses and the Nrf2 signaling pathway plays a
pivotal role in defending cells against lipid peroxidation and
ferroptosis. Nrf2 needs to be activated to function as an
antioxidant (79). The deficiency
or mutation of Kelch-like ECH-associated protein 1 (KEAP1),
mediated by p62, leads to the activation and upregulation of Nrf2.
In KEAP1-deficient lung cancer cells, the upregulated Nrf2
translocates to the nucleus and binds to antioxidant response
elements, activating genes critical for redox homeostasis.
Inhibition of the p62-KEAP1-Nrf2 antioxidant signaling pathway can
trigger ferroptosis, emphasizing the importance of this pathway in
ferroptosis regulation (80,81).
Nrf2 further plays a role in regulating the regeneration of the
redox electron donor NADPH. By blocking the NADPH redox cycle, Nrf2
mitigates the inhibitory effect of FSP1, thus limiting its ability
to induce ferroptosis (82). In
addition to Nrf2, p53 is another transcription factor for FSP1 that
regulates the level of FSP1 by binding to its promoter to exert its
role in suppressing ferroptosis (83).
Acetyltransferase 10 (NAT10) regulates the stability
and protein expression of FSP1 via modulation of the mRNA of the
N4-acetylcysteine site, which in turn affects the stability and
translation efficiency of the mRNA. Aberrant expression of NAT10
has been associated with tumorigenesis and progression. Of note,
NAT10 knockdown has been found to promote ferroptosis and inhibit
tumor proliferation and metastasis, indicating its significance in
tumor biology (84,85). These findings indicate that NAT10
regulates the stability of FSP1 mRNA to suppress
ferroptosis, with potential for the treatment of tumors.
Non-coding RNA (ncRNA) has a pivotal role in
regulating the expression and activity of FSP1 in cancer cells.
Various types of ncRNAs, including small, long and circular RNAs,
have been identified as targeting key genes and signaling pathways
that govern ferroptosis (86).
MicroRNA, a type of small ncRNA that regulates gene expression at
the post-transcriptional level, has been shown to repress the
expression of methyltransferase-like 3 in non-small-cell lung
carcinoma (NSCLC). This repression leads to the methylation of
N6-methyladenosine (m6A) and ultimately upregulation of FSP1
expression and inducing ferroptosis (87,88).
Ferroptosis-associated long ncRNA can bind directly to FSP1,
abrogating FSP1 ubiquitination degradation. This interaction lowers
the vulnerability of liver cancer cells to ferroptosis and reduces
the induction of ferroptosis (64,89,90).
Furthermore, the nuclear reader YTH
N6-methyladenosine RNA binding protein C1 (YTHDC1), which possesses
the YTH domain, has a role in nuclear m6A-tagged mRNAs and is
crucial for normal physiological development processes (91). Recently, Yuan et al (92) demonstrated that YTHDC1 levels are
inversely associated with the development and progression of lung
cancer, with this association being mediated by FSP1-induced
ferroptosis. When YTHDC1 was knocked down in A549 and H1299 cells,
they exhibited resistance to RSL3-induced ferroptosis but remained
sensitive to the FSP1 inhibitor iFSP1. This suggests that knockdown
of the YTHDC1 gene did not result in the upregulation of
both GPX4 and FSP1 mRNA expression in A549 and H1299
cells. Instead, it significantly increased the protein levels of
FSP1. However, in YTHDC1-knockdown cells, FSP1 mRNA levels
displayed a slight elevation, indicating that the regulation of
FSP1 protein levels by YTHDC1 occurs beyond the transcriptional
stage. Given that YTHDC1 functions as an m6A reader, analysis of
FSP1 mRNA levels revealed that most m6A sites reside in the
3′-untranslated region (UTR) of FSP1 mRNA. In both A549 and
H1299 cells, FSP1 mRNA expression is modulated by m6A, which
is recognized by YTHDC1. This interaction results in the formation
of an unstable FSP1 mRNA subtype with a truncated 3′-UTR.
Consequently, when YTHDC1 is downregulated, the FSP1 mRNA
subtype with a longer 3′-UTR predominates, leading to elevated FSP1
protein levels. This mechanism highlights the role of YTHDC1 in
suppressing FSP1 at the post-transcriptional level through
m6A-dependent modulation, thereby attenuating resistance to
ferroptosis and facilitating therapeutic intervention in lung
cancer (92).
FSP1 expression poses a significant impediment to
tumor treatment, as evidenced by the finding that its deletion in
cells results in slower tumor growth and proliferation. These
results suggest that the loss of FSP1 activity is sufficient to
trigger ferroptosis under specific in vivo conditions,
indicating its potential as a target for effective tumor cell
treatment (18,93,94).
Relationship between FSP1 inhibitors,
ferroptosis and tumor growth
Multiple studies have shown that inhibition of FSP1
enhances the antitumor efficacy of GPX4 inhibitors (95–97).
For instance, FSP1-mediated ferroptosis can be effectively targeted
for the treatment of challenging cancers, including triple-negative
breast cancer (TNBC), hepatocellular carcinoma (HCC), colon cancer,
acute lymphocyte leukemia and NSCLC. Laboratory studies have
further demonstrated that FSP1 inhibitors can increase cancer
cells' susceptibility to ferroptosis. Thus, FSP1 holds significant
potential as a target for tumor therapy, offering hope for more
effective treatment options.
FSP1 inhibitors are associated with
ferroptosis and cancer
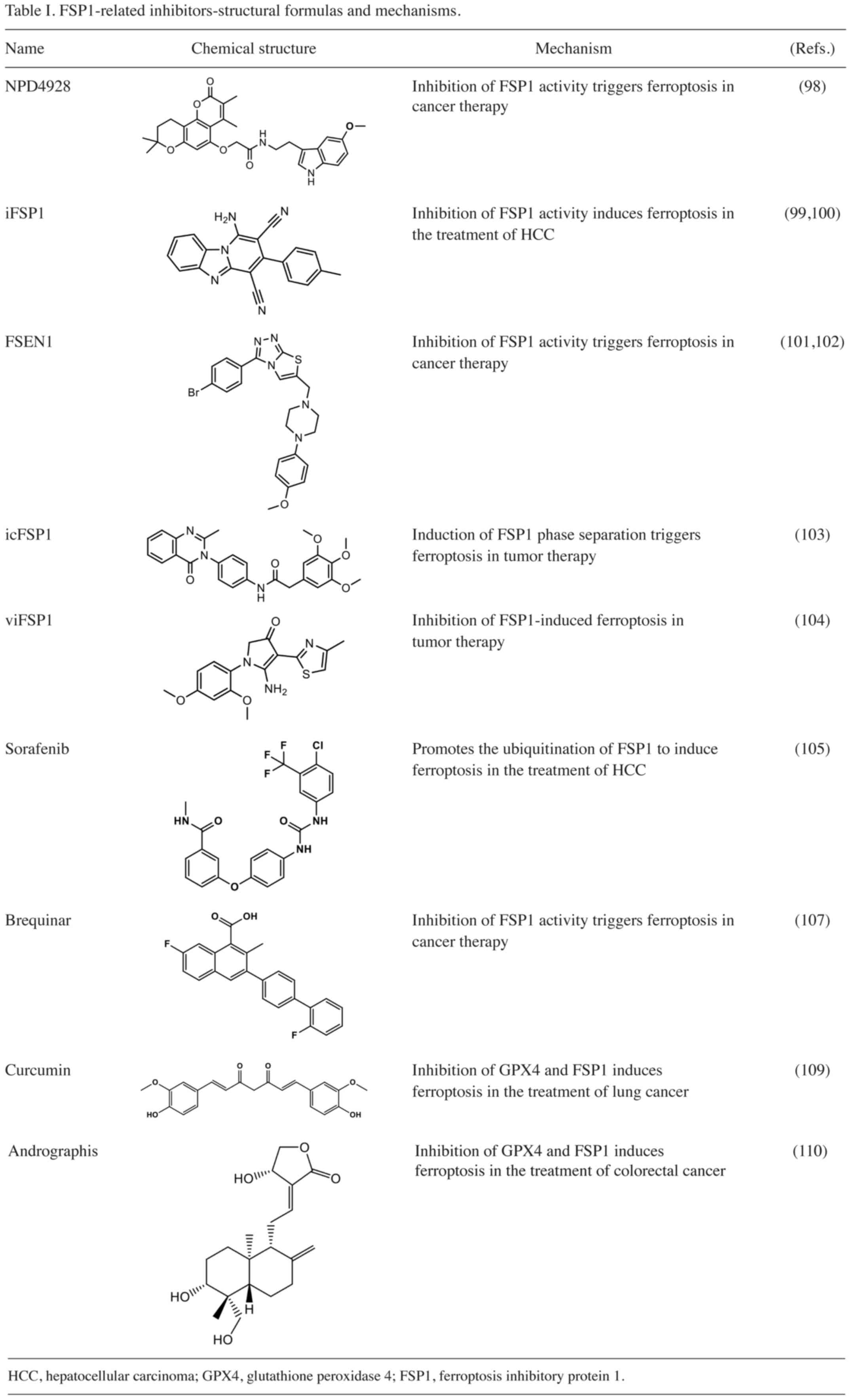
NPD4928 targets FSP1 to trigger ferroptosis in
cancer cells
Yoshioka et al (98) discovered that the combination of
NPD4928 and RSL3 enhances GPX4 inhibitor-induced ferroptosis,
similar to that observed with FSP1 knockdown (Table I). Of note, NPD4928 stabilizes
endogenous FSP1 in a dose-dependent manner by consuming NADH in
vitro. Thus, NPD4928 stabilizes endogenous FSP1 in a
dose-dependent manner, indicating that NPD4928-dependent
ferroptosis is primarily mediated through the inhibition of FSP1
activity. In addition, NPD4928 does not show any cytotoxicity
against the HCT116 cell line. However, it enhances ferroptosis in
cancer cell lines when used to treat colon cancer, indicating its
efficacy towards cancer cells. These findings underscore the
potential of the FSP1 inhibitor NPD4928 to overcome resistance to
ferroptosis in various types of cancer cell, thus emerging as a
promising candidate for the treatment of multiple drug-resistant
cancers.
iFSP1 targeting FSP1 triggers
ferroptosis in HCC cells
Lee et al (99) proposed a strong connection between
FSP1 overexpression and HCC, indicating that elevated FSP1 levels
are associated with shorter overall survival among patients. iFSP1,
a specific inhibitor of FSP1, effectively triggers ferroptosis in
hepatoma cells by attracting immune cells. This approach has been
shown to reduce HCC cell counts and enhance immune infiltration of
dendritic cells, macrophages and T cells (Table I). When tested on MHCC97L cells at
various concentrations, iFSP1 was shown to effectively suppress
tumor-cell proliferation without causing significant changes in
body weight. Treatment with iFSP1 also resulted in modifications to
the immune landscape of HCC tumor cells. Therefore, iFSP1, as an
FSP1 inhibitor, holds immense potential as a superior therapeutic
agent for the treatment of HCC (99,100).
Ferroptosis sensitizer 1 (FSEN1)
triggers ferroptosis in cancer cells by mediating FSP1
Hendricks et al (101) discovered that FSEN1 is an
inhibitor of FSP1 and exerts synergistic effects with multiple
ferroptosis inducers (Table I).
Their findings demonstrated that FSEN1 acts specifically through
FSP1. To induce ferroptosis in H460C Cas9 cells, an
optimal dosage of FSEN1 at 0.55 mM and RSL3 at 0.55 mM was
identified. This dosage achieved EC50 values of 69.363
nM in H460C GPX4 knockdown cells. Pharmacokinetics
analysis conducted in mice revealed a plasma half-life of 8 h and
an intrinsic clearance rate of 11.54 ml/min/mg in mouse liver
microsomes. These findings suggest improved metabolic stability of
FSEN1 in vivo (102).
Evaluation of the impact of FSEN1 on cell ferroptosis triggered by
GPX4 inhibitors across diverse cancer cell lines, including lung,
liver and breast cancer cells, revealed that FSEN1 enhances the
sensitivity of cancer cells, to varying degrees, towards
RSL3-induced ferroptosis. Furthermore, preclinical studies have
demonstrated synergistic effects when FSEN1 was used alongside
endoperoxide-derived ferroptosis inducers, such as
dihydroartemisinic acid (101).
These findings provide further evidence supporting the potential of
FSEN1 as an FSP1 inhibitor in cancer therapy.
icFSP1 targets phase separation of
FSP1 to trigger ferroptosis in cancer cells
Nakamura et al (103) found that icFSP1 exhibits
specificity towards ferroptosis and does not stimulate the
myristylation of FSP1 in vitro (Table I). Their findings suggest that
icFSP1 may regulate FSP1-membrane interactions, inducing the
production of condensates that lead to reduced membrane-binding
affinity of FSP1. Furthermore, the induction of FSP1 condensate
formation within tumor cells triggers the phase separation of FSP1,
ultimately inducing ferroptosis in cancer cells. the
EC50 value of icFSP1, measured in Pfa1 cells, was found
to be 0.21 µM. Treatment with the FSP1 inhibitor icFSP1 impaired
tumor-cell growth and significantly reduced tumor weight. However,
at higher concentrations, there was no significant impact on tumor
weight, indicating no off-target effects. Furthermore, icFSP1
significantly improved microsomal stability and maximum tolerated
dose in mouse plasma compared with iFSP1. The stronger
pharmacokinetic and metabolic stability of icFSP1 renders it
suitable for use in vivo (103), making it a promising candidate for
targeting FSP1-dependent phase separation in anticancer therapy.
These findings provide the basis for targeting FSP1-dependent phase
separation as an effective anticancer therapy.
viFSP1 targets FSP1 to treat
tumor
Nakamura et al (104) proposed that the treatment of Pfa1
cells expressing mouse FSP1 or human FSP1 with viFSP1, a
multifunctional inhibitor of FSP1, leads to ferroptosis, which can
be reversed by administration of the ferroptosis inhibitor
liproxstatin-1 (Table I),
confirming that the dependence of viFSP1 on FSP1 causes
ferroptosis. Specifically, viFSP1 targets the NADH binding pocket
around residues A328, F294, M327 and T1 in FSP1. In addition, both
human and mouse FSP1 cells exhibited similar sensitivity to viFSP1
with no significant difference in the calculated IC50
values. Thus, viFSP1 emerges as a species-independent direct
inhibitor of FSP1, with an EC50 value of 170 nM in Pfa1
cells. In multiple human and mouse cancer cell lines, as well as in
rat fibroblasts and Pfa1 cells overexpressing FSP1, the concurrent
use of viFSP1 and the GPX4 inhibitor RSL3 synergistically enhanced
ferroptosis induction, suggesting its potential therapeutic utility
in tumor treatment (104).
Sorafenib mediates the ubiquitination
and degradation of FSP1 to induce ferroptosis in the treatment of
HCC
Ubiquitination, a form of posttranslational
modification, has a crucial role in regulating cellular processes.
Sorafenib, a multi-target kinase inhibitor, inhibits cell
proliferation by interfering with serine-threonine kinase-related
signaling pathways and hinders tumor angiogenesis by targeting
specific tyrosine kinases (Table
I). In HCC cells, sorafenib has been demonstrated to induce
ferroptosis (105). Lai et
al (106) revealed that
sorafenib regulates the interaction between tripartite motif
containing 54 and FSP1 through the MAPK/ERK kinase pathway,
promoting the ubiquitination of FSP1. Elevated FSP1 expression
partially reverses sorafenib-induced ferroptosis, whereas FSP1
inhibition enhances HCC cell sensitivity to sorafenib-induced
ferroptosis (106). Thus, high
levels of FSP1 inhibit sorafenib-induced ferroptosis in HCC cells
(95). This suggests that
downregulation of FSP1 in HCC cells may promote sorafenib-induced
ferroptosis to facilitate therapeutic effects against HCC.
Brequinar, a DHODH inhibitor, inhibits
FSP1-induced ferroptosis to treat tumors
Mishima et al (107), proposed that the DHODH inhibitor
brequinar suppresses FSP1 activity at elevated dosages, thereby
mitigating resistance to ferroptosis (Table I). Even in the absence of DHODH,
high concentrations of brequinar were shown to effectively induce
ferroptosis in Pfa1 cells derived from GPX4-deficient mouse
fibroblasts. However, in FSP1-knockdown cells, ferroptosis was not
induced, which was in contrast to the effects observed with
BAY-2402234, another DHODH inhibitor that lacks inhibitory activity
against FSP1 (108). These
findings suggest that the ferroptosis-inducing effect of brequinar
is mediated by the inhibition of FSP1 rather than DHODH.
Curcumin inhibits apoptosis of lung
cancer cells induced by GPX4 and FSP1
Zhou et al (109) demonstrated that curcumin has the
capacity to induce ferroptosis in highly tumorigenic A549 CD133+
cells via the GSH-GPX4 and FSP1-COQ10-NADH signaling pathways
(Table I). Treatment with curcumin
resulted in a dose-dependent decrease in the activity of
CD133+ cells and the expression levels of GPX4 and FSP1
protein. The levels of ROS, GSH, CoQ10 and NAD+/NADH in the
curcumin-treated group were significantly lower than in the control
group; this effect was attenuated by ferroptosis inhibitor Fer-1.
Furthermore, the IC50 value of curcumin was determined
to be 36 µM, indicating its high potency. In addition, curcumin
demonstrated robust stability in vivo. Upon curcumin
treatment, tumor growth was effectively suppressed, tumor pathology
improved and the expression of Ki-67, a marker of tumor
proliferation, was notably downregulated. Furthermore, the
inhibitory effect of curcumin was higher than that of RSL3 and
iFSP1. However, Fer-1 significantly inhibited the antitumor effects
of curcumin (109). Thus, curcumin
can inhibit GPX4 and FSP1 and promote ferroptosis to treat
tumors.
Andrographis inhibits GPX4 and FSP1 to
induce ferroptosis in colorectal cancer cells
Miyazaki et al (110) demonstrated that Andrographis
exhibited comparable pharmacological effects to curcumin,
effectively suppressing the activities of GPX4 and FSP1, thereby
inducing ferroptosis in colorectal cancer cells (Table I). Treatment of SW480 and HCT116
cells with Andrographis significantly reduced cell viability as
well as GPX4 and FSP1 expression; these effects were comparable to
those elicited by RSL3 or iFSP1. In addition, the IC50
of Andrographis in both cell lines was 40 µg/ml, indicating its
stability and reliability in vivo. Treatment with
Andrographis was shown to inhibit the growth of cancer cells, with
a reduction in invasiveness. Furthermore, the drug had no adverse
effect on body weight. Fer-1 was found to mitigate the inhibitory
action of Andrographis on colorectal cancer. The combination of
Andrographis and curcumin exhibited a superior inhibitory effect
against GPX4 and FSP1 to either drug alone.
Material complexes acting on
FSP1-related pathways to treat tumors
Metabolic intervention nanoparticles Cu-silk
fibroin (SF)-Rosuvastatin (RSV) nanoparticles (NPs) for treating
TNBC through FSP1
Yang et al (111) effectively utilized SF to form a
complex in coordination with Cu2+ ions. Rosuvastatin
(RSV) was then encapsulated within this complex, yielding Cu-SF-RSV
NPs, which were used to treat TNBC by overcoming FSP1-mediated
ferroptosis. RSV was shown to hinder valproic acid metabolism,
resulting in reduced CoQH2 levels and disruption of the redox
balance. This, in turn, attenuated FSP1′s inhibitory effect on
ferroptosis. In addition, Cu2+ triggered the Fenton
reaction to generate ROS, and SF consumed GSH in cells, thus
promoting redox stress and amplifying the effect of
Cu2+. The accumulation and retention of Cu-SF-RSV NPs in
tumor tissues markedly suppressed TNBC growth and metastasis. In
addition, the plasma half-life of Cu-SF-RSV NPs in vivo was
prolonged (7.03±1.18 h) compared with that of free RSV (0.42±0.02
h), indicating an extended blood circulation time for these
nanomaterials. These results suggest that Cu-SF-RSV NPs may
effectively eradicate TNBC tumors and inhibit tumor metastasis. Of
note, compared to RSV, they do not induce histological damage or
morphological alterations in various organs, with less systemic
toxicity (111). Therefore,
metabolic intervention with Cu-SF-RSV NPs may be a promising
treatment for TNBC.
Multienzyme-like reactivity
cooperatively impairs GPX4 and FSP1 pathways to induce ferroptosis
in TNBC
Li et al (112) employed [Fe (III)] Cu-tetra
(4-carboxyphenyl) porphyrin chloride [Cu-TCPP (Fe)] metal organic
framework (MOF) nanosheets as substrates for the deposition of Au
NPs through in situ nucleation. Subsequently, they
successfully deposited the ferroptosis inducer RSL3 onto the
surface of these NPs via π-π stacking interactions to obtain NPs
capable of targeting ferroptosis for TNBC therapy. Furthermore,
they enhanced the system by integrating a long-circulating
polyvinyl sugar segment and a tumor-targeting iRGD ligand (112). Liu et al (113) demonstrated that Au NPs possess
similar activity to glucose oxidase, which oxidized glucose into
gluconic acid, effectively depleting glucose levels, disrupting the
pentose phosphate pathway and inhibiting GSH biosynthesis. This
prevents the conversion of CoQ10 to CoQ10H2, leading to an
elevation in the NADP+/NADPH ratio and effectively suppressing the
FSP1-CoQ10H2 pathway. At the same time, increased
NADP+/NADPH ratios further inhibit the conversion of
cystine to cysteine, reducing GSH and GPX4 biosynthesis.
Furthermore, Yuan et al (114) showed that Cu2+
immobilized within the TCPP MOF nanoplates exhibited the ability to
oxidize GSH into GSH oxide, depleting the cofactor essential for
GPX4 activity. This process attenuated GPX4′s inhibitory function
against ferroptosis, effectively blocking the GPX4/GSH pathway. In
addition, this nanosystem possessed the ability to release
Cu2+, further enhancing its inhibitory effect against
the GPX4/GSH pathway. Studies have shown that
Au/Cu-TCPP-Fe-polyethylene glycol (PEG) exhibits a prolonged blood
circulation time and an enhanced tumor-targeting effect in
vivo. Mice treated with Au/Cu-TCPP (Fe)@RSL3-PEG-iRGD had the longest
survival with a median survival time of 60 days, and only a small
number of tumor cells retaining proliferative activity. In
addition, these nano-tablets demonstrate excellent tolerability
in vivo, even at very high dosages. Thus, multienzyme-like
reactions of NPs can simultaneously inhibit GPX4 and the
FSP1-CoQ10H2 pathway in TNBC cells to promote ferroptosis,
providing a promising avenue for the clinical treatment of
drug-resistant tumors.
Photo-enhanced synergistic induction
of ferroptosis used in anti-tumor immunotherapy
Yang et al (115) utilized photodynamic therapy to
target FSP1-mediated ferroptosis. They employed a photoresponsive
nanocomposite composed of boron-dipyrromethene (BODIPY)-modified
polyamide (amidoamine) (BMP), which was used to encapsulate iFSP1
and chlorin e6 (Ce6); this was facilitated by π-π stacking and
hydrophobic interactions between Ce6 and BODIPY. Thus, BMP and
ferroptosis inducers were integrated into NPs. Under light
irradiation, these NPs effectively triggered ferroptosis both in
vitro and in vivo, leading to a significant slowing of
tumor growth. Ferroptosis directly promotes immunogenic cell death,
a process in which cells release ATP and high mobility group box 1
proteins, while exposing calreticulin on their surface. During this
process damage-associated molecular patterns mediate the
recruitment and activation of dendritic cells, thereby facilitating
T-cell infiltration and cytotoxicity. It was suggested that IFN-γ
secreted by T cells can trigger LPO of tumor cells; however, in the
absence of NPs, cell death is not elicited, particularly when GPX4
and FSP1 are inhibited. In addition, pharmacokinetics studies have
shown that the half-life of Ce6 is prolonged when it is
encapsulated within NPs, and furthermore, during treatment, the
drug did not accumulate in normal organs, with stable body weights
maintained across groups. Of note, no significant histological
damage or morphological alterations were observed in any of the
organs. Therefore, light-responsive nanocomposites have the
potential to synergistically induce ferroptosis in cancer
immunotherapy.
Discussion and outlook
The exploitation of ferroptosis, a type of
programmed cell death, holds great promise for various cancer
therapies. Ferroptosis inducers heighten tumor cell sensitivity
towards chemotherapeutic agents and facilitate the elimination of
tumor cells that are refractory to other programmed cell death
modalities. Consequently, the induction of ferroptosis has emerged
as a promising approach for treating refractory tumors. Although
numerous ferroptosis inducers have been identified, the primary
focus has been limited to targeting the system xc-/GSH/GPX4
pathway. This limited perspective has somewhat hindered the
exploration of the ferroptosis regulatory system as a target for
cancer therapy.
FSP1, a linchpin anti-ferroptosis factor, enhances
cancer-cell resistance to ferroptosis, making it an attractive
target for cancer therapy. The current review reported on FSP1
inhibitors that boost cancer-cell sensitivity to ferroptosis. These
inhibitors exhibit pharmacokinetic and metabolic stability,
suitable for in vivo studies, and have been shown to
effectively block the FSP1-mediated cellular defense mechanism
against ferroptosis. Among them, the FSP1 inhibitors mentioned
above have demonstrated remarkable potency in overcoming
ferroptosis resistance in several cancer cell lines, potentially
enabling the development of more effective cancer therapies.
Although FSP1 inhibitors may potentially have a
significant role in cancer treatment, several challenges remain to
be tackled. First, the specific mechanism by which FSP1 regulates
ferroptosis remains unclear, necessitating further research.
Although FSP1 is highly expressed in a variety of tumors, it is
also expressed in normal tissues, making target selection a
challenge. Furthermore, the association between FSP1 expression
levels and tumor malignancy as well as poor prognosis further
complicates treatment strategies. Third, research pertaining to
FSP1 inhibitors is currently in its early stages. Although
preliminary experimental studies have yielded encouraging outcomes,
extensive further exploration is imperative to establish their
clinical utility. Fourth, the number of reported FSP1 inhibitors is
limited and it is crucial to develop more drug molecular structures
through natural drug screening and AI technology. The goal is to
develop inhibitors that are highly selective, exhibit low toxicity
and possess desirable pharmaceutical characteristics. In addition,
further research is required to investigate the potential clinical
application of combinations of FSP1 inhibitors with other
antineoplastic agents and immune checkpoint inhibitors. Moreover,
nanomaterials hold immense promise in this domain, offering novel
avenues for research.
In conclusion, although FSP1 inhibitors
demonstrated tremendous potential in tumor therapy, further
research is necessary to address existing challenges and optimize
the application of these compounds in clinical practice.
Acknowledgements
Not applicable.
Funding
This review was supported by grants from Shaanxi Provincial
Health Research (grant no. 2022E010), Shaanxi Provincial Education
Department Scientific Research Program (grant nos. 22JK0612 to XLL
and 23JK0731 to YX), the Natural Science Foundation of Shaanxi
Province (grant no. 2023-JC-QN-0125 to XLL) and Yan'an Science and
Technology Bureau (grant nos. 2023-SFGG-141 to LXL and
2023-SFGG-091 to YX).
Availability of data and materials
Not applicable.
Authors' contributions
QD and XL were responsible for writing and revising
the manuscript. YL was responsible for the preparation of figures
and revisions of this article. XW, JZ and YY were responsible for
the revisions of this article. DZ was responsible for data
collection. YX was responsible for revising the article. XL was
responsible for the conceptualization of the study, obtained
funding and provided guidance in the preparation of the article.
All authors contributed to the article and have read and approved
the submitted version. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD, Wagle NS and Jemal
A: Cancer statistics, 2023. CA Cancer J Clin. 73:17–48. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee S, Hwang N, Seok BG, Lee S, Lee SJ and
Chung SW: Autophagy mediates an amplification loop during
ferroptosis. Cell Death Dis. 14:4642023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kinowaki Y, Taguchi T, Onishi I, Kirimura
S, Kitagawa M and Yamamoto K: Overview of ferroptosis and synthetic
lethality strategies. Int J Mol Sci. 22:92712021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alborzinia H, Chen Z, Yildiz U, Freitas
FP, Vogel FCE, Varga JP, Batani J, Bartenhagen C, Schmitz W, Büchel
G, et al: LRP8-mediated selenocysteine uptake is a targetable
vulnerability in MYCN-amplified neuroblastoma. EMBO Mol Med.
15:e180142023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Floros KV, Cai J, Jacob S, Kurupi R,
Fairchild CK, Shende M, Coon CM, Powell KM, Belvin BR, Hu B, et al:
MYCN-amplified neuroblastoma is addicted to iron and vulnerable to
inhibition of the system Xc-/glutathione axis. Cancer Res.
81:1896–7908. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu Y, Yang Q, Su Y, Ji Y, Li G, Yang X, Xu
L, Lu Z, Dong J, Wu Y, et al: MYCN mediates TFRC-dependent
ferroptosis and reveals vulnerabilities in neuroblastoma. Cell
Death Dis. 12:5112021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lei G, Zhuang L and Gan B: Targeting
ferroptosis as a vulnerability in cancer. Nat Rev Cancer.
22:381–396. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Labrie M, Brugge JS, Mills GB and
Zervantonakis IK: Therapy resistance: Opportunities created by
adaptive responses to targeted therapies in cancer. Nat Rev Cancer.
22:323–339. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cai Y, Lv L, Lu T, Ding M, Yu Z, Chen X,
Zhou X and Wang X: α-KG inhibits tumor growth of diffuse large
B-cell lymphoma by inducing ROS and TP53-mediated ferroptosis. Cell
Death Discov. 9:1822023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhu X and Li S: Ferroptosis, necroptosis,
and pyroptosis in gastrointestinal cancers: The chief culprits of
tumor progression and drug resistance. Adv Sci (Weinh).
10:e23008242023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu L, Jin H, Dong M, Tian J, Li H, Liu Q,
Chen Y and Zou Z: Identification of ferroptosis-related signature
with potential implications in prognosis and immunotherapy of renal
cell carcinoma. Apoptosis. 27:946–960. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang W, Jiang B, Liu Y, Xu L and Wan M:
Bufotalin induces ferroptosis in non-small cell lung cancer cells
by facilitating the ubiquitination and degradation of GPX4. Free
Radic Biol Med. 180:75–84. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang X, Niu Y, Jian J, Guo Y, Wang Y, Zhu
Y and Liu B: Potential applications of ferroptosis inducers and
regulatory molecules in hematological malignancy therapy. Crit Rev
Oncol Hematol. 193:1042032024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang L, Hobeika CS, Khabibullin D, Yu D,
Filippakis H, Alchoueiry M, Tang Y, Lam HC, Tsvetkov P, Georgiou G,
et al: Hypersensitivity to ferroptosis in chromophobe RCC is
mediated by a glutathione metabolic dependency and cystine import
via solute carrier family 7 member 11. Proc Natl Acad Sci USA.
119:e21228401192022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Alborzinia H, Flórez AF, Kreth S, Brückner
LM, Yildiz U, Gartlgruber M, Odoni DI, Poschet G, Garbowicz K, Shao
C, et al: MYCN mediates cysteine addiction and sensitizes
neuroblastoma to ferroptosis. Nat Cancer. 3:471–485. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li W, Liang L, Liu S, Yi H and Zhou Y:
FSP1: A key regulator of ferroptosis. Trends Mol Med. 29:753–764.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Emmanuel N, Li H, Chen J and Zhang Y:
FSP1, a novel KEAP1/NRF2 target gene regulating ferroptosis and
radioresistance in lung cancers. Oncotarget. 13:1136–1139. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
He X, Liang SM, Wang HQ, Tao L, Sun FF,
Wang Y, Zhang C, Huang YC, Xu DX and Chen X: Mitoquinone protects
against acetaminophen-induced liver injury in an FSP1-dependent and
GPX4-independent manner. Toxicol Appl Pharmacol. 465:1164522023.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang H, Zhang Z, Ruan S, Yan Q, Chen Y,
Cui J, Wang X, Huang S and Hou B: Regulation of iron metabolism and
ferroptosis in cancer stem cells. Front Oncol. 13:12515612023.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dixon SJ and Pratt DA: Ferroptosis: A
flexible constellation of related biochemical mechanisms. Mol Cell.
83:1030–1042. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Guo R, Duan J, Pan S, Cheng F, Qiao Y,
Feng Q, Liu D and Liu Z: The road from AKI to CKD: Molecular
mechanisms and therapeutic targets of ferroptosis. Cell Death Dis.
14:4262023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Feng H, Schorpp K, Jin J, Yozwiak CE,
Hoffstrom BG, Decker AM, Rajbhandari P, Stokes ME, Bender HG, Csuka
JM, et al: Transferrin receptor is a specific ferroptosis marker.
Cell Rep. 30:3411–3423.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen H, Wang C, Liu Z, He X, Tang W, He L,
Feng Y, Liu D, Yin Y and Li T: Ferroptosis and Its multifaceted
role in cancer: Mechanisms and therapeutic approach. Antioxidants
(Basel). 11:15042022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Q, Sun T, Yu F, Liu W, Gao J, Chen
J, Zheng H, Liu J, Miao C, Guo H, et al: PAFAH2 suppresses
synchronized ferroptosis to ameliorate acute kidney injury. Nat
Chem Biol. Jan 29–2024.(Epub ahead of print).
|
|
26
|
Wang D, Tang L, Zhang Y, Ge G, Jiang X, Mo
Y, Wu P, Deng X, Li L, Zuo S, et al: Regulatory pathways and drugs
associated with ferroptosis in tumors. Cell Death Dis. 13:5442022.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tian X, Li S and Ge G: Apatinib promotes
ferroptosis in colorectal cancer cells by targeting ELOVL6/ACSL4
signaling. Cancer Manag Res. 13:1333–1342. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Y, Zheng L, Shang W, Yang Z, Li T,
Liu F, Shao W, Lv L, Chai L, Qu L, et al: Wnt/beta-catenin
signaling confers ferroptosis resistance by targeting GPX4 in
gastric cancer. Cell Death Differ. 29:2190–2202. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ye Y, Chen A, Li L, Liang Q, Wang S, Dong
Q, Fu M, Lan Z, Li Y, Liu X, et al: Repression of the antiporter
SLC7A11/glutathione/glutathione peroxidase 4 axis drives
ferroptosis of vascular smooth muscle cells to facilitate vascular
calcification. Kidney Int. 102:1259–1275. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Krümmel B, Plötz T, Jörns A, Lenzen S and
Mehmeti I: The central role of glutathione peroxidase 4 in the
regulation of ferroptosis and its implications for pro-inflammatory
cytokine-mediated beta-cell death. Biochim Biophys Acta Mol Basis
Dis. 1867:1661142021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu S, Zhang HL, Li J, Ye ZP, Du T, Li LC,
Guo YQ, Yang D, Li ZL, Cao JH, et al: Tubastatin A potently
inhibits GPX4 activity to potentiate cancer radiotherapy through
boosting ferroptosis. Redox Biol. 62:1026772023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nishida Xavier da Silva T, Friedmann
Angeli JP and Ingold I: GPX4: Old lessons, new features. Biochem
Soc Trans. 50:1205–1213. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen T, Leng J, Tan J, Zhao Y, Xie S, Zhao
S, Yan X, Zhu L, Luo J, Kong L and Yin Y: Discovery of novel potent
covalent glutathione peroxidase 4 inhibitors as highly selective
ferroptosis inducers for the treatment of triple-negative breast
cancer. J Med Chem. 66:10036–10059. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li D, Zhang M and Chao H: Significance of
glutathione peroxidase 4 and intracellular iron level in ovarian
cancer cells-‘utilization’ of ferroptosis mechanism. Inflamm Res.
70:1177–1189. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ursini F and Maiorino M: Lipid
peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic
Biol Med. 152:175–185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Rochette L, Dogon G, Rigal E, Zeller M,
Cottin Y and Vergely C: Lipid peroxidation and iron metabolism: Two
corner stones in the homeostasis control of ferroptosis. Int J Mol
Sci. 24:4492022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhang Y, Swanda RV, Nie L, Liu X, Wang C,
Lee H, Lei G, Mao C, Koppula P, Cheng W, et al: mTORC1 couples
cyst(e)ine availability with GPX4 protein synthesis and ferroptosis
regulation. Nat Commun. 12:15892021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen M, Shi Z, Sun Y, Ning H, Gu X and
Zhang L: Prospects for anti-tumor mechanism and potential clinical
application based on glutathione peroxidase 4 mediated ferroptosis.
Int J Mol Sci. 24:16072023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jiang Y, Zhao J, Li R, Liu Y, Zhou L, Wang
C, Lv C, Gao L and Cui D: CircLRFN5 inhibits the progression of
glioblastoma via PRRX2/GCH1 mediated ferroptosis. J Exp Clin Cancer
Res. 41:3072022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hu Q, Wei W, Wu D, Huang F, Li M, Li W,
Yin J, Peng Y, Lu Y, Zhao Q and Liu L: Blockade of GCH1/BH4 axis
activates ferritinophagy to mitigate the resistance of colorectal
cancer to erastin-induced ferroptosis. Front Cell Dev Biol.
10:8103272022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kraft VAN, Bezjian CT, Pfeiffer S,
Ringelstetter L, Müller C, Zandkarimi F, Merl-Pham J, Bao X,
Anastasov N, Kössl J, et al: GTP cyclohydrolase
1/tetrahydrobiopterin counteract ferroptosis through lipid
remodeling. ACS Cent Sci. 6:41–53. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Soula M, Weber RA, Zilka O, Alwaseem H, La
K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA and Birsoy K:
Metabolic determinants of cancer cell sensitivity to canonical
ferroptosis inducers. Nat Chem Biol. 16:1351–1360. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lv Y, Wu M, Wang Z and Wang J:
Ferroptosis: From regulation of lipid peroxidation to the treatment
of diseases. Cell Biol Toxicol. 39:827–851. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang S, Kang L, Dai X, Chen J, Chen Z,
Wang M, Jiang H, Wang X, Bu S, Liu X, et al: Manganese induces
tumor cell ferroptosis through type-I IFN dependent inhibition of
mitochondrial dihydroorotate dehydrogenase. Free Radic Biol Med.
193:202–212. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Amos A, Amos A, Wu L and Xia H: The
Warburg effect modulates DHODH role in ferroptosis: A review. Cell
Commun Signal. 21:1002023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee
H, Koppula P, Wu S, Zhuang L, Fang B, et al: DHODH-mediated
ferroptosis defence is a targetable vulnerability in cancer.
Nature. 593:586–590. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wang F and Min J: DHODH tangoing with GPX4
on the ferroptotic stage. Signal Transduct Target Ther. 6:2442021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Desler C, Durhuus JA, Hansen TL, Anugula
S, Zelander NT, Bøggild S and Rasmussen LJ: Partial inhibition of
mitochondrial-linked pyrimidine synthesis increases tumorigenic
potential and lysosome accumulation. Mitochondrion. 64:73–81. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Tarangelo A, Rodencal J, Kim JT, Magtanong
L, Long JZ and Dixon SJ: Nucleotide biosynthesis links glutathione
metabolism to ferroptosis sensitivity. Life Sci Alliance.
5:e2021011572022. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yang C, Zhao Y, Wang L, Guo Z, Ma L, Yang
R, Wu Y, Li X, Niu J, Chu Q, et al: De novo pyrimidine biosynthetic
complexes support cancer cell proliferation and ferroptosis
defence. Nat Cell Biol. 25:836–847. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liu Y, Lu S, Wu LL, Yang L, Yang L and
Wang J: The diversified role of mitochondria in ferroptosis in
cancer. Cell Death Dis. 14:5192023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liang D, Feng Y, Zandkarimi F, Wang H,
Zhang Z, Kim J, Cai Y, Gu W, Stockwell BR and Jiang X: Ferroptosis
surveillance independent of GPX4 and differentially regulated by
sex hormones. Cell. 186:2748–2764.e22. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sun S, Shen J, Jiang J, Wang F and Min J:
Targeting ferroptosis opens new avenues for the development of
novel therapeutics. Signal Transduct Target Ther. 8:3722023.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zeng F, Chen X and Deng G: The
anti-ferroptotic role of FSP1: Current molecular mechanism and
therapeutic approach. Mol Biomed. 3:372022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang Y, Wu X, Ren Z, Li Y, Zou W, Chen J
and Wang H: Overcoming cancer chemotherapy resistance by the
induction of ferroptosis. Drug Resist Updat. 66:1009162023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen Z, Wang W, Abdul Razak SR, Han T,
Ahmad NH and Li X: Ferroptosis as a potential target for cancer
therapy. Cell Death Dis. 14:4602023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Novo N, Ferreira P and Medina M: The
apoptosis-inducing factor family: Moonlighting proteins in the
crosstalk between mitochondria and nuclei. IUBMB Life. 73:568–581.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zheng J and Conrad M: The metabolic
underpinnings of ferroptosis. Cell Metab. 32:920–937. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Nguyen HP, Yi D, Lin F, Viscarra JA,
Tabuchi C, Ngo K, Shin G, Lee AY, Wang Y and Sul HS: Aifm2, a NADH
oxidase, supports robust glycolysis and is required for cold- and
diet-induced thermogenesis. Mol Cell. 77:600–617.e4. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mishima E, Ito J, Wu Z, Nakamura T, Wahida
A, Doll S, Tonnus W, Nepachalovich P, Eggenhofer E, Aldrovandi M,
et al: A non-canonical vitamin K cycle is a potent ferroptosis
suppressor. Nature. 608:778–783. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hadian K: Ferroptosis suppressor protein 1
(FSP1) and coenzyme Q10 cooperatively suppress
ferroptosis. Biochemistry. 59:637–638. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lee J and Roh JL: Unleashing ferroptosis
in human cancers: Targeting ferroptosis suppressor protein 1 for
overcoming therapy resistance. Antioxidants (Basel). 12:12182023.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yuan J, Lv T, Yang J, Wu Z, Yan L, Yang J
and Shi Y: HDLBP-stabilized lncFAL inhibits ferroptosis
vulnerability by diminishing Trim69-dependent FSP1 degradation in
hepatocellular carcinoma. Redox Biol. 58:1025462022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang S, Gou S, Zhang Q, Yong X, Gan B and
Jia D: FSP1 oxidizes NADPH to suppress ferroptosis. Cell Res.
33:967–970. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lv Y, Liang C, Sun Q, Zhu J, Xu H, Li X,
Li YY, Wang Q, Yuan H, Chu B and Zhu D: Structural insights into
FSP1 catalysis and ferroptosis inhibition. Nat Commun. 14:59332023.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Guo J, Chen L and Ma M: Ginsenoside Rg1
suppresses ferroptosis of renal tubular epithelial cells in
sepsis-induced acute kidney injury via the
FSP1-CoQ10-NAD(P)H pathway. Curr Med Chem. 31:2119–2132.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yang M, Tsui MG, Tsang JKW, Goit RK, Yao
KM, So KF, Lam WC and Lo ACY: Involvement of FSP1-CoQ(10)-NADH and
GSH-GPx-4 pathways in retinal pigment epithelium ferroptosis. Cell
Death Dis. 13:4682022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Santoro MM: The antioxidant role of
non-mitochondrial CoQ10:. Mystery solved! Cell Metab. 31:13–15.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Koppula P, Lei G, Zhang Y, Yan Y, Mao C,
Kondiparthi L, Shi J, Liu X, Horbath A, Das M, et al: A targetable
CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1
inactive lung cancers. Nat Commun. 13:22062022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shen G, Li C, Cao Q, Megta AK, Li S, Gao
M, Liu H, Shen Y, Chen Y, Yu H, et al: Structural features
determining the vitamin K epoxide reduction activity in the VKOR
family of membrane oxidoreductases. FEBS J. 289:4564–4579. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mladěnka P, Macáková K, Kujovská Krčmová
L, Javorská L, Mrštná K, Carazo A, Protti M, Remião F and Nováková
L; OEMONOM researchers collaborators, : Vitamin K-sources,
physiological role, kinetics, deficiency, detection, therapeutic
use, and toxicity. Nutr Rev. 80:677–698. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jin DY, Chen X, Liu Y, Williams CM,
Pedersen LC, Stafford DW and Tie JK: A genome-wide CRISPR-Cas9
knockout screen identifies FSP1 as the warfarin-resistant vitamin K
reductase. Nat Commun. 14:8282023. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Mishima E, Wahida A, Seibt T and Conrad M:
Diverse biological functions of vitamin K: From coagulation to
ferroptosis. Nat Metab. 5:924–932. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ward NP and DeNicola GM: Long-sought
mediator of vitamin K recycling discovered. Nature. 608:673–674.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Pfitzner AK, Mercier V, Jiang X, Moser von
Filseck J, Baum B, Šarić A and Roux A: An ESCRT-III polymerization
sequence drives membrane deformation and fission. Cell.
182:1140–1155.e18. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liu J, Kang R and Tang D:
ESCRT-III-mediated membrane repair in cell death and tumor
resistance. Cancer Gene Ther. 28:1–4. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Dai E, Meng L, Kang R, Wang X and Tang D:
ESCRT-III-dependent membrane repair blocks ferroptosis. Biochem
Biophys Res Commun. 522:415–421. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Shakya A, McKee NW, Dodson M, Chapman E
and Zhang DD: Anti-ferroptotic effects of Nrf2: Beyond the
antioxidant response. Mol Cells. 46:165–175. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Anandhan A, Dodson M, Shakya A, Chen J,
Liu P, Wei Y, Tan H, Wang Q, Jiang Z, Yang K, et al: NRF2 controls
iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv.
9:eade95852023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
He F, Ru X and Wen T: NRF2, a
transcription factor for stress response and beyond. Int J Mol Sci.
21:47772020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Müller F, Lim JKM, Bebber CM, Seidel E,
Tishina S, Dahlhaus A, Stroh J, Beck J, Yapici FI, Nakayama K, et
al: Elevated FSP1 protects KRAS-mutated cells from ferroptosis
during tumor initiation. Cell Death Differ. 30:442–456. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chen L, Cai Q, Yang R, Wang H, Ling H, Li
T, Liu N, Wang Z, Sun J, Tao T, et al: GINS4 suppresses ferroptosis
by antagonizing p53 acetylation with Snail. Proc Natl Acad Sci USA.
120:e22195851202023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zheng X, Wang Q, Zhou Y, Zhang D, Geng Y,
Hu W, Wu C, Shi Y and Jiang J: N-acetyltransferase 10 promotes
colon cancer progression by inhibiting ferroptosis through
N4-acetylation and stabilization of ferroptosis suppressor protein
1 (FSP1) mRNA. Cancer Commun (Lond). 42:1347–1366. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang N, Ma T and Yu B: Targeting
epigenetic regulators to overcome drug resistance in cancers.
Signal Transduct Target Ther. 8:692023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wu J, Zhu S, Wang P, Wang J, Huang J, Wang
T, Guo L, Liang D, Meng Q and Pan H: Regulators of epigenetic
change in ferroptosis-associated cancer (review). Oncol Rep.
48:2152022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hao M, Jiang Y, Zhang Y, Yang X and Han J:
Ferroptosis regulation by methylation in cancer. Biochim Biophys
Acta Rev Cancer. 1878:1889722023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang Y, Hu J, Wu S, Fleishman JS, Li Y, Xu
Y, Zou W, Wang J, Feng Y, Chen J and Wang H: Targeting epigenetic
and posttranslational modifications regulating ferroptosis for the
treatment of diseases. Signal Transduct Target Ther. 8:4492023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Wu X, Xu M, Geng M, Chen S, Little PJ, Xu
S and Weng J: Targeting protein modifications in metabolic
diseases: Molecular mechanisms and targeted therapies. Signal
Transduct Target Ther. 8:2202023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Lee J and Roh JL: Epigenetic modulation of
ferroptosis in cancer: Identifying epigenetic targets for novel
anticancer therapy. Cell Oncol (Dordr). 46:1605–1623. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Widagdo J, Anggono V and Wong JJL: The
multifaceted effects of YTHDC1-mediated nuclear m6A
recognition. Trends Genet. 38:325–332. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yuan S, Xi S, Weng H, Guo MM, Zhang JH, Yu
ZP, Zhang H, Yu Z, Xing Z, Liu MY, et al: YTHDC1 as a tumor
progression suppressor through modulating FSP1-dependent
ferroptosis suppression in lung cancer. Cell Death Differ.
30:2477–2490. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Brewer G: FSP1 in cancer: Not just a
phase. Nat Rev Cancer. 23:5782023. View Article : Google Scholar
|
|
94
|
Zhang Q, Li N, Deng L, Jiang X, Zhang Y,
Lee LTO and Zhang H: ACSL1-induced ferroptosis and platinum
resistance in ovarian cancer by increasing FSP1 N-myristylation and
stability. Cell Death Discov. 9:832023. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Liu MR, Shi C, Song QY, Kang MJ, Jiang X,
Liu H and Pei DS: Sorafenib induces ferroptosis by promoting
TRIM54-mediated FSP1 ubiquitination and degradation in
hepatocellular carcinoma. Hepatol Commun. 7:e02462023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gotorbe C, Durivault J, Meira W, Cassim S,
Ždralević M, Pouysségur J and Vučetić M: Metabolic rewiring toward
oxidative phosphorylation disrupts intrinsic resistance to
ferroptosis of the colon adenocarcinoma cells. Antioxidants
(Basel). 11:24122022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Pontel LB, Bueno-Costa A, Morellato AE,
Carvalho Santos J, Roué G and Esteller M: Acute lymphoblastic
leukemia necessitates GSH-dependent ferroptosis defenses to
overcome FSP1-epigenetic silencing. Redox Biol. 55:1024082022.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Yoshioka H, Kawamura T, Muroi M, Kondoh Y,
Honda K, Kawatani M, Aono H, Waldmann H, Watanabe N and Osada H:
Identification of a Small molecule that enhances ferroptosis via
inhibition of ferroptosis suppressor protein 1 (FSP1). ACS Chem
Biol. 17:483–491. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Cheu JW, Lee D, Li Q, Goh CC, Bao MH, Yuen
VW, Zhang MS, Yang C, Chan CY, Tse AP, et al: Ferroptosis
suppressor protein 1 inhibition promotes tumor ferroptosis and
anti-tumor immune responses in liver cancer. Cell Mol Gastroenterol
Hepatol. 16:133–159. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Xavier da Silva TN, Schulte C, Alves AN,
Maric HM and Friedmann Angeli JP: Molecular characterization of
AIFM2/FSP1 inhibition by iFSP1-like molecules. Cell Death Dis.
14:2812023. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Hendricks JM, Doubravsky CE, Wehri E, Li
Z, Roberts MA, Deol KK, Lange M, Lasheras-Otero I, Momper JD, Dixon
SJ, et al: Identification of structurally diverse FSP1 inhibitors
that sensitize cancer cells to ferroptosis. Cell Chem Biol.
30:1090–1103.e7. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Xavier da Silva TN and Friedmann Angeli
JP: Sabotaging the breaks: FSEN1 expands the toolbox of FSP1
inhibitors. Cell Chem Biol. 30:1006–1008. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nakamura T, Hipp C, Santos Dias Mourão A,
Borggräfe J, Aldrovandi M, Henkelmann B, Wanninger J, Mishima E,
Lytton E, Emler D, et al: Phase separation of FSP1 promotes
ferroptosis. Nature. 619:371–377. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Nakamura T, Mishima E, Yamada N, Mourão
ASD, Trümbach D, Doll S, Wanninger J, Lytton E, Sennhenn P, Nishida
Xavier da Silva T, et al: Integrated chemical and genetic screens
unveil FSP1 mechanisms of ferroptosis regulation. Nat Struct Mol
Biol. 30:1806–1815. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B,
Wu F, Wang Q, Wang S, Rong D, Reiter FP, et al: The mechanisms of
sorafenib resistance in hepatocellular carcinoma: Theoretical basis
and therapeutic aspects. Signal Transduct Target Ther. 5:872020.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Lai YL, Wang KH, Hsieh HP and Yen WC:
Novel FLT3/AURK multikinase inhibitor is efficacious against
sorafenib-refractory and sorafenib-resistant hepatocellular
carcinoma. J Biomed Sci. 29:52022. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Mishima E, Nakamura T, Zheng J, Zhang W,
Mourão ASD, Sennhenn P and Conrad M: DHODH inhibitors sensitize to
ferroptosis by FSP1 inhibition. Nature. 619:E9–E18. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhang L, Zhang J, Wang J, Ren C, Tang P,
Ouyang L and Wang Y: Recent advances of human dihydroorotate
dehydrogenase inhibitors for cancer therapy: Current development
and future perspectives. Eur J Med Chem. 232:1141762022. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Zhou J, Zhang L, Yan J, Hou A, Sui W and
Sun M: Curcumin induces ferroptosis in A549 CD133+ cells
through the GSH-GPX4 and FSP1-CoQ10-NAPH pathways. Discov Med.
35:251–263. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Miyazaki K, Xu C, Shimada M and Goel A:
Curcumin and andrographis exhibit anti-tumor effects in colorectal
cancer via activation of ferroptosis and dual suppression of
glutathione peroxidase-4 and ferroptosis suppressor protein-1.
Pharmaceuticals (Basel). 16:3832023. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Yang J, Jia Z, Zhang J, Pan X, Wei Y, Ma
S, Yang N, Liu Z and Shen Q: Metabolic intervention nanoparticles
for triple-negative breast cancer therapy via overcoming
FSP1-mediated ferroptosis resistance. Adv Healthc Mater.
11:e21027992022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Li K, Lin C, Li M, Xu K, He Y, Mao Y, Lu
L, Geng W, Li X, Luo Z and Cai K: Multienzyme-like reactivity
cooperatively impairs glutathione peroxidase 4 and ferroptosis
suppressor protein 1 pathways in triple-negative breast cancer for
sensitized ferroptosis therapy. ACS Nano. 16:2381–2398. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Liu C, Xing J, Akakuru OU, Luo L, Sun S,
Zou R, Yu Z, Fang Q and Wu A: Nanozymes-engineered metal-organic
frameworks for catalytic cascades-enhanced synergistic cancer
therapy. Nano Lett. 19:5674–5682. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Ding Y, Xu H, Xu C, Tong Z, Zhang S, Bai
Y, Chen Y, Xu Q, Zhou L, Ding H, et al: A nanomedicine fabricated
from gold nanoparticles-decorated metal-organic framework for
cascade chemo/chemodynamic cancer therapy. Adv Sci (Weinh).
7:20010602020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhou Y, Chen K, Lin WK, Liu J, Kang W,
Zhang Y, Yang R, Jin L, Cheng Y, Xu A and Wang W: Photo-enhanced
synergistic induction of ferroptosis for anti-cancer immunotherapy.
Adv Healthc Mater. 12:e23009942023. View Article : Google Scholar : PubMed/NCBI
|