1. Introduction
Prostate cancer (Pca) has emerged as the most common
type of cancer affecting males worldwide, holding the 4th position
among all types of cancer, with a total of 1,466,680 new cases and
396,792 related deaths worldwide in 2022(1). A variety of modifiable and
non-modifiable risk factors are linked to the development of Pca,
rendering its etiology complex and challenging to understand,
including age, ethnicity, family history and lifestyle management
(2). Therefore, it is critical to
diagnose Pca in its early stages and begin treatment as soon as
possible. Multiple treatment options are available for patients
with Pca, including active surveillance regimens for low-grade Pca,
radiation therapy and prostatectomy surgery for localized disease,
or a combination of several therapies [chemotherapy, immunotherapy,
androgen deprivation therapy (ADT), or agents targeting androgen
receptors] for metastatic disease (3). In this therapy, the testosterone
level is reduced in the body to near castrate levels using
gonadotropin hormone-releasing hormone agonists or antagonist
drugs, which block the production of testosterone in the body and
arrest the cell cycle of cancer cells (3). Among the aforementioned treatment
options, ADT is commonly used for the treatment of metastatic
Pca.
It has been demonstrated that ~30-50% of patients
receiving ADT develop therapeutic resistance that leads to
castrate-resistant Pca (CRPC) (4).
In these patients, the levels of prostate-specific antigen (PSA)
are high even if they have low levels of testosterone. CRPC may
develop due to the reactivation of androgen receptor (AR) through
the upregulation of the P450 enzymes, CYP11A1 and CYP17A1(5). Chemotherapy, along with ADT, is
usually administered to patients with CRPC. For example,
FDA-approved drugs, such as apalutamide and darolutamide are
currently being incorporated with ADT to decrease the progression
of CRPC (6,7). Both drugs competitively inhibit the
binding of androgens to ARs and the transcription of genes
regulated by AR signaling. However, they have been shown to have
various side-effects (8).
Similarly, taxanes such as docetaxel and cabazitaxel are commonly
used in the treatment of metastatic CRPC (mCRPC). However, despite
the improved survival rates, almost each patient eventually
develops resistance, which is a critical barrier to long-term
survival (9).
The varied mechanisms of action of drugs in PCa
necessitate the identification of clinically valuable biomarkers to
guide precision-oriented therapies. However, this remains
challenging due to tumor heterogeneity at genetic, epigenetic and
phenotypic levels, resulting in diverse molecular profiles even
within the same tumor. Not all mutations are clinically relevant,
further complicating biomarker identification. Sequential biopsies,
essential for capturing tumor heterogeneity and dynamics, are often
impractical due to limitations associated with both solid and
liquid biopsies, as previously described (4).
Despite challenges in identifying biomarkers and
characterizing PCa at the molecular level, biomarkers are
invaluable for assessing treatment responses and stratifying
patients into risk groups (high, intermediate or low). This
stratification allows clinicians to customize treatments, opting
for aggressive approaches for high-risk tumors and more
conservative strategies for low-risk cases. Approved biomarkers,
such as AR splice variant 7-V7 (AR-V7), have exhibited utility in
predicting therapeutic outcomes. For instance, patients with high a
expression of AR-V7 exhibit improved overall survival rates with
taxane therapy compared to treatment with AR signaling (ARS)
inhibitors (10). A number of
genetic biomarkers, such as phosphatase and tensin homolog
(PTEN), poly(ADP-ribose) polymerase (PARP) and
homologous recombination repair (HRR), have emerged as
potential treatment targets for Pca (11). Numerous biomarkers have surfaced
that could eventually be proven reliable indicators of how
different treatments would affect metastatic patients with Pca
(12). However, comparisons in
different studies are challenging due to differences in the type of
therapy, the patient population size and prior treatments received.
While biomarkers are being used to differentiate between metastatic
and non-metastatic Pca, their clinical implications, for instance,
and their effects on overall survival or disease-free survival,
remain limited. Therefore, therapies should not only focus on the
basis of the high or low expression of biomarkers, but should also
be based on their effects on survival or other factors to establish
genotype-phenotype associations (13,14).
This will markedly improve the precision of disease treatment. It
is necessary to address the challenges associated with current
biomarkers in order for risk stratification and treatment efficacy
to be improved for a specific patient (15).
The present discusses the advancements that have
been made in therapeutic biomarkers that are used in Pca to assess
the response to therapy, progress being made in therapy and
associated challenges. The present review included studies
assessing the efficacy of biomarker-based therapies in all types of
Pca. An extensive search of the literature was performed using
three online databases: PubMed, Embase and Web of Science. The
search was carried out from the beginning of the databases up until
August 31, 2024. To identify ongoing clinical trials for Pca
treatments, a systematic search was also conducted on ClinicalTrials.gov. A combination of the following key
words was used: targeted therapy, therapeutic biomarkers,
inhibitors, drugs, prostate cancer, PARP inhibitors, cancer, AR-V7,
PI3K/AKT, PSMA, HDAC and clinical trial. The inclusion criteria
required participants with pathologically or cytologically
confirmed prostate adenocarcinoma and radiographic evidence of
metastases. Studies were excluded if they met any of the following
criteria: i) Studies not containing sufficient data for analysis;
and ii) case reports.
2. Androgen receptor splice variant 7
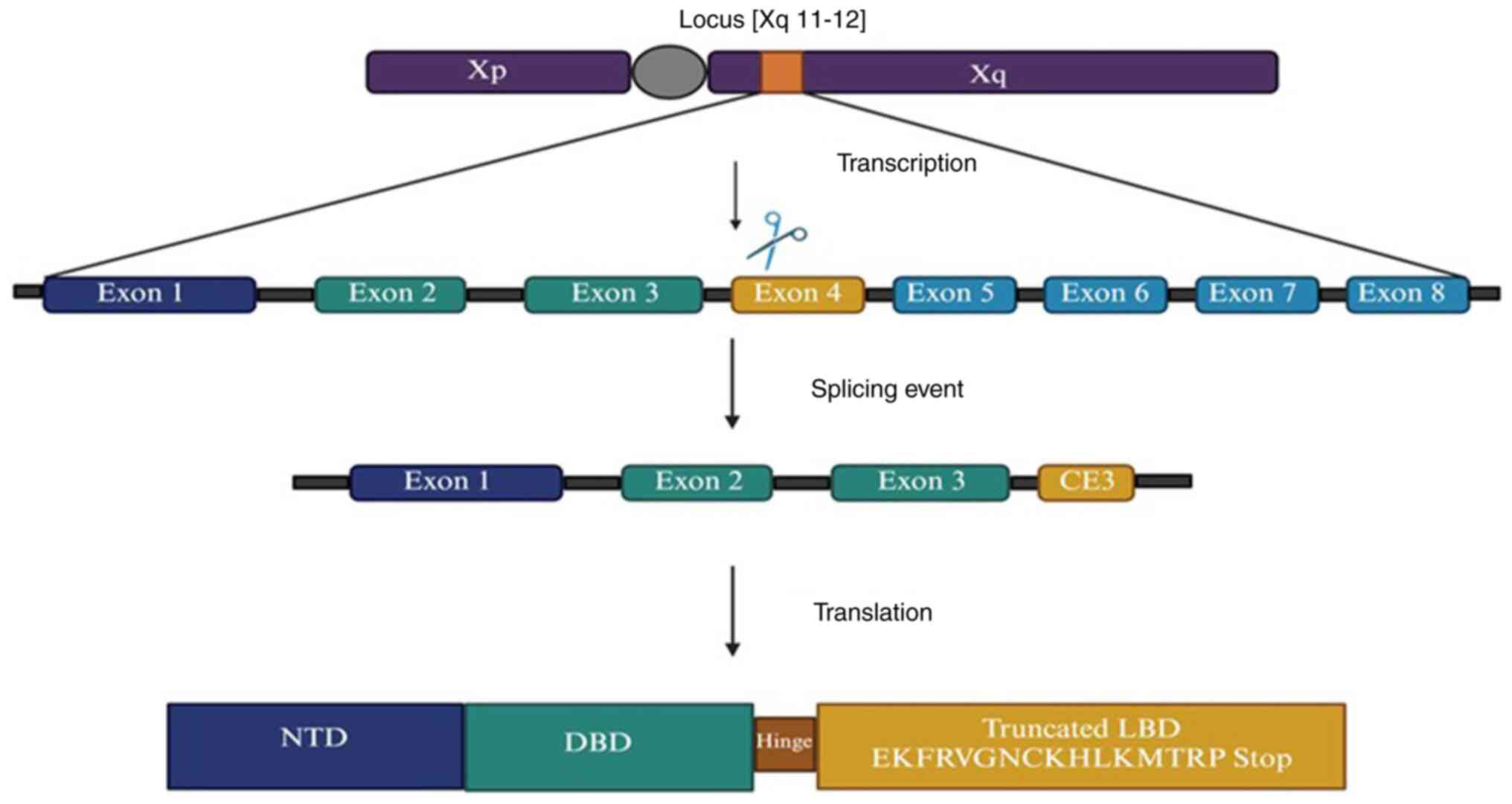
AR-V7 is a mutation that arises due to the
truncation of the ligand-binding domain (LBD) of the AR, which
allows the receptor to remain in an active state independent of
androgen ligand binding (16).
Wild-type ARs are 110 kDa or 919 amino acids in length. It consists
of four functional domains and contains eight exons: i) The
N-terminal transactivation domain (exon 1), which has an AF1 domain
with two transactivation units (TAU-1 and TAU-5) that can lead to
the aberrant activation of the AR in CRPC. This region is
responsible for the full activation of the AR. In the presence of
the ligand, 50% of AR activation is contributed by TAU-1, whereas
in the absence of a ligand-binding domain, TAU-5 mediates
activation. ii) The DNA-binding domain (DBD) (exons 2 and 3): It
has two zinc fingers that promote receptor dimerization. iii) The
hinge region (exon 4), which separates the DBD into a
ligand-binding domain and has a nuclear localization signal. iv)
The LBD (exon 5-8), which has an AF2 domain and is responsible for
binding to the ligand. Several splice variants of AR have been
identified in cell lines and xenograft models. However, due to the
availability of AR-V7 specific antibody, AR-V7 is the most
well-characterized AR variant (17). AR-V7 can also form dimers with
full-length AR (AR-FL). The mRNA of AR-V7 includes the first three
canonical exons, followed by a variant-specific cryptic exon, known
as CE3. A splicing event at CE3 resulted in the production of a
truncated LBD for AR-V7. Truncation occurs due to the premature
termination of translation after 16 variant-specific amino acids
(EKFRVGNCKHLKMTRP stop) (Fig. 1)
(18). Certain splicing factors
and chaperone proteins regulate the expression of AR-V7. For
example, the recruitment of splicing factors hnRNPA1, U2AF65 and
ASF/SF2 to AR-SV mRNA splice junctions increased. The same was not
true for AR-FL. By contrast, HSP90 inhibition disrupted AR-V7
splicing. The expression of non-coding RNA PCGEM1 increases under
conditions of androgen deprivation, which facilitates the
interaction of this RNA with the splicing factor (U2AF565). Thus,
the splicing mechanisms of AR-V7 are likely cell-context-specific
and regulated by numerous factors (19). AR signaling has been linked to the
development of certain types of cancer beyond Pca. AR-V7 can be
present in primary breast cancer (BC) even in patients who have
never received treatments, such as anti-estrogen therapy or drugs
that block the AR. For patients with BC being considered for
treatment with AR inhibitors, having certain features in the tumor
(such as apocrine changes) and testing positive for AR on
laboratory slides may suggest the need for further testing to test
for AR-V7. It has been suggested that AR-V7 may be a helpful marker
to predict how well patients with BC may respond to treatments that
block the AR. This could be useful for future clinical trials
testing these treatments (20).
Studies using laboratory-grown cells and animal models have
demonstrated that androgens and AR play a role in cancer
development (21). For example, it
was shown that AR signaling helps liver cancer cells grow and
spread, and it also makes these cells resistant to treatments that
block the AR (22). As with liver
cancer and BC, AR-V7 has also been shown to play a role in bladder
and kidney cancers (19). However,
the role of AR-V7 in other types of cancer is not yet fully
understood. Research is still in the early stages, and its effects
may differ depending on the type of cancer and the specific
circumstances.
AR-V7 expression in Pca
The expression of AR-V7 has been found to be
markedly higher in metastatic Pca than in primary Pca specimens.
For instance, in a previous study, the expression of AR-V7 in
metastatic biopsy specimens was >75% following ADT, whereas its
expression was only <1% in primary Pca tissues (23). In that study, 651 Pca samples
(primary Pca samples, 358; metastatic biopsies, 293) were included
to observe the expression of AR-V7. However, a wide heterogeneity
was observed, such as the expression of AR-V7, which differed in
different metastatic sites of the same patient, thus rendering it
an unreliable biomarker (23).
These changes necessitate the need for further biopsies on
progression. However, it is challenging to collect tissue samples
multiple times.
The levels of AR-V7 are generally higher in
castration-resistant tumors than in androgen-dependent tumors
(24). Its high expression can
also be used to prognose the severity of disease. For instance,
patients with AR-V7-positive hormone-sensitive Pca have been shown
to have a worse prognosis following first-line hormonal therapy and
prostatectomy (25). AR-V7
overexpression facilitates the development of lesions that resemble
prostatic interstitial neoplasia. Of note, AR-V7 also alters the
expression of genes associated with prostate stem and progenitor
cells (19). These findings
suggest that AR-V7 expression increases following ADT and may lead
to therapeutic resistance. In mCRPC, AR-V7 is primarily nuclear,
but is also present in the cytoplasm and cytoplasmic granules. A
previous study on 410 patients with Pca revealed that cytoplasmic
AR-V7 staining was associated with a shorter relapse-free survival
(RFS), while granular cytoplasmic AR-V7 staining was linked to a
longer RFS and was negatively associated with aggressive disease
features, such as higher Gleason scores, tumor stage and metastasis
(26). By contrast, nuclear and
cytoplasmic AR-V7 staining was positively associated with Gleason
scores and tumor invasiveness. Granular AR-V7 staining emerges as
an independent prognostic marker for RFS, emphasizing the need to
assess AR-V7 localization for accurate prognostic evaluation in
prostate cancer patients (26).
The expression of AR-V7 has also been reported in
circulating tumor cells (CTCs). In a previous study,
immunofluorescence staining demonstrated the presence of
AR-V7-positive CTCs in 34 out of 161 samples (18%) (P<0.001).
During AR-axis-targeted therapies (AATT), patients with positive
AR-V7 staining demonstrate a poorer overall survival (OS) and
progression-free survival (PFS) than AR-V7-negative patients.
However, in patients treated with taxanes, no association was
observed between AR-V7 staining and the high risk of mortality
(10). That study concluded that
AR-V7-positive CTCs were negatively associated with the benefits of
AATT therapy and could predict therapeutic resistance in patients
with mCRPC. Similarly, patients treated with AATT demonstrated
worse outcomes if they had a high expression of AR-V7. These
findings confirm that AR-V7 positivity is associated with adverse
outcomes. These factors include a Gleason score ≥8, metastasis and
prior therapeutic outcomes. Therefore, the nuclear localization of
AR-V7 can help clinicians make wise decisions, such as opting for
taxane therapy instead of AATT.
AR-V7 and regulated genes
It has been shown that genes induced by AR-V7 and
AR-FL differ markedly in terms of tumor promoting properties. For
instance, AR-V7 induces the expression of cell cycle-related genes.
By contrast, AR-FL promotes the expression of genes associated with
metabolism, differentiation and macromolecular synthesis (17). It has also shown that the presence
of AR-FL is not required for inducing cell cycle-related genes in
cells transiently transfected with AR-V7. However, the suppression
of AR-FL expression using siRNA or drugs induces the expression of
AR-V7-regulated genes (27). It
was also previously shown that the overexpression of AR-V7 induced
the expression of 407 genes and reduced the expression of 80 genes.
Of these 407 positively associated genes, only 59 were found to be
significantly associated with nuclear AR-V7 staining. Of these 59
genes, 33% had zinc finger domains and were associated with
chromatin-binding regions proximal to the transcriptional start
site. These genes included HOXB13, ELL2, STEAP2 and
BAZ2A. Due to the absence of the LBD in AR-V7, it can either
expose or mask regions that are capable of interacting with
distinct coregulators in comparison to FL-AR (28,29).
These results suggest that FL-AR and AR-V7 target unique genes;
however, a few genes may be common.
In Pca, specifically in phase II mCRPC trials, the
levels of PSA contribute significantly to determining the response
to any given therapy. It is a serum biomarker that can be measured
easily. In a previous study, a ≥50% reduction in PSA level from
baseline was shown to be associated with increased survival rates.
That study demonstrated that patients who were AR-V7-negative had
significantly better PSA response rates (100 vs. 54%; P=0.03) and a
longer OS compared to AR-V7-positive patients [74 vs. 24 months;
hazard ratio (HR), 0.23; P=0.02]. This suggests that AR-V7
negativity is associated with more favorable treatment outcomes in
Pca (23).
AR-V7 as a predictor of the
therapeutic response. Clinical relevance and challenges
The hypothalamus of the brain releases
gonadotropin-releasing hormone (which stimulates the pituitary
gland to produce follicle-stimulating hormone and luteinizing
hormone). These hormones signal the gonads to produce testosterone,
which is then converted to dihydrotestosterone (DHT) by the enzyme
5-alpha-reductase. DHT binds to the intracellular AR, forming a
DHT-AR complex that migrates to the nucleus and regulates the
transcription and translation of cancer-related genes. AR
antagonists, such as enzalutamide, apalutamide and darolutamide
(AATT) inhibit DHT from binding to the LBD of AR, which prevents
the translocation of AR to the nucleus, transcription of target
genes and its ability to bind to chromatin (11).
There are several mechanisms that can lead to
chemoresistance following AATT: i) The upregulation of
steroidogenesis, leading to the synthesis of endogenous androgens
within the prostate tumor; ii) single point mutations in the LBD
domain of the AR; iii) the silencing of androgen inactivation
enzymes, such as HSD17B2 by methylation; and iv) the upregulation
of the glucocorticoid receptor; these receptors are upregulated in
metastatic Pca and provide resistance to AATT by surpassing AR
blockade; v) the emergence of AR splice variants such as AR-V7; vi)
the higher expression of AR due to AR gene amplification (30,31).
The absence of the LBD in AR-V7 results in the constitutive
activation of the AR and its target genes, even in the absence of
androgens (32). Research has
shown that AR-V7 can form heterodimers with AR-FL, activate
canonical AR target genes such as PSA, and promote cell growth in
castration-resistant conditions (33). It has been suggested that a
positive correlation between AR-V7 and AR-FL expression and copy
number. However, conflicting cases of AR-V7 and AR-FL suggest that
AR-V7 expression following ADT is not simply a consequence of AR
gene amplification (33). The
inhibition of AR-V7 has also been shown to confer sensitivity to
AATT. For instance, the inhibition of AR-V7 protein by the
anthelmintic drug, niclosamide, inhibits Pca cell growth and
restores sensitivity to enzalutamide in enzalutamide-resistant
tumors (34). Notably, a higher
expression of AR-V7 confers chemoresistance to AATT, but not to
taxane therapy. It has been observed that the presence of AR-V7
results in significantly improved outcomes in taxane-treated
patients with Pca compared to abiraterone- or enzalutamide-treated
patients (35,36). These findings indicate that AR-V7
expression in CTCs serves as a marker of AATT resistance, but not
taxane-based treatments. By comparison, although AR-V7 is less
extensively studied in BC, its expression may contribute to
resistance in hormone receptor-positive cases by circumventing
estrogen receptor signaling or influencing AR activity. However,
its role in BC is less well-defined and requires further research
to establish its exact mechanisms and clinical relevance (37). Researchers have also explored the
expression of AR-V7 in salivary gland cancers; however, the
evidence is limited, largely exploratory and context-dependent
(38). In another PROPHECY trial,
PSA or soft tissue responses were lower in AR-V7-positive mCRPC
(PSA, 0 to 11%; soft tissue response, 0 to 6%), confirming the
association between AR-V7 expression, and a shorter PFS and OS with
abiraterone or enzalutamide, and such patients with mCRPC should be
offered alternative treatments (39). Similarly, the EXCAAPE (NCT03002220)
phase IIa trial was conducted on 52 patients with mCRPC and
asymptomatic bone metastases who had progressed on abiraterone
acetate or enzalutamide up to six doses of 223Ra
(40). It was shown that
AR-V7-positive patients had a shorter radiographic PFS and OS than
AR-V7-negative patients (rPFS-AR-V7-negative, 5.5 months;
AR-V7-positive, 2.2 months, P=0.056) (OS: AR-V7-negative, 14.8
months; AR-V7-positive, 3.5 months, P<0.01). The incidence of
grade 3/4 adverse events was lower in AR-V7-negative patients when
compared to AR-V7-positive patients (5.8 and 11.5%, respectively).
These results suggest that AR-V7 expression can lead to therapeutic
resistance to 223Ra therapy in patients with mCRPC
(40).
Immune checkpoint inhibitors (nivolumab and
ipilimumab) combined with androgen-axis-targeted therapies have
exhibited limited efficacy in AR-V7-positive patients with mCRPC.
In the STARVE-PC trial, 30 patients with progressive mCRPC and
detectable AR-V7 transcripts received either nivolumab and
ipilimumab (cohort 1) or the same combination with enzalutamide
(cohort 2). Outcomes, including PSA response rates, PFS and overall
response rates (ORR), exhibited no significant differences between
cohorts. The median OS was higher in cohort 2 (14.2 vs. 8.2
months), but the results were not statistically significant. These
findings highlight the need for novel immune targets and biomarkers
to improve outcomes in AR-V7-positive Pca (41).
In ongoing clinical trials, a phase 3, randomized,
open-label study will compare opevesostat treatment with
alternative abiraterone acetate or enzalutamide in participants
with mCRPC based on the LBD mutation of AR for each drug
(NCT06136624 and NCT06136650). Similarly, a biomarker-driven study
in patients with metastatic prostate cancer (ProBio) will treat
patients based on biomarker signatures, such as androgen receptor,
DNA repair deficiency, TP53, TMPRSS2-ERG gene fusion, and PI3K
pathway alterations (NCT03903835).
Patients with positive AR-V7 expression should avoid
AATT and 223Ra therapy, as these treatments may be less
effective. Instead, they should consider alternative therapeutic
options.
Future research directions. Nuclear staining
is a hallmark of AR-V7. The inclusion of cytoplasmic and
cytoplasmic granule staining should also be included to confirm the
presence of AR-V7. Taken together, AR-V7 expression in granular
cytoplasmic structures is an independent prognostic factor for RFS
in patients with Pca. AR-V7 binds to various co-regulators.
Similarly, several genes have been found to be significantly
associated with nuclear AR-V7 staining. These genes included
HOXB13, ELL2, STEAP2 and BAZ2A. Therefore, the
inclusion of such co-regulators and genes will add to accurately
dividing the patients into two groups: i) AR-V7-positive and ii)
AR-V7-negative, and may ultimately help clinicians to make precise
decisions. Notably, not all AR-V7-positive patients demonstrate
chemoresistance to AATT. Therefore, more accuracy is required to
make an accurate decision.
3. Histone deacetylases
Positively charged lysine residues are present at
the N-terminal site of histone proteins. In the absence of
acetylation, lysine interacts tightly with negatively charged DNA
molecules, leading to chromatin condensation. This interaction
prevents the access of the transcriptional machinery to the
transcription site. The acetylation of lysine residues prevents
this interaction and thus leads to transcription initiation. In
addition to histones, non-histone proteins are capable of
undergoing acetylation. Non-histone proteins (NHPs) are a diverse
group of proteins found in the nucleus that are distinct from
histones. These NHPs are involved in various processes, such as
gene regulation, cell cycle regulation, DNA repair and
differentiation. Examples of NHPs include transcription factors and
DNA polymerases. The acetylation of transcription factors can
either activate or repress their DNA-binding ability of
transcriptional factors. If acetylation occurs in the DNA-binding
region of the transfection factor, it will reduce their binding to
DNA, such as FoxO1 and HMGI. On the contrary, if acetylation occurs
near the DNA binding region, it will enhance the binding of TF to
DNA such as p53, NF NF-κB. These findings suggest that the
acetylation of both histone proteins and NHPs regulates various
activities, including cell cycle, DNA repair, apoptosis, autophagy
and metabolism. An imbalance in the transcription of tumor
promoters and suppressor genes can lead to the development of
cancer (42).
HDACs are enzymes that deacetylate histones and
NHPs. To date, 18 different HDACs have been identified and are
categorized into four classes based on their homology to yeast and
co-factor dependencies. Class I, II and IV HDACs contain a
zinc-binding domain and therefore require zinc ions for their
catalytic activity, whereas class III HDACs have an NAD+ binding
domain and thus require NAD+ as a co-factor (2). Class I HDACs (HDACs 1, 2, 3, and 8)
have an acetylase domain located in the N-terminal region and are
primarily localized in the nucleus. By contrast, class II HDACs
(HDACs 4, 5, 6, 7, 9 and 10) apart from HDAC6 have an acetylase
domain at the C-terminal site and are located in both the nucleus
and cytoplasm. HDAC6 has two acetylase domains located in its
N-terminal region. Class II HDACs are comparatively longer than the
other three classes of HDACs. Class III HDACs are silent
information regulator 2 proteins with seven homologues (sirtuins).
These sirtuins can deacetylate many proteins such as p53 and
tubulin. Therefore, class III prevents differentiation and promote
tumorigenesis (43). Class IV
HDAC, specifically HDAC 11, possess characteristics of both class I
and class II enzymes, exhibiting HDAC activity in both its N- and
C-terminal regions (44).
Research has shown that the levels of HDACs vary
significantly in cancer cells, with differences depending on the
cancer type. For instance, HDAC1 is found in high levels in
cancers, such as prostate, stomach, lung, esophageal, colon cancers
and BC. HDAC2 is more prevalent in colorectal, cervical and stomach
cancers. HDAC6, HDAC8 and HDAV11 are overexpressed in breast
tumors, neuroblastoma and rhabdomyosarcoma, respectively and can be
used as biomarkers (45,46). These increased levels of HDACs in
different cancers are caused by various factors, which may
influence how effective HDAC inhibitors are in the treatment of
these cancers.
HDACs expression in Pca
The regulation of ARs can be mediated at the
epigenetic level. Therefore, HDAC inhibitors are also considered as
therapeutic options for the treatment of mCRPC. HDACs have been
shown to be upregulated in Pca. For instance, Wang et al
(47) compared HDAC expression in
primary Pca (42 cases with undetectable PSA levels), recurrent Pca
(n=37), metastatic tumors (n=8) and benign prostatic tissue (n=11,
did not show cancer under a microscope) using DNA microarray
analysis. The expression of HDAC1, 3, 4 and 5 was significantly
enhanced in benign and malignant human prostate tissue and various
PCa cell lines (47). In another
study conducted on 192 patients who underwent radical
prostatectomy, it was shown that HDAC1 (69.8%), 2 (74%) and 3
(94.8%) levels were significantly enhanced in the majority of cases
and were found to be associated with dedifferentiation and tumor
cell proliferation. Furthermore, the high expression of HDAC2 has
been shown to be associated with a shorter PSA relapse time; this
suggests that HDAC2 is linked to more aggressive tumor behavior and
faster disease recurrence following surgery (48). The increased expression of HDAC1,
HDAC2 and HDAC3 in Pca, as observed in a previous study on 192
patients, is unfavorable. Weichert et al (48) investigated the expression patterns
of HDACs 1, 2 and 3 in a cohort of 192 patients with Pca who had
undergone radical prostatectomy. Their findings revealed that HDACs
1 and 2 were positively associated with Gleason scores, with higher
expression levels in high-grade tumors. Additionally, the
expression of HDAC1, HDAC2 and HDAC3 was significantly associated
with a positive proliferative fraction of Ki-67 in Pca cells. The
probability of post-operative 7-year disease-free survival (DFS)
was lower in the HDAC1 high and HDAC2 high groups than in the HDAC1
low and HDAC2 low groups, respectively (HDAC1: median DFS
probability, 0.6 vs. 0.8 and in the HDAC2 high vs. HDAC2 low group)
(48). The inhibition of class I
HDACs mediates the re-expression of maspin, which suppresses the
proliferation and migration of Pca cells (LNCaP and DU145)
(49). In prostate tissues, HDAC1
is exclusively expressed in the cell nucleus and its expression is
usually lower in stromal cells. Unexpectedly, HDAC8 was not
detected in epithelial cells, but was uniquely expressed in the
cytoplasm of stromal cells. Taken together, these findings indicate
that epithelial and stromal cells exhibit distinct class I HDAC
expression profiles (50). In Pca,
HDACs, particularly HDAC1, HDAC2 and HDAC3, play a crucial role in
driving disease progression by modifying chromatin structure and
regulating gene expression to promote tumor growth, survival and
metastasis (51). These changes
can also lead to resistance to therapies, such as ADT and
chemotherapy. The differential expression of HDACs may provide new
insight for targeted therapies, such as isoform specific HDAC
inhibitors, which may help overcome resistance, reduce tumor
aggressiveness and improve patient outcomes by selectively
modulating the epigenetic landscape. Of note however, only a
limited number of studies have been conducted to determine the
expression of HDACs in Pca tissues and their association with OS or
PFS (47,52). However, the majority of studies
have focused on reducing the uncontrolled growth of Pca cells using
various HDAC inhibitors or other natural compounds. These
inhibitors or compounds mediate their antitumor effects by
inhibiting the expression of HDACs (53). For example, rosmarinic acid, a
phenolic component of rosemary tea, induces cell cycle arrest and
apoptosis by modulating HDAC2 expression in Pca cell lines
(54). Similarly, apigenin
inhibits HDAC and remodels chromatin to induce growth arrest and
apoptosis in prostate cancer cells (55).
HDAC inhibitors and challenges
Class I, II and IV HDACs require zinc ions for their
catalytic activities. Therefore, HDAC inhibitors targeting these
classes of HDACs possess a zinc-binding group (ZBG). ZBGs are
mainly of two types: i) Classic ZBGs, such as carboxylates,
hydroxamic acids, benzamides and thiols; and ii) novel ZBGs, such
as tropolone derivatives, 3-hydroxypyridin-2-thione, chelidamic
derivatives, and benzoyl hydrazide scaffolds (56). Classic ZBGs are characterized by
high activity, selectivity, off-target effects, potential
toxicities, and instability in vivo. It is well known that
SAHA-containing hydroxamic acid acts as a pan-HDAC inhibitor with
competitive binding and fast on/off profiling. Substituting the
hydroxamic acid group in SAHA with benzamide results in a class
I-selective HDAC inhibitor with a fast-on/fast-off profile and
competitive binding mode for HDAC1, HDAC2 and HDAC3. This differs
from the properties of other benzamide inhibitors, such as MS275
and 106, described in previous studies (57). These findings indicate that the
kinetic profile of benzamide-based HDAC inhibitors is influenced
not only by the ZBG, but also by the cap and linker regions.
Hydroxamic acid and benzamide are the most frequently utilized ZBGs
among HDAC inhibitors. Hydrazide-based HDAC inhibitors show
slow-binding class I selective (HDAC3) activity in competitive and
non-competitive modes. The kinetic profiles of these inhibitors are
not dependent on the cap or linker groups (57). HDAC inhibitors with hydrazide
motifs are not dependent on zinc-binding interactions, and thus
exert fewer off-target effects (56). These findings suggest that
designing a variety of ZBGs can overcome the limitations of the
currently available HDAC inhibitors, such as off-target effects and
toxicity. HDAC inhibitors can be isoform-specific or pan
inhibitors. Pan inhibitors can be used for all types of HDACs.
These inhibitors are mainly divided into five classes: i)
Hydroxamates, (Practinostat approved by the FDA against Pca); ii)
short chain fatty acids; iii) benzamides; iv) cyclic tetrapeptides;
and v) sirtuin inhibitors, including the pan-inhibitor nicotinamide
and the specific SIRT1 and SIRT2 inhibitors sirtinol and cambinol,
respectively. HDAC inhibitors induce various effects on cancer
cells, including cell cycle arrest, stem cell renewal,
differentiation and apoptosis. Additionally, they decrease
angiogenesis and modulate the immune response. Several HDAC
inhibitors also affect the activity of cytochrome P450 (CYP)
enzymes, potentially enhancing the effects of certain anticancer
drugs in combination with HDAC inhibitors (45). Several dual or multimodal HDAC
inhibitors have been investigated; however, practinostat has gained
FDA approval for Pca treatment. Developing newer and more specific
HDAC inhibitors could provide advantages in managing drug-resistant
CRPC, where earlier HDAC inhibitors have shown limited efficacy.
For instance, compound 13, a class II HDAC inhibitor from the
trifluoromethyl oxadiazole and hydroxamic acid series, exhibits
favorable drug-like characteristics, potent anti-proliferative
effects, and significant anti-migratoru activity in Pca cells
(58). These advancements may
improve treatment options for patients with CRPC.
HDAC inhibitors in Pca
Currently, 13 clinical trials have been reported on
clinicaltrials.gov for the treatment of
metastatic prostate cancer using HDAC inhibitors. Of these 13
clinical trials, seven were completed, two trials are recruiting
patients, three trials were terminated and one was not recruiting
any patients. Of the seven completed clinical trials, three trials
were completed before 2010 and did not publish the results on Pca
(NCT00045006, NCT00005634 and NCT00020579). The results of the
other four trials were not promising. For instance, a phase II
study (NCT00106418) on 35 patients with metastatic CRPC revealed a
partial response to romidepsin (class I) in 2 patients (DoR, >6
months; PSA level decline of >50%). A total of 11 patients
discontinued the treatment due to of toxicity. The clinical trial
NCT01075308 was conducted on 32 patients with CRPC using SB939
(class I, II and IV HDACs), which is an inhibitor of HDAC. SB939
did not exhibit sufficient activity based on the PSA response rate
(2 patients responded, 7 patients had stable disease at 1.6-8
months) (59). Similarly, a phase
II trial of intravenous panobinostat (NCT00667862) demonstrated a
decrease in PSA levels in only 14% of patients with metastatic
hormone refractory Pca (n=35); however, the decrease was not
>50%. The major adverse effects observed were fatigue and
thrombocytopenia. Furthermore, in a phase II clinical trial
(NCT00330161), 27 patients with progressive metastatic Pca received
vorinostat 400 mg (class I, II) daily continuously, and all
patients had previously received chemotherapy. Of the 27 patients,
11 discontinued therapy due to toxicity and 13 patients had
progressive disease. Stable disease was observed in 2 patients
(7%), which was the optimal objective response in that trial. Data
analysis indicated that elevated levels of IL-6 contributed to drug
failure in patients with progressive CRPC. A >50% decline in
PSMA levels was not observed in any patient. These clinical trials
exhibit differences in the type of HDAC inhibitors used, the number
and type of patients, and the outcome of the trials. They also
suggest that PAN inhibitors are comparatively and slightly more
effective than inhibitors of individual classes. Overall, the
results of clinical trials suggest that HDAC inhibitors such as
practinostat (SB939), panobinostat, romidepsin and vorinostat are
ineffective against metastatic Pca as a monotherapeutic agents.
Relatively insufficient responses have been observed in patients
with mCRPC, which explains why no phase 3 studies for these drugs
in patients with CRPC have been completed to date; therefore,
combinatorial treatment therapies are ongoing. For example, a phase
2 trial involving 55 patients with CRPC treated with oral
panobinostat and bicalutamide demonstrated improved radiographic
progression-free (rPF) survival in those who had developed
resistance to second-line antiandrogen therapy (60). However, adverse events were higher
with a high dose of panobinostat (40 mg) (27.5%) than at a low dose
of 20 mg (11.5%). These events require low-dose panobinostat for
mCRPC. Notably, a low dose of the drug also reduced the PSA
response from 24 weeks to 5.9 weeks (60). These results suggest that
panobinostat effectively overcomes bicalutamide-induced androgen
resistance in a significant number of patients with CRPC.
Additionally, panobinostat is regarded to be less toxic to patients
compared to other HDAC inhibitors, such as romidepsin and
vorinostat (61). Similarly,
trials on talazoparib in combination with belinostat against mCRPC
are ongoing to observe dose-limiting toxicities in the first 10
weeks or two cycles of treatment (NCT04703920). The study will be
completed in December, 2024. Overall, the results suggest that
grade 3 and 4 toxicities are the main reasons for discontinuing
treatment with HDAC inhibitors, either alone or in combination
therapies. While HDAC inhibitors are approved for certain
hematologic malignancies, their role in prostate cancer remains
less well-defined.
Future research directions
A number of studies have suggested the significance
of HDAC inhibition using various drugs, suggesting that less toxic
HDAC inhibitors need to be developed. For instance, in a previous
study, the adamantyl-capped HDAC inhibitor, CN133, was shown to
have low IC50 values against HDAC1 (IC50, 0.6 nM), 2 (IC50, 2 nM)
and 3 (IC50, 0.3 nM) in comparison to vorinostat (IC50, 4, 11 and 3
nM) (62). Vorinostat was more
effective against HDAC6 (IC50, 2 nM) than CN133 (IC50, 4.1 nM).
CN133 suppressed the proliferation, migration and invasion of 22Rv1
CRPC cells more effectively than vorinostat. CN133 also suppressed
AR signaling (62). Tumor volume
and weight in CN133 treated mice with tumor xenografts were also
effectively reduced by 50% in comparison with the placebo group.
They also developed a PET/CT imaging method to directly observe the
therapeutic effects of HDAC inhibitors (62). These results suggest the following:
i) Every HDAC inhibitor is not effective against all HDACs, and
therefore, it is essential to first characterize HDACs inhibitors
against all HDACs at the preclinical level; and ii) to then use
them precisely after obtaining biomarker panel reports of patients.
PAN inhibitors targeting all HDAC inhibitors may not be effective,
but may rather have toxic effects.
Hybrid molecules of enzalutamide and SAHA were
designed to have weak pan-anti-HDAC activity to target HSP90 and AR
in enzalutamide-resistant PCa cells. The hybrid molecule was named
2-75. In comparison to enzalutamide, the hybrid molecule induced
the degradation of both AR and HSP90 with more potency than SAHA in
Pca cells by preventing its translocation to the nucleus. The
cytoplasmic retention property allows this inhibitor to selectively
inhibit HDAC6 and has a limited impact on other HDACs. In addition
to targeting the AR, 2-75 also downregulated the expression of
AR-V7, suggesting enhanced AR degradation in 2-75-treated cells
(63). In vivo studies
demonstrated that compared to enzalutamide, 2-75 significantly
reduced tumor growth, increased apoptosis, and inhibited AR nuclear
accumulation in LNCaP tumor xenograft models. These studies suggest
that 2-75 can selectively inhibit HDAC6 and overcome the drug
resistance and toxicity properties associated with classical AR
antagonists, HDAC inhibitors, and Hsp90 inhibitors (63,64).
Similarly, a CUDC-101 hybrid molecule with a HDAC inhibitory
fragment and an EGFR inhibitory scaffold from erlotinib (an EGFR
inhibitor) was designed. It suppressed AR, AR-V7 and HER2
expression, and upregulated p21 expression. It also significantly
reduced tumor growth in xenograft mice model having aggressive
22Rv1. However, the ethinylphenyl residue of erlotinib can be
oxidized by cytochrome P450 enzymes to phenol, potentially causing
toxic effects. Similarly, CUDC-101 is a substrate for ABC
transporters, which can contribute to drug resistance (65-67).
The EGFR inhibitor, gefitinib, can reverse drug resistance to
CUDC-101, leading to the development of chimeric HDAC inhibitors
with gefitinib-derived cap scaffolds. The chimeric molecules
3ClQuin-SAHA and 3BrQuin-SAHA have been shown to inhibit HDAC,
reduce EGFR expression similar to vorinostat and exhibit only
marginal non-specific toxicity. They have also been shown to induce
apoptosis and inhibit angiogenesis in DU145 cells (68). Therefore, this chimeric compound
may be a more suitable candidate for anticancer therapy in Pca, as
it aims to reduce both toxicity and drug resistance. Additionally,
CUDC-907 is a promising HDAC/kinase inhibitor that targets HDAC
enzymes along with PI3K. Chimeric compound induced apoptosis in Pca
by suppressing myc expression. Therefore, c-Myc expression should
be measured before beginning any trials on patients with Pca.
CUDC-907 was not found to be a substrate of ABCB1 transporters, but
was a substrate for ABCB2 transporters; therefore, the drug could
be combined with ABCB2 inhibitors in patients with Pca having
altered myc status (69,70). These studies strongly suggest that
more specific HDAC inhibitors should be developed and further
characterized to reduce the toxicities associated with pan-HDAC
inhibitors. Inhibitors should be tested as combinatorial
treatments. It is suggested that with the development of
bioinformatics tools (AdmetSAR, Osiris, molecular docking, MD
simulation, MM-GBSA, C-ImmSim, etc.), it would be beneficial to
design and screen multiple drugs at the bioinformatics level before
conducting in vitro or in vivo experiments. Certain
phytochemical drugs (caffeic acid, gallic acid, quercetin,
apigenin, luteolin, resveratrol, grape seed, curcumin, etc.) also
have anti-HDAC activity and, therefore, should be further
characterized due to their low toxicity profile (71). The active ingredients of these
phytochemicals need to be isolated and characterized to achieve
improved outcomes and low IC-50 values.
4. Prostate-specific membrane antigen
PSMA is encoded by the folate hydrolase 1
(FOLH1) gene and has three domains: i) An extracellular
domain of 707 amino acids; ii) a transmembrane domain of 24 amino
acids; and iii) an intracellular cytoplasmic domain of 19 amino
acids. The intracellular domain contains a motif (MWNLL)
responsible for the internalization of bound PSMA via
clathrin-coated pits. The deletion of this motif significantly
reduced PSMA internalization. Therefore, the fusion of drugs to
these 5 amino acids will result in drug-bound PSMA internalization
and, thus, the presence of concentrated drug within tumor cells.
FOLH1 is located on the short arm of chromosome 11. In addition to
its expression in benign and malignant prostate epithelium, it is
also expressed in a variety of other tissues, such as the proximal
renal tubules, salivary glands, liver, esophagus, stomach, small
intestine, colon, breast, fallopian tubes, testicular seminiferous
tubules, hippocampal neurons and astrocytes, ependyma, cortex and
medulla of the adrenal gland and ovarian stroma. The varying
expression of PSMA across different types of cancer, such as lung,
breast, ovarian, renal, glioblastoma and colon cancer, along with
its relatively low levels in non-prostate malignancies, poses
challenges for developing PSMA-targeted therapies beyond Pca
(72). The amount of PSMA in
cancer cells can vary depending on the type of cancer; it sometimes
it can be higher, lower, or may remain the same. As PSMA is found
in several types of cancer, it has become a promising target for
novel cancer treatments. Scientists are developing treatments, such
as specific antibodies, radioactive agents, or immune-based
therapies that specifically target PSMA. These treatments are being
tested in clinical trials to help attack cancer cells with PSMA,
while causing less harm to healthy cells (73). Of note, >70% of primary tumors
exhibited PSMA expression on the new blood vessels associated with
the tumors. It was shown that medullary thyroid carcinomas and
hepatocellular carcinomas were the most likely to express PSMA in
their neovasculature. On the other hand, adenoid cystic carcinoma
and papillary renal cell carcinoma showed PSMA expression on the
neovasculature in only a few cases. Notably, while the majority of
solid cancers do not exhibit PSMA expression on tumor cells, it has
been observed in salivary gland tumors and, to a lesser extent, in
tissues of hepatocellular carcinoma, lung cancer and BC (73). In summary, while PSMA is most
well-known for its role in Pca, it is also expressed in several
other types of cancer, and its presence is being studied for its
potential as both a diagnostic biomarker and a therapeutic target
in those cancers. In the case of Pca, PSMA is detected at a modest
level, but demonstrates a 100-1,000-fold increased expression in
prostate adenocarcinoma when compared to normal prostate tissues.
It is positively associated with various measures of tumor
aggressiveness, such as Gleason grade, tumor stage, biochemical
recurrence and castration resistance. Therefore, higher levels of
PSMA are associated with the worse clinical outcomes (74). PSMA is an attractive target for
theragnostic purposes for several reasons, such as a selective high
expression in Pca with a limited expression in benign prostate
tissue, targeting of the extracellular domain by antibodies or
small-molecule ligands, and the internalization of drug-bound PSMA
by the specific motif present in the cytoplasmic domain. The
physiological role of PSMA is to support glutamate synthesis in the
brain and folate absorption in the intestines. However, its
physiological role in Pca cells remains poorly understood. With the
rapid advancement of PSMA-targeted therapies and imaging agents,
these are currently used in prostate cancer screening, risk
stratification for recurrence and ongoing therapy monitoring. Thus,
it is essential to clarify the regulation and function of PSMA in
Pca to enhance the precision and maximize the benefits of
PSMA-targeted therapies.
PSMA as a therapeutic target and
challenges. Clinical relevance
PSMA is used as a therapeutic antigenic target for
antibodies and small molecule inhibitors. When combined with
radionuclides, these molecules can be used in the treatment of
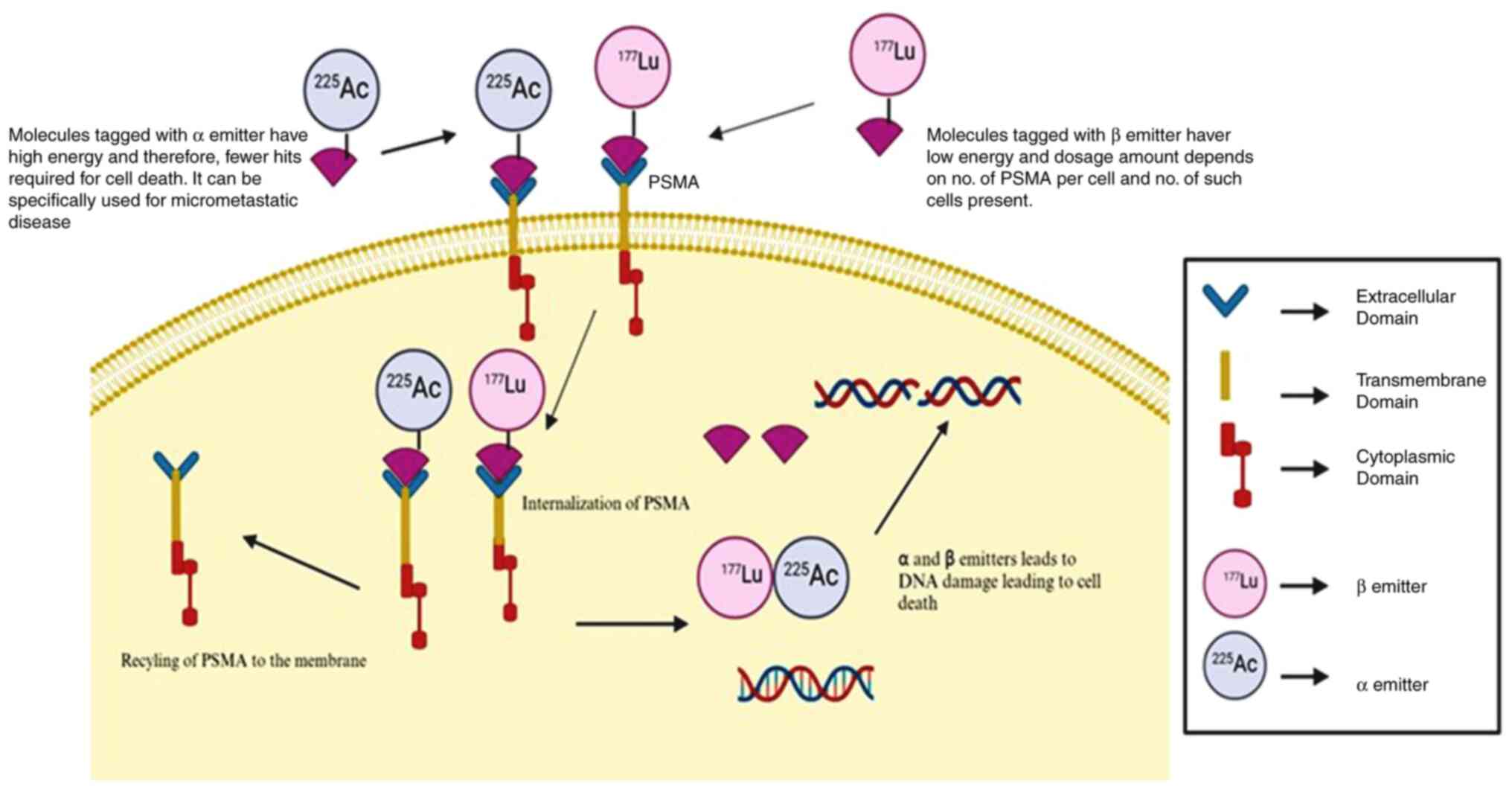
mCRPC (Fig. 2). The radionuclides
that have been used in clinical trials of mCRPC are β-particle
emitters (177Lu, 90Y) and α-particle emitters
225Ac and 227Th. However, the majority of
trials have used β-particle emitters linked to PSMA domains either
alone or in combination with chemotherapies. Alpha particles
possess significantly higher energy, meaning that they require
fewer hits to kill cancer cells compared to β-emitters. The use of
alpha particles may be particularly effective for treating
micrometastatic disease, as a single alpha particle in close
proximity to a microdeposit of prostate cancer could be sufficient
to destroy cancer cells. By contrast, the amount of drug delivered
to a specific metastatic site depends on the density of PSMA
coverage (PSMA expression level in one cancer cell and the number
of cancer cells expressing PSMA) (Fig.
2). Given the physical properties of alpha particles, an alpha
emitter specifically targeting prostate cancer cells is likely to
be the most effective treatment for micrometastatic disease.
However, the optimal approach could be to combine different
emitters with various targets, including PSMA.
Clinical trials and challenges. Combinations
of α- and β-emitting agents have been developed. Clinical trials
for combination therapy with the α-ray-emitting agents
223Ra and 177Lu-PSMA-I&T (NCT05383079,
AlphaBet), 225Ac-J591 antibody, and 177Lu-PSMA-I&T
(NCT04886986) are underway. In the case of Pca, there are currently
no clinical trials that compare alpha and β emitters that have been
linked to targeted molecules. However, in the case of colorectal
carcinoma, 213Bi-IMP288 (alpha emitter) was feasible and
at least as effective as 177Lu-IMP288 (beta emitter).
IMP288 is an anti-histamine-succinyl-glycine hapten peptide. The
survival rate was comparatively higher with a low dose of alpha
emitters (45 days) than with the β emitter (42 days). However,
higher doses of alpha emitters lead to kidney toxicity and require
dose optimization (75). Clinical
trial studies have utilized alpha- or β-emitting radionuclides,
such as NCT03545165, NCT03805594, NCT03658447, NCT04343885,
NCT03874884, NCT04419402, NCT00195039 and NCT00538668 for
177Lu, NCT03276572, and NCT03724747 for alpha emitters.
The results of these trials have been discussed elsewhere (76) and are not included in the present
review. Overall, it was shown that β-radionucleotides or
PSMA-targeted monoclonal antibodies (177Lu-PSMA-617) can
reduce PSMA levels by ≥50% in 59-66% of mCRPC. For
225Ac-PSMA-I&T, PSA50 was observed in 50% (7/14) of
patients (77). These patients
received prior AR treatment. The results of 225Ac-PSMA-617 in
chemotherapy-naive patients with advanced-stage Pca were more
promising (78). PSA90 was
observed in 14/17 (82%) patients. Of note, 1 patient developed
grade 4 renal toxicity. The notable therapeutic efficacy reported
in that study could be achieved with reduced toxicity to salivary
glands due to de-escalation of administered activities in
subsequent treatment cycles (78).
Efforts have been made to reduce these effects by using several
approaches, such as conjugation with an antibody to reduce salivary
gland distribution (may have less exposure to tubules), dose
fractionation, dose titration, and salivary gland protective
measures (76), although their
effectiveness remains unclear. For instance, clinical trials on
alpha emitter-based therapy have been conducted using PSMA-targeted
antibodies. Owing to their larger size, antibodies require a longer
circulation time. In a previous study, 32 patients who were
refractory to or who refused standard treatment options (including
androgen receptor pathway inhibitors) and had received or been
deemed ineligible for taxane chemotherapy were included. A total of
46.9% had at least 50% PSA decline at any time, with the majority
of patients demonstrating hematology-based high-grade adverse
events (79). Further trials are
required on different types of alpha emitters (such as
213Bi), and head-to-head comparisons of alpha and beta
emitters (specifically177 Lu-PSMA-617, and 177Lu-J591 or
90Y-J591) are also required.
Likewise, modifications to the PSMA-binding domains
can affect the pharmacokinetic and pharmacodynamic properties,
thereby influencing both antitumor activity and toxicity profile.
For instance, a 177Lu-labeled PSMA ligand
[DOTAGA-(I-y)fk(Sub-KuE or PSMA-I&T)] leads to a PSA decline of
>50% with no clinically significant hematological toxicity,
nephrotoxicity, thrombocytopenia, or xerostomia. However, similar
to studies involving 177Lu-PSMA-617, the most frequently
reported adverse events were grade 1-2 anemia, leukopenia, and
transient xerostomia (80).
PSMA-directed immunotherapy can involve various
approaches: i) The creation of artificial T-cell receptors
containing PSMA-specific monoclonal antibodies; and ii) the
creation of bispecific T-cell engagers (BiTEs), such as targeting
both PSMA and CD3 T-cell receptors to induce T-cell activation.
These agents include AMG160, pasotuxizumab/AMG212/BAY2010112
(NCT03792841 and NCT01723475); iii) the infusion of dendritic cells
with PSMA peptide; iv) recombinant soluble PSMA protein as a
vaccine; v) targeting pathways that crosstalk with PSMA, such as
the PI3K/Akt/mTOR pathway; vi) trispecific T-cell-activating
constructs, such as HPN424, which includes a PSMA-targeting domain,
a CD3-targeting domain, and a third domain that noncovalently binds
to serum albumin to extend its half-life, is currently in phase 1
clinical development (NCT03577028).
Of note, eight studies have been reported on
clinicaltrials.gov and are currently working on
PSMA-directed immunotherapies. Of these eight studies, four studies
have been completed, one is in an inactive state, one is withdrawn,
and the remaining two are currently recruiting patients. The first
completed study was a phase I trial of 177Lu-PSMA-617 and
pembrolizumab in patients with mCRPC. In this trial, 37 patients
were selected based on their high PSMA expression levels. These
patients received prior treatment with docetaxel and an androgen
receptor-targeted agent; PSA50-RR was 76%. The 12-month rPFS and OS
rates were 38 and 83%, respectively. Common treatment-related
adverse events were mainly grade (G) 1-2, including xerostomia,
fatigue, pruritus, nausea, rash, diarrhea, anorexia,
thrombocytopenia, anemia, neutropenia, elevated ALT, arthralgia and
a flare in bone pain. Five patients (14%) discontinued
pembrolizumab because of toxicity (81). The second completed study included
133 patients with mCRPC refractory to AR pathway inhibitor therapy
and taxane-based chemotherapy received targeted acapatamab.
Acapatamab (AMG160) is a prostate-specific membrane antigen (PSMA)
x CD3 bispecific T-cell engager. PSA50 was observed in 30.4% of
patients, and radiographic partial responses in 7.4% with a median
PSA PFS of 3.3 months. The results of that study did not mention
anything regarding the toxicity profile (82). The third completed study included
31 and 9 mCRPC patients were treated with subcutaneous (SC) or
intravenous injections of pasotuxizumab, respectively. PSA50 was
observed in 28 and 9.6% of the patients in the SC and continuous
intravenous cohorts, respectively. All SC cohort patients developed
anti-drug antibodies; Therefore, continuous intravenous injections
were administered. The change in the sponsor terminated the
continuous intravenous study (83). The completed study 4 was an
open-label, phase 1/2a study of HPN424 as a monotherapy aimed to
evaluate the safety, tolerability, and pharmacokinetic profile of
drugs in patients with advanced-stage Pa who were refractory to
androgen therapy. In comparison to other bispecific platforms,
HPN424 was optimized for its small size and increased stability.
That study involved 80 patients who received targeted doses ranging
from 1.3 to 160 ng/kg as a fixed dose and up to 300 ng/kg with step
dosing following an initial priming dose. The most frequently
observed grade >3 treatment-emergent adverse events were
increased AST levels, anemia and ALT levels. Dose-limiting
toxicities included grade 3 cytokine release syndrome, elevated
lipase levels and seizures. However, these events did not hinder
the dose escalation. A total of 63 patients continued the treatment
for >24 weeks; 3 of these 63 patients had PSA50 responses. That
study was terminated early (84).
These studies vary significantly in several aspects, including the
type of therapy, patient population size, prior treatments received
and PSA50 responses, making direct outcome comparisons
challenging.
As regards the recruiting studies, the first
recruiting study evaluated the safety and tolerability of AMG 509
in adult mCRPC participants (NCT04221542). The second recruiting
study was a phase 1 trial of 177Lu-PSMA-617 and olaparib in
patients with mCRPC. In that study, 48 patients were recruited and
received prior treatment with docetaxel and an androgen receptor
pathway inhibitor. The PSA50-RR was 62% (18/29) and PSA90-RR was
48% (14/29). Of note, 5 of the 7 patients (71%) with measurable
RECIST disease exhibited a partial response. Common
treatment-related adverse events included xerostomia, anemia,
thrombocytopenia, neutropenia, nausea, fatigue, constipation,
anorexia, vomiting and diarrhea. These toxicities were transient
and without clinical sequelae (85).
PSMA-directed immunotherapies. The results of
PSMA-directed immunotherapies were not as significant as those of
radionucleotide-based therapies. For instance, PSA50 is in the
range of 59-66% in the case of radionucleotide-based therapies
(76), while for PSMA-directed
immunotherapies, it is 28-30.4%. The 62-76% response was achieved
when 177Lu-PSMA-617 was combined with pembrolizumab or olaparib.
Additionally, adverse outcomes were not mentioned in the majority
of studies. Toxicities were manageable in the trispecific
T-cell-activating construct (HPN424). However, PSA50 was only
observed in 3 out of 63 patients. These patients had already
received two prior systemic therapies. However, a description of
these therapies has not yet been provided. PSMA-directed
immunotherapies indirectly target prostate tumor cells, which can
also affect nearby cells that have low levels of PSMA. Even a small
amount of PSMA can activate T-cell-mediated immunotherapies.
Therefore, clinical trials have not selected patients based on PSMA
expression levels.
Future research directions. i) Alpha and beta
emitters have different properties; therefore, head-to-head
comparisons of these emitters would be beneficial in the treatment
of patients with mCRPC. Therefore, additional alpha and beta
emitters should be characterized for mCRPC. Alpha emitters will
require lower doses. In the case of lutetium PSMA, the primary
sites of overexpression in healthy tissues include the salivary and
lacrimal glands, which account for the symptoms of dry eyes and
mouth. Due to its ability to target cancer cells directly, the
lutetium has the potential to induce cytotoxic effects in various
tissues. Therefore, combinatorial therapies, including both alpha
and beta emitters with dose optimization or fractionation, may
benefit mCRPC. A small number of trials are underway, and the
results will shed light on this issue. The PSMA-targeted alpha
emitter (225Ac) has shown promising results in
chemotherapy-naïve mCRPC patients (n=17). The results suggest that
alpha emitters could be more effective in treating
chemotherapy-naïve patients, and thus, more recruitment of patients
is required at the global level. ii) Additionally, linking these
radionuclides with different PSMA domains should also be attempted
at preclinical and clinical levels. iii) The results of
PSMA-directed immunotherapies suggest that they should be combined
with other therapies such as AR blockade, PARP inhibitors, and
other cross-talk chemotherapies. iv) Patients with bone metastases
should receive anti-resorptive therapy. A balance between toxicity
and efficacy is a requirement for clinical studies. Patients should
be divided into two cohorts: a) They have not received prior
treatments; and b) have received prior standard treatments. This
should be the minimal requirement, as radionuclide therapy shows
promising results in naïve patients. Therefore, a collaborative
approach should be used to include a greater number of patients.
The collaborative approach will provide broad spectrum and early
outcome of the studies.
5. Immune checkpoint proteins
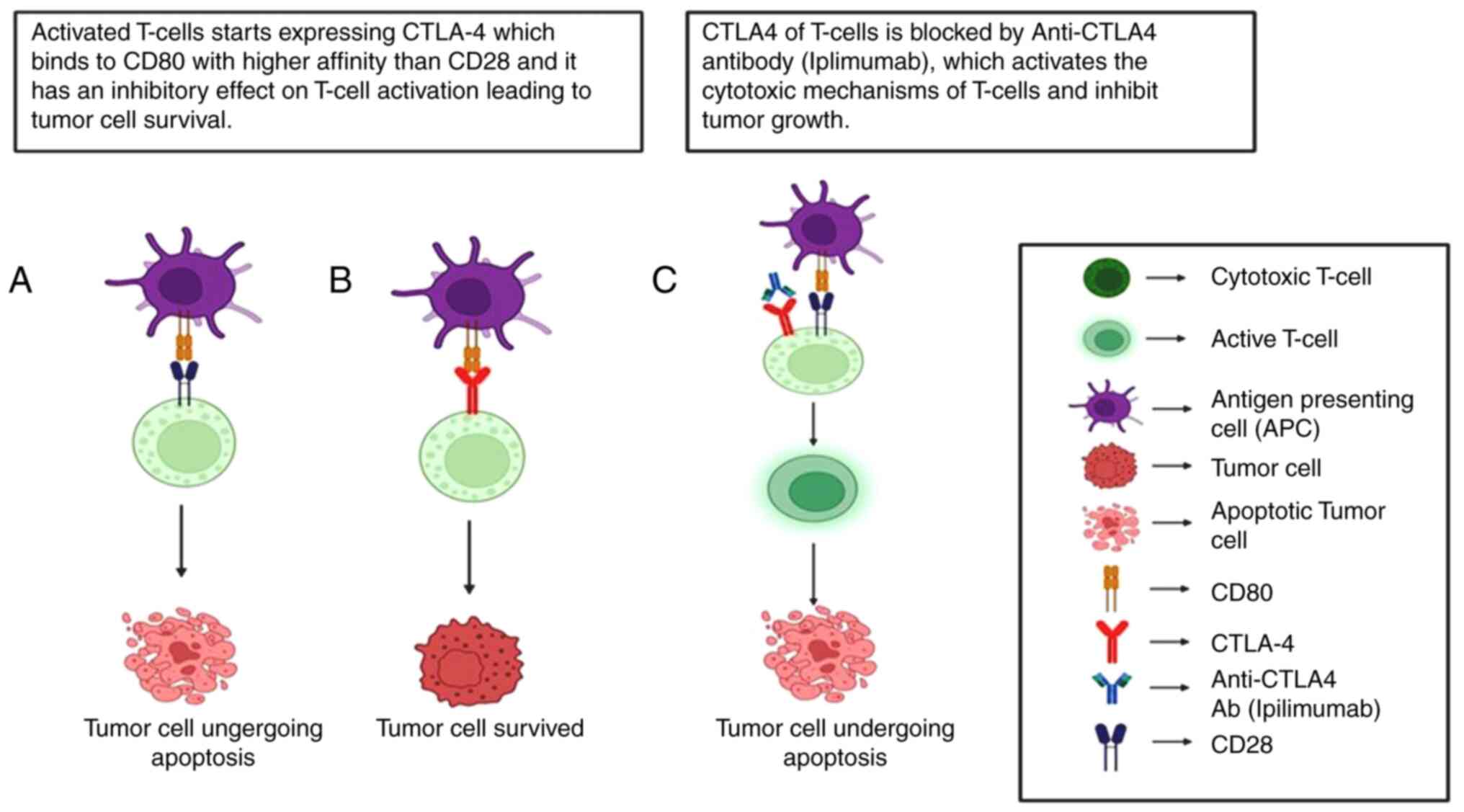
Cytotoxic T-lymphocyte associated protein 4 (CTLA-4)
and programmed cell death protein 1 (PD-1) are among the most
widely studied immune checkpoints. CTLA-4 functions early in the
immune response, whereas PD-1 is expressed later on mature
T-effector cells in peripheral tissues, including tumors. PD-1
binds to its ligands, PD-L1 and PD-L2, inhibiting T cell-mediated
tumor cell killing. Blocking PD-1/PD-L1 interaction enhances the
ability of T-cells to destroy tumor cells. T-cell activation occurs
when CD28 on T-cells interacts with CD80 and CD86 on
antigen-presenting cells. Once activated, T-cells begin to express
CTLA-4, which binds to CD80 and CD86 with greater affinity than
CD28. This competition between CD28 and CTLA-4 exerts an inhibitory
effect on T-cell activation, helping to prevent over-activation and
autoimmunity (Fig. 3).
Consequently, various immune checkpoint inhibitor therapies
targeting the PD-1/PD-L1 and CTLA-4 interactions have been
developed for cancer. The role of ICIs is generally to suppress the
immune response, but how it affects cancer outcomes is not
straightforward. For example, higher levels of CTLA-4 and PD-1 in
tumor samples have been linked to worse survival in nasopharyngeal,
liver cancer, but interestingly, better survival in non-small cell
lung cancer (86-88).
In summary, both CTLA-4 and PD-1 can suppress the immune system in
the tumor environment; however, their roles and effects can vary
depending on the type of cancer and the cells they are acting
on.
Clinical trials
Immune checkpoint inhibitors enhance the life
expectancy of patients with several solid tumors. However, CTLA-4
and PD-1 as monotherapy or in combination therapy achieve only
limited benefits in terms of biochemical and radiological responses
in advanced-stage Pca. For instance, in monotherapies, overall
survival ranges between 7.9-28.7 months while for combinatorial
therapies, it ranges between 8.5-23 months. However, it is very
difficult to compare clinical trials due to the different
characteristics of the patients, different drugs used, number of
patients and different primary endpoints. Overall, the results
suggest that patients with mCRPC should be treated after evaluating
certain parameters and biomarkers. For example, it was previously
shown that patients with mCRPC with alkaline phosphatase
concentration <1.5 ULN, a hemoglobin concentration ≥110 g/l and
no visceral metastases (n=146) had a significantly higher mOS in
the treatment group (22.7 months) when compared with placebo (15.8
months) (P=0.0038) (89).
Similarly, another study demonstrated that patients with high a
expression of PD-L1 IC2/3, CD8, CXCL9 and TAP1 experienced a longer
PFS in the atezolizumab and enzalutamide arm. This was also
observed alongside other potentially relevant biomarkers, including
alterations in phosphatase and tensin homolog (PTEN) (90). In another study, the ORR was higher
in chemotherapy-naïve patients treated with nivolumab plus
ipilimumab with PD-L1 ≥1%, homologous recombination deficiency, or
above-median tumor mutational burden (91). All three studies (89-91)
included patients with different characteristics who were treated
with different drugs (Table I)
(89-100).
In a single arm, phase II trial, it was shown that the efficacy of
ICIs, including PD-1 and CTLA-4 inhibitors, varied across subgroups
of patients with Pca on the basis of mismatch repair deficiency
(dMMR), non-synonymous tumor mutational burden (hTMB), a BRCA2
mutation (BRCAm), or biallelic CDK12 inactivation (CDK12i). It was
shown that dual ICIs exhibited modest responses in the hTMB,
BRCAm,and CDK12i subgroups, but demonstrated exceptional efficacy
in dMMR (101). Thus, careful
patient selection based on biomarkers may be essential for the
effective use of immune checkpoint inhibitors, helping to identify
specific subgroups of patients who are likely to benefit from these
treatment strategies. Heterogeneity in Pca encompasses genetic,
phenotypic and microenvironmental variations, posing challenges to
treatment and contributing to resistance. Addressing this
complexity is crucial for advancing precision therapies, enhancing
patient outcomes, and overcoming resistance in advanced stages of
the disease. For example, significant heterogeneity was observed in
the ipilimumab treated group. No significant heterogeneity was
found in the pembrolizumab and nivolumab plus ipilimumab subgroups
(102). Aligning patients with
the most suitable treatment is essential, especially in overcoming
the challenges of inter- and intra-patient heterogeneity and tumor
evolution. Advanced tools, such as imaging technologies,
next-generation sequencing, circulating tumor DNA, CTCs and
artificial intelligence provide promising avenues to enhance
patient selection and optimize treatment outcomes.
 | Table IClinical trials data of immune
checkpoint inhibitors in prostate cancer. |
Table I
Clinical trials data of immune
checkpoint inhibitors in prostate cancer.
| A, Monotherapy |
|---|
| Serial no. | Drug | Trial | No. of
patients | Eligibility of
patients | Outcome |
|---|
| 1. | Bone-directed
radiotherapy + ipilimumab 10 mg/kg | Phase III
NCT00861614 | 399: T 400: PC | Bone metastasis
from castration-resistant prostate cancer that had progressed after
docetaxel treatment | Patients with
alkaline phosphatase concentration (<1.5 ULN), haemoglobin
concentration ≥110 g/l, and no visceral metastases (n=146) had
significantly higher mOS in the treatment group (22.7 months) when
compared with placebo (15.8 months); P=0·0038. Overall mOS, PSA
reductions, and mPFS 11·2 months, 13.1, and 30.7% for ipilimumab
and 10·0 months, 5.2, and 18.1% for the placebo |
| 2. | Ipilimumab 10 mg/kg
vs. placebo | Phase III
NCT01057810 | 399: T 199: PC | -Asymptomatic or
minimally symptomatic-Chemotherapy-naive -No visceralmetastases
mCRPC | Treatment group
mOS:28.7 months, mPFS: 5.6 months, PSA response rate: 23%; deaths:
9 (2%), immune-related grade 3 to 4 AEs: 31% Placebo arm mOS, 29.7
months; mPFS, 3.8 months; PSA response rate, 8%; deaths: 0 arm.
Immune-related grade 3 to 4 AEs, 2% |
| 3. | Pembrolizumab 10
mg/kg | Phase II
NCT02054806 | 23 | -Advanced prostate
adenocarcinoma -unsuccessful standard therapy -measurabledisease
(RECIST v1.1) -PD-L1 expression: ≥1% of tumor or | mOS, 7.9 months;
mPFS, 3.5 months; DoR, 13.5 months; R, 4/23 patients; stable
disease, 8/23; AEs, 14 (60.9%) patients with no
pembrolizumab-related deaths or discontinuations occurred |
| 4. | Pembrolizumab 200
mg | Phase II
NCT02787005 | 258; 252
discontinued due to progression C1 group: 133 patients,
PD-L1-positive C2 group: 67 patients, PD-L1-negative C3:
bone-predominant disease, irrespective of PD-L1 | stromal cells.
Patients previously received ≥1 next generation hormonal agents and
1 or 2 chemotherapies, including docetaxel. | C1: Median follow
up, 9.5 months; ORR, 6%; DCR, 11%; PSA response, 6%; mOS, 10
months; OS at 24 months, 22%; AEs, 57% C2: Median follow up, 7.9
months; ORR, 3%; DCR, 6%; PSA response, 8%; mOS. 8 months; OS at 24
months. 16%; AEs, 60% C3: Median follow up, 14.2 months; DCR, 21%;
PSA response, 0%; mOS, 14 months; OS at 24 months, 21%; AEs,
71% |
| B, Combined
therapy |
| Serial no. | Drug | Trial | No. of
patients | Eligibility of
patients | Outcome |
| 1. | Pembrolizumab plus
enzalutamide | II NCT02787005 | n=126 C4:
RECIST-measurable (n=81) C5: bone-predominant (n=45) | -Previously receive
abiraterone and enzalutamide resistant | C4: mOS, not
reached; rPFS rates, 17%; AEs, 75% 12-month OS rate: 70% C5: mOS,
19 months; rPFS rates, 23%; AEs, 69%; 12-month OS rate, 75% |
| 2. | Pembrolizumab plus
olaparib | Phase 1b/2
NCT02861573 | n=84; 42 patients
discontinued due to progression; 57% of patients had measurable
disease |
-Docetaxel-pre-treated, molecularly
unselected patients with mCRPC with progression within 6 months of
screening per PSA or radiologic bone/soft tissue progression
enrolled.-may have received 1 other chemotherapy and ≤2
2nd-generation hormone therapy (HT) | OS: 14 months PSA
response rate, 7/82; rPFS, 4 months; ORR in measurable disease
(24/42 patients), 2/24 |
| 3. | Pembrolizumab plus
enzalutamide | Phase 1b/2
NCT02861573 | n=102; 73 patients
discontinued due to progression 39% patients measurable
disease | -Patients who
failed or became intolerant to ≥4 weeks of abiraterone in
prechemotherapy mCRPC state -whose disease progressed within 6
months of screening per PSA progression or radiologic bone or soft
tissue progression enrolled | Median follow-up:
13 months OS: 20.4 months PSA response rate, 22/101; rPFS, 6.1
months; ORR in measurable disease, 3/25 (12%) |
| 4. | Atezolizumab +
enzalutamide vs. enzalutamide | Phase III
NCT03016312 | 759 | mCRPC whose disease
progressed on abiraterone | -Longer
progression-free survival was seen with atezolizumab arm in
patients with high PD-L1 IC2/3, CD8 expression and established
immune gene signatures. -Exploratory analysis linked
progression-free survival in the atezolizumab arm with immune genes
such as CXCL9 and TAP1, together with other potentially relevant
biomarkers including phosphatase and tensin homolog alterations.
-No role for atezolizumab incombination with enzalutamide in
unselected patients with CRPC -Careful patient selection may be
required for immune checkpoint inhibitors to identify subgroups of
patients who may benefit from this treatment approach. |
| 5. | Nivolumab (NIVO)
plus ipilimumab | II NCT02985957 | 78 | Cohort 1:
Asymptomatic/minimally symptomatic patients who progressed after
2nd-generation hormone therapy and have not received chemotherapy
for mCRPC Cohort 2: same as cohort 1 and patients who progressed
after taxane-based chemotherapy | Cohort I: ORR, 26%;
PSA, 21%; Cohort 2: ORR, 10%; PSA, 13% ORR was higher in patients
with PD-L1 ≥1%, DNA damage repair (DDR), homologous recombination
deficiency (HRD), or above-median tumor mutational burden. |
| 6. | ADXS-PSA with
pembrolizumab vs. ADXS-PSA | Phase I/II
NCT02325557 | n=50 mCRPC, ≥18
years who received ≤2 prior chemo-/targeted-/immunotherapies or ≤1
prior chemotherapy in a metastatic setting Part A (PA; n=13):
ADXS-PSA every 3 weeks Part B (PB; n=37) ADXS-PSA, 200 mg
pembrolizumab every 3 weeks with a 4th pembrolizumab dose 3 weeks
later (in 12 weeks cycles), for up to 2 years or until
progression/toxicity | mCRPC, ≥18 years
who received ≤2 prior chemo-/targeted-/immunotherapies or ≤1 prior
chemotherapy in a metastatic setting Part A (PA; n=13): ADXS-PSA
every 3 weeks Part B (PB; n=37) ADXS-PSA, 200 mg pembrolizumab
every 3 weeks with a 4th pembrolizumab dose 3 weeks later (in 12
weeks cycles), for up to 2 years or until progression/toxicity | PA: PSA reduction,
14%; mOS, 8.5 months PB: PSA reduction, 43%; PSA 50, 7/15 patients
mOS, 23 months ADXS-PSA + pembro reduced PSA ≥50% and prolonged
OS |
| 7. | DNA vaccine
encoding prostatic acid phosphatase (PAP) + pembrolizumab | Pilot Trial | n=26 | Group 1: receive
vaccine and pembrolizumab together (n=13) Group 2: receive vaccine
for 12 weeks and then pembrolizumab for 12 weeks (n=13) | Group 1: PSA
decline, 62% Group 2: PSA decline, 8% PSA declines were associated
with the development of PAP-specific Th1-biased T cell immunity and
CD8+ T-cell infiltration in metastatic tumor biopsy
specimens. |
| 8. | Ipilimumab and a
poxviral vaccine targeting prostate-specific antigen | Phase I
dose-escalation trial, NCT00113984 | 30; 24 of whom had
not been previously treated with chemotherapy | mCRPC | Of the 24 patients
who were chemotherapy-naive, 14 (58%) had PSA declines from
baseline, of which six were greater than 50%. Of the remaining 6
patients, PSA decline was observed in 1 patient. The use of a
vaccine targeting PSA that also enhances co-stimulation of the
immune system did not seem to exacerbate the immune-related adverse
events associated with ipilimumab. |
| 8. |
Granulocyte-macrophage colony-stimulating
factor-transduced allogeneic prostate cancer cells vaccine (GVAX) +
ipilimumab | Phase 1
dose-escalation trial NCT01510288 | 28: 16 patients
with 3·0 mg/kg level of GVAX | Eligible patients
had documented mCRPC and had not been previously treated with
chemotherapy. | GVAX combined with
3·0 mg/kg ipilimumab is tolerable and safe for patients with mCRPC.
PSA50: 25% (7/28) |
| 9. | Pembrolizumab plus
prostatic cryotherapy | Pilot Trial
study | 12 | Newly diagnosed
oligometastatic prostate cancer between 2015 and 2016 | PSA decline: 42%
(5/12); mPFS:14 months, and mSTFS: 17.5 months. PD-L1 expression:
Not detectable by IHC. AEs: grade ≤2, and there were no apparent
complications from cryotherapy. |
Vaccines or cryotherapy, in combination with
immunotherapies, have shown promising results (Table I) (103,104). However, the number of patients
included in these studies was small; therefore, the inclusion of
more patients in subsequent trials is necessary. BiTEs are designed
to bind to both T-cells and tumor cells, thereby facilitating tumor
cell destruction by activated T-cells. One such BiTE, AMG 160,
targets PSMA on tumor cells and CD3 on T-cells. The role of AMG-160
has been studied in combination with the PD-1 monoclonal antibody
AMG 404 (NCT04631601) and with pembrolizumab (NCT03792841). The
NCT04631601 trial has been terminated, and trial NCT03792841 has
only been completed for a monotherapy arm that involves 43
PSMA-positive patients. In a trial involving 43 PSMA-positive
patients, 27.6% had a confirmed PSA response, 13.3% achieved a
confirmed partial response (PR), and 53.3% experienced stable
disease (42). Additionally,
XmAb®22841 is a bispecific antibody that targets both
CTLA-4 and LAG-3, aiming to enhance tumor-selective T cell
activation to improve therapeutic outcomes. This clinical trial was
designed to assess the maximum tolerated dose and/or recommended
dose of XmAb22841, both as a standalone treatment and in
combination with pembrolizumab. The study will focus on evaluating
the safety, tolerability, pharmacokinetics, immunogenicity, and
antitumor activity in patients with selected advanced solid tumors
(NCT03849469). To the best of our knowledge, the results of that
study have not been reported.
Overall, immune checkpoint inhibitor monotherapy for
Pca has not demonstrated a significant survival benefit. The
possible reasons could be innate resistance, the activation of
immune suppressive mechanisms by tumor cells, the response by both
PD-1-positive or -negative patients (105,106), and the lack of characterization
of tumor-infiltrating lymphocytes (TILs). For instance,
CD8+ TILs can suppress tumor growth in early-stage Pca.
However, the presence of both CD8+ TILs and PD-L1 in
node-positive PCa cases has been linked to disease progression
(107-109).
However, the results remain controversial. Similarly, immune
checkpoint inhibitor therapeutic responses in bone metastases were
lower than those in soft-tissue metastases due to TGF-β-mediated
signaling that favored Th17 and restrained Th1 lineage expansion.
As patients with mCRPC have bone metastasis, the optimization of
immunotherapies is warranted (110). Despite the FDA approval of
pembrolizumab for microsatellite-high tumors, the response rate of
microsatellite-high prostate cancer patients to anti-PD-1 therapy
has not been well documented (111).
Future research directions
i) The efficacy of immunotherapy can be enhanced by
selecting patients on the basis of biomarker panels, such as
mutations in BRCA1 and/or BRCA2, ATM, and CHEK2. ii)
The subgrouping of patients should be mandatory in every clinical
trial to provide high benefits to patients with mCRPC. iii)
Additionally, patient selection should be performed not only on the
basis of one biomarker, but also on the basis of
treatment-associated biomarker panels. For example, both
PD-1-positive and -negative patients may benefit from treatment, as
shown in previous studies (105,106). Therefore, it would be beneficial
to perform sequencing-based experiments at a preclinical level. iv)
The differentiation of therapy-resistant patients from
therapy-sensitive patients may prevent the side-effects of
therapies. v) Combined therapies including vaccines along with
immune checkpoint inhibitors should be performed at a larger scale
due to their beneficial effects on advanced-stage Pca. vi)
Collaborative clinical trials should be conducted using the same
drugs and similar types of patients. vii) Combined therapies
involving cross-reactive targets should also be evaluated. viii)
The evaluation of the vaccines with T-cell amplifiers and anti PD-1
therapy. Currently, one study is evaluating the effect of SLT-10
and GX-17 either alone or in combination with pembrolizumab in
patients with mCRPC. SLT-10 was developed by VaxiGen and is a DNA
vaccine, whereas GX-17 is a T-cell immunity amplifier by
interleukin-7(IL-7) fused to hyFc (developed by Genexine)
(NCT06344715).
6. PARP
PARP inhibitors have emerged as promising cancer
therapies. Single-strand breaks (SSB) are repaired by PARP enzymes
in the DNA. However, PARP inhibitors bind to PARP enzymes and
prevent SSB repair, leading to the generation of double-strand
breaks. Patients with mutations in homologous recombination repair
genes such as BRCA are unable to repair these breaks,
resulting in cell death through apoptosis. In addition to
BRCA, other DNA repair genes, such as ATM, FANCA, RAD51B,
RAD51C, MLH1, and MSH2 can also be used as markers in
the selection of patients with prostate cancer for PARP
inhibitor-based therapy. Of note, two PARP inhibitors, olaparib
(Lynparza) and rucaparib (Rubraca), have received FDA approval for
the treatment of mCRPC. These agents have slightly different
indications, dosages (olaparib, 300 mg twice daily; rucaparib, 600
mg twice daily) and adverse effect (AE) profiles. For example,
olaparib was selected for patients with mCRPC who showed resistance
to enzalutamide and abiraterone. However, rucaparib has been
selected for patients with mCRPC who have been treated with
AR-directed or taxane therapy. FDA breakthrough therapy designation
has been provided to expedite drug development. Niraparib (Zejula)
has received this designation by FDA for mCRPC. In addition to
niraparib, talazoparib (Talzenna) has emerged as a PARP inhibitor
for mCRPC treatment. The pharmacokinetic properties of these
inhibitors differ, as discussed elsewhere, and are not included in
this review (112). Approximately
18 drugs have been approved for the treatment of HER2-positive BC
by the FDA. However, the development of novel small-molecule drugs
that specifically target chemoresistance in triple-negative BC
(TNBC) remains challenging. PARP inhibitors are particularly
effective in treating TNBC, BCs and ovarian cancers that carry
BRCA1 or BRCA2 mutations. Emerging evidences also suggest their
role in ovarian and oral cancer (113). However, the development of novel
small-molecule drugs that specifically target chemoresistance in
various types of cancer remains challenging.
Clinical relevance
After the release of data from the phase 3 PROfound
study (NCT02987543), olaparib received FDA approval for the
treatment of mCRPC in 2020. PROfound study (NCT02987543). The
PROfound study was conducted on patients with mCRPC who had disease
progression while receiving a new hormonal agent (e.g.,
enzalutamide or abiraterone). Patients received olaparib,
enzalutamide, or abiraterone plus prednisone. Patients were divided
into two cohorts: i) Cohort A included genetic mutations in the
BRCA1, BRCA2, or ATM genes, ii) and cohort B included
mutations in 12 other prespecified genes. In both cohorts, patients
were treated with either olaparib or AR-therapy of the physician's
choice (cohort A: Olaparib-162 patients and AR-therapy-83 patients;
cohort B: Olaparib-94 patients and AR-therapy-48 patients). The
median OS in cohort A was higher than that in cohort B (19.1 months
vs. 14.7 months). Additionally, in the olaparib arm of cohort B,
the median OS was higher than that in the AR arm (14.1 vs. 11.5
months). Anemia and nausea were the main toxic effects in patients
who received olaparib. Recently, a PROfound study also showed the
superior effect of olaparib on improving quality of life by
reducing pain compared with either enzalutamide or abiraterone
(114). In the olaparib arm of
cohort A, pain progression was not reached, while it was 9.92
months in the control arm. The findings of that study indicate that
in patients with mCRPC who experienced disease progression while on
enzalutamide or abiraterone and had alterations in homologous
recombination repair, olaparib was associated with longer
progression-free survival and improved response measures as well as
improved patient-reported outcomes compared to either enzalutamide
or abiraterone (115).
In the phase 3 PROpel study, patients with mCRPC
were randomly assigned to receive three drugs, namely: i)
abiraterone, ii) prednisone/prednisolone, and iii) olaparib (300
mg) twice daily or the placebo. In that study, patients did not
receive any prior chemotherapy and were not selected based on HRR
mutation status. That study was conducted at 126 centers in 17
countries. Patients were divided into two groups: Group 1 received
abiraterone, prednisone/prednisolone, and olaparib, whereas group 2
received abiraterone, prednisone/prednisolone, and the placebo. The
median rPFS was higher in group 1 than in group 2 (24.8 vs. 16.6
months, P<0.0001). The most frequent adverse event of anemia in
cohort 1 was managed by applying two strategies: i) Dose reduction
or ii) temporary cessation of treatment (116,117). These findings suggest that,
irrespective of the HRR markers, olaparib had superior benefits in
cohort 1. However, other studies have shown the benefits of PARP
inhibitors in the presence of HRR mutations and therefore, these
findings need to be validated in subsequent studies (115,118). These studies suggest that it
would be beneficial to identify the importance of HRR biomarkers in
the selection of patients for PARP therapy.
Based on the results of the phase 2 TRITON2 trial
(NCT02952534) on mCRPC, rucaparib has received accelerated FDA
approval. In that study, the eligibility criteria for patients were
the presence of somatic or germline alterations in the HRR genes.
The patients received 600 mg of rucaparib twice daily. That study
included 115 patients with deleterious BRCA alterations. The
confirmed PSA response rate was 54.8% (95% CI, 45.2-64.1%)
(119). ORRs were similar for
patients with a germline or somatic BRCA alteration and for
patients with a BRCA1 or BRCA2 mutation, whereas a higher PSA
response rate was observed in patients with a BRCA2 mutation. The
results revealed a beneficial effect of the drug on patients with
Pca. Two ongoing phase 3 trials will assess the effect of this drug
either alone (TRITON3 study: NCT02975934) or in combination with
enzalutamide as first-line treatment for mCRPC (CASPAR study:
NCT04455750) (120). The results
from the previous phase 1b CASPAR trial demonstrated that 4 out of
8 patients (50%) had a confirmed PSA response (≥50%). Additionally,
1 patient with measurable disease achieved a confirmed complete
radiographic response following treatment with a combination of
rucaparib 600 mg twice daily and enzalutamide 160 mg once daily,
which was observed even in the presence of AR alterations and
absence of DNA damage repair gene alterations (121). The breakthrough therapy
designation of niraparib was based on data from the phase 2 GALAHAD
study (NCT02854436) (122). In
that study, 223 patients were divided into the BRCA (n=142) and
non-BRCA (n=81) cohorts. In the final analysis, the ORR in the
measurable BRCA cohort (n=76) was 34.2%. The median follow-up
duration was 10.0 months, and the median duration of response was
5.55 months (95% CI, 3.91-7.20). The phase 3 MAGNITUDE trial
(NCT03748641) is currently evaluating the combinatorial effect of
niraparib with abiraterone acetate and prednisone in men with
mCRPC. Patients with or without HRR gene mutations were eligible
for enrollment in this trial. To date, patients with HRR gene
mutations have shown survival benefits such as enhanced rPFS, ORR,
time to initiation of cytotoxic chemotherapy, symptomatic
progression, and PSA progression (122,123). Talazoparib is also being
evaluated for mCRPC in the phase 3 TALAPRO-2 study (NCT03395197).
TALAPRO-2 compares rPFS in two arms: arm 1, talazoparib plus
enzalutamide (with and without HRR mutations), and arm 2,
enzalutamide plus placebo (with and without HRR mutations). A total
of 20 secondary outcome measures, including OS and ORR, were also
included (124). In a
non-randomized phase I/II clinical trial, 17 patients had
previously received enzalutamide and/or abiraterone. Patients
received durvalumab 1,500 mg every 28 days and olaparib 300 mg
tablets every 12 h until disease progression or unacceptable
toxicity was observed. An rPFS of 16.1 months was achieved in
patients with mCRPC treated with olaparib and durvalumab, with an
rPFS rate of 51.5% within 1 year. Furthermore, patients with HRR
mutations exhibited a high rPFS probability (rPFS rate of 83.3%
within 1 year) compared to patients with no mutations in HRR
(36.4%) (125). The combinatorial
therapeutic regimen of olaparib and pembrolizumab also improved the
response in patients with mCRPC (regardless of HRR state; arm I)
when compared with olaparib (arm 2) or pembrolizumab (arm 3)
monotherapy (phase Ib/II KEYNOTE-365). The ORRs and PSA response
rates in arm 1 were higher in the patients with BRCA mutations than
in the BRCA-non-mutated cohorts (BRCA-mutated patients: ORR, 33%;
PSA response rate, 50%; BRCA-non-mutated cohort, ORR: 6%; PSA
response rate: 14%) (126). The
ORR for pembrolizumab and olaparib were 5 and 6%, respectively.
Overall, these results suggest that pembrolizumab + olaparib may
improve the PSA response rate regardless of HRR mutation
status.
Overall, the results of clinical trials suggest that
PARP inhibitors can exert beneficial effects with or without HRR
mutation status in patients. Therefore, an important question that
remains is whether biomarkers proofing is important in patients
treated with PARP inhibitors. PARP inhibitors also impact
downstream expression of antigen-regulated genes. Thus,
combinatorial therapies with PARP inhibitors may be advantageous in
certain populations. However, further studies are necessary.
Future research directions
Notably, the combination of PARP inhibitors with
novel hormonal therapies is expected to provide advantages to a
broader population of patients with prostate cancer. The following
considerations specific to PARP inhibitors can be advantageous for
patients with Pca: i) Identifying treatment-specific biomarkers to
PARP inhibitors both before and after beginning therapy; ii) using
a combination of various PARP inhibitors in combination therapies;
iii) identifying resistance-related biomarkers and targeting these
biomarkers in combination with PARP inhibitors; and iv) the
selection of patients on the basis of biomarker profiling. These
considerations could lead to improved survival rates for patients
with mCRPC.
7. PI3K/AKT/mTOR
The PI3K/AKT/mTOR signaling pathway is essential for
regulating cell growth and survival. It begins with activation of
the PI3K enzyme, which recruits and activates the AKT subfamily
(AKT1, AKT2 and AKT3). Once activated, AKT phosphorylates various
downstream effectors, including mTOR. In normal cells, the PI3K
pathway is controlled by negative feedback mechanisms, such as the
dephosphorylation of signaling molecules by PTEN. However, the
hyperactivation of this pathway has been observed in 49% of
patients with mCRPC (127). Of
these patients, 41% have loss-of-function aberrations in PTEN and
activating alterations in PIK3CA and AKT1. Moreover, the PI3K
pathway is activated by the inhibition of the AR pathway in
PTEN-deficient Pca, and can thus function as a resistance mechanism
to ARS inhibitors (128). The
PI3K/AKT/mTOR pathway is essential for regulating cell growth,
survival and metabolism, thus rendering it a critical target for
cancer therapy across several cancer types beyond pancreatic
cancer, including breast, gastric, ovarian, colorectal,
glioblastoma and endometrial cancers. The hyperactivation of the
PI3K/AKT/mTOR pathway can lead to treatment resistance in cancer
cells. Consequently, this pathway and its associated components
have emerged as promising targets for the development of anticancer
therapeutics aimed at overcoming resistance and improving treatment
efficacy (129). Therefore,
co-targeting the AR axis and PI3K pathways may overcome resistance
to AR-mediated therapy.
This concept was evaluated in the phase 3
IPATential150 trial, which randomized 1,101 patients with mCRPC to
receive first-line treatment with ipatasertib (400 mg once daily
orally) in combination with abiraterone (1,000 mg once daily
orally) and prednisolone (5 mg twice daily orally) or a placebo
alongside the same regimen of abiraterone and prednisolone.
Ipatasertib is a selective inhibitor of all three AKT isoforms
(130). The trial was conducted
at 200 sites across 26 countries or regions. In this trial, 554
(50%) patients were assigned to the placebo-abiraterone group and
547 (50%) to the ipatasertib-abiraterone group. All patients had
asymptomatic or mildly symptomatic mCRPC with progressive disease
and an ECOG score of 0 or 1. The median follow-up period was 19
months. The treatment group demonstrated a higher median PFS and
ORR compared with the placebo group (PFS, 19.2 months vs. 16.6
months; ORR, 61 vs. 44%). Immunohistochemical analysis indicated
that 521 patients experienced a loss of PTEN. Among these patients,
the median PFS was 18.5 months for the ipatasertib-abiraterone
group compared to 16.5 months for the placebo-abiraterone group
(HR, 0.77; P=0.034), with ORRs of 61 vs. 39%, respectively
(130). These data indicate that
a combinatorial approach targeting both the AKT and AR signaling
pathways with ipatasertib and abiraterone may serve as a promising
treatment for males with mCRPC and a loss of PTEN, a group that
typically has a poor prognosis. Other inhibitors of the PI3K
pathway, both as monotherapy and in combination, have also been
explored in early phase trials (Table
II) (131-140).
The majority of studies have completed phase 1 trials to determine
the recommended dose for phase 2 trials. One phase 2 trial revealed
a comparative beneficial effect on naïve patients with CRPC
(NCT00814788), whereas in another study, the presence of AKT was
considered a detrimental factor for disease progression
(NCT01051570) (Table II). In
patients with advanced-stage CRPC with abiraterone resistance,
samotolisib (LY3023414), along with the absence of AR-V7, may be a
better strategy (NCT02407054). By contrast, in patients with CRPC
with abiraterone resistance, apalutamide (ARN509) and everolimus
were not found to exhibit any beneficial effects (NCT02106507)
(Table II). Ongoing clinical
trials are listed in Table II. It
is suggested that careful patient selection is necessary to
understand their treatment-related genomic profiles.
| Table IIClinical trials data of PI3K/AKT/mTOR
inhibitors in prostate cancer. |
Table II
Clinical trials data of PI3K/AKT/mTOR
inhibitors in prostate cancer.
| A, Completed
trials |
|---|
| Clinical trials
ID/no. of patients | AKT/PI3K/mTOR
inhibitor | Phase | Treatment plan | Prior therapy | Outcomes |
|---|
|
NCT02215096/n=36 | PIK3 inhibitor
GSK2636771 in combination with enzalutamide | I | Fixed dose of
enzalutamide (160 mg) once daily received GSK2636771 in a dose
escalation manner to evaluate the long-term safety of the
combination as well as the 12-week non-PD rate-200 mg (n=22) 300 mg
(n=12) 400 mg (n=2) | Enzalutamide | Dose-limiting
toxicities (DLTs) occurred least at the dose of 200 mg and
therefore, this was considered the recommended dose |
| | | | | | -(DLTs: 200 mg:
n=1; 300 mg: n=2,400 mg: n=2). |
| | | | | | -PSA50: 4/34 (12%)
-radiographic partialresponse: 1/34 (3%) |
| | | | | | -12-week
non-progressive disease rate: 50% |
| | | | | | -limited antitumor
efficacy but adequate safety and tolerability with no new or
unexpected AEs. |
|
NCT02064608/n=20 | mTOR inhibitor
AZ12729279 | I | This is a single
arm study whereby a cohort of 20 patients with early high risk
prostate cancer were treated with a 15-day course of AZD2014 (mTOR
inhibitor) treatment prior to radical prostatectomy. | Information not
provided | -PSA decline: 3/20
(15%) over two-week dosing period. |
| | | | | | -Mean fasting
plasma glucose before treatment: 4.88±0.56 mmol/l |
| | | | | | -Mean fasting
plasma glucose after treatment: 6.16±0.78 mmol/l |
| | | | | | -mTOR1/2 (pS6,
4EBP1 and NDRG1 were inhibited |
|
NCT02091531/n=9 | mTOR inhibitor
MLN0128 | II | Patients were
treated with the established phase II dose of MLN0128 (4 mg p.o.
daily continuously; 1 cycle=4 weeks) to assess mechanisms of
sensitivity and enzalutamide and/or abiraterone resistance | Abiraterone acetate
and/or enzalutamide | -All patients had a
rise in PSA on treatment, with a median 159% increase from baseline
(range: 12-620%). |
| | | | | | -Eight patients
discontinued treatment early because of radiographic progression,
grade 3 toxicity, or investigator discretion. |
| | | | | | -AR activation
result from mTOR inhibition, and poor inhibition of mTOR signaling
targets (pAKT, 4EBP1 phosphorylation, and eIF4E
activity) |
| NCT03618355/n=14; 2
patients withdraw due to dose limiting toxicity | eRapamycin
(encapsulated rapamycin) | I | Experimental:
Cohort 1: 0.5 mg weekly (n=3), Cohort 2; 1 mg weekly (n=3); Cohort
3: 05 mg daily (n=6) oral administration of encapsulated rapamycin
Patients were treated for 3 months and followed for 6 months to
assess safety, PSA, pharmacokinetics, immune response, and quality
of life (QOL) | Immunosuppressed
and ADT by CYP3A4 a cytochrome inhibitor | -Central memory CD8
T-cells and CD3+/CD56+ NK cells were more
prevalent in cohort 3 than other cohorts at 1 month and 3
months. |
| | | | | | -dose of 0.5 mg
daily produced stable serum rapamycin through the duration of
treatment and resulted in a positive immune impact. |
| | | | | | -no significant
change in PSA level |
| | | | | | -no patients
clinicallyprogressed on therapy |
| NCT01548807/n=16: 1
withdraw | mTOR inhibitor
everolimus and radiation therapy | I | 14 days everolimus
daily therapy (5,7.5 or 10 mg/daily) followed by prostate bed
irradiation (15 IMRT, 1 proton) with daily cone beam CT
localization in 37 fractions of 1.8 Gy each (total dose 66.6 Gy)
for the treatment of BCR following prostatectomy. | Prostatectomy and
salvage radiotherapy | -No dose-limiting
toxicities were observed. |
| | | | | | -five patients(33%)
developed secondary biochemical recurrence (sBCR). sBCR was defined
as a rise in PSA of >0.5 ng/ml above nadir following the
completion of protocol therapy. |
| | | | Median follow-up
time: 20.1 months | | -Median time to
sBCR: 33.8 months |
|
NCT02407054/n=129 | Samotolisib
(LY3023414) | II | Participants
received 200 mg of LY3023414 BID in combination with 160 mg of
enzalutamide QD beginning Cycle 1 day 1. A treatment cycle was
defined as 28 days | Advanced CRPC with
progression on prior abiraterone | -Median rPFS in
samotolisib/enzalutamide vs. placebo/enzalutamide arm: 10.2 vs. 5.5
months; P=0.03 |
| | | | | | -rPFS inpatients
without AR-V7 in the samotolisib/enzalutamide vs.
placebo/enzalutamide arm: 13.2 months vs. 5.3 months; P=0.03 |
|
NCT00459186/n=12 | Everolimus and
docetaxel | I | Everolimus 10 mg
daily for 2 weeks, followed by Everolimus + docetaxel at one of
three doses: 5 mg everolimus and docetaxel at 60 mg/m2,
10 mg everolimus and docetaxel at 60 mg/m2, and 10 mg
everolimus and docetaxel at 70 mg/m2. everolimus was
given daily. Docetaxel was given every 3 weeks by intravenous
infusion. Patients also received prednisone 5 mg by mouth twice
daily. | Eligible patients
had progressive, metastatic, chemotherapy-naive CRPC | -The combination of
everolimus 10 mg/day and docetaxel 60 mg/m2 was
comparatively well tolerated. |
| | | | | | -PSA50: 5/12 |
| | | | | | -Four patients had
partial metabolic response (PMR) and PSA50 was observed in 3 of 4
patients. |
|
NCT00814788/n=24 | Bicalutamide (an
mTOR) inhibitor | II | Patients receive
oral Bicalutamide (50 mg) and oral Everolimus (10 mg) once daily on
days 1-28. Courses repeat every 28 days in the absence of disease
progression or unacceptable toxicity. | Eligible patients
should have progressive CRPC with serum testosterone <50
ng/dl | -PSA response:
18/24 (75%) |
| | | | | | -PSA50: 15/24
(62.5%) |
| | | | | | -mOS: 28
months |
| | | | | | -Grade 3 (13
patients) or Grade 4 (1 patient with sepsis): 14 (54%) |
| | | | -Meanlength of
treatment: 8 cycles | | |
|
NCT01051570/n=26 | Carboplatin,
everolimus, and Prednisone (RAD 001) | II | Carboplatin: AUC=4
by Calvert's formula (max dose 600 mg)*IV over 30-60 min, Day 1 of
a 21 day cycle everolimus: 10 mg Orally daily, starting from Day 2
continuously Prednisone 5 mg Orally twice daily, continuously | -Docetaxel | -Median time to
progression: 2.5 months |
| | | | | | -mOS:12.5
months |
| | | | | | -Disease
progression in AKT +ve patients within 9 weeks |
| | | | | | -Minimal clinical
efficacy |
|
NCT02106507/n=9 | Apalutamide
(ARN509) and everolimus | I | Apalutamide, 240
mg/day Cohort 1: everolimus 5 mg (n=3) Cohort 2: everolimus 10 mg
(n=6). | Abiraterone acetate
and prednisone (AAP) | -SD: 9/9 patients.
Patients came off study for: progression (n=3), investigator choice
(n=3) and toxicity unrelated to treatment (n=2). |
| | | | | | -The treatment
responsewas similar to historical data of AAP followed by
apalutamide alone |
| | | | -The median time on
treatment was 17 weeks | | -Study closed in
favour of evaluating novel AR/PI3K pathway combinations in patients
who have not yet been exposed to AAP |
| B, Ongoing clinical
trials |
| Clinical trials
ID/no. of patients | AKT/PI3K/mTOR
inhibitor | Phase | Treatment plan | Outcomes |
| NCT05593497 | Capivasertib in
combination with abiraterone and leuprolide | II | Four-week
intensified androgen deprivation drugs (iADT; abiraterone and
leuprolide), 16-week treatment with Capivasertib in combination
with iADT | Prior to radical
prostatectomy and resistant to standard treatments |
| NCT05593497 | Capivasertib in
combination with abiraterone and leuprolide | II | Four-week
intensified androgen deprivation drugs (iADT; abiraterone and
leuprolide), 16-week treatment with Capivasertib in combination
with iADT | Prior to radical
prostatectomy and resistant to standard treatments |
| NCT03903835 | Treatments will be
given on the basis of biomarker profile of patients | III | Biomarker
signatures will include: Androgen receptor | Information not
provided |
| | | |
DNA-repair
deficiency | |
| | | |
TP53 | |
| | | |
TMPRSS2-ERG
gene fusion | |
| | | |
PI3K pathway
alterations | |
| NCT06190899 | Gedatolisib
(inhibitor of PIK3/mTOR) in combination with darolutamide
(AR-inhibitor) | I/II | Experimental: Phase
1 Arm 1 | Chemotherapy plus
ADT for castration-sensitive disease, including docetaxel plus
darolutamide |
| | | | Arm 1-120 mg of
Gedatolisib (administered once weekly for 3 weeks on/1 week off) in
combination with darolutamide 600 mg orally administered twice
daily (equivalent to a total daily dose of 1200 mg on Days 1-28 of
each cycle) | |
Future research directions
Several molecules within the PI3K/AKT/mTOR signaling
pathway are involved in the pro-survival activity in prostate
cancer. The inhibition of these molecules may be advantageous in
terms of bypassing the pro-survival pathways associated with drug
resistance. Therefore, combination treatment with AR inhibitors and
PI3K/AKT/mTOR signaling pathway inhibitors may be beneficial in
overcoming AR-mediated resistance. However, biomarker analysis may
play an indispensable role in the identification of a suitable
inhibitor. For instance, if a clinician knows about the biomarker
that will eventually be enhanced owing to drug resistance, then
they can wisely select the inhibitor that will decrease the
expression of drug resistance-associated biomarkers. This would
allow for specific personalized therapy for patients. Additionally,
the development of isoform-specific AKT inhibitors could avoid some
of the toxicities of pan-AKT inhibitors and improve the side-effect
profile in patients (141).
8. Implications and future directions
Although multiple treatment options with varying
mechanisms are available, careful patient selection should be
performed to maximize the treatment response with minimal toxicity.
Effective biomarkers could revolutionize clinical care by making
treatments more personalized and reducing unnecessary treatments.
Biomarkers can identify specific molecular and genetic traits to
classify patients by their risk levels. This helps provide
aggressive treatments to those who need them most while avoiding
side effects in others. Emerging technologies, such as liquid
biopsies, offer non-invasive alternatives to traditional tissue
sampling, allowing for real-time monitoring of tumor dynamics and
treatment responses. These advancements could lead to the earlier
detection of resistance mechanisms and more adaptive treatment
strategies. With advancements in the field of whole-genome
sequencing and next-generation sequencing, the detection of
circulating tumor cells and circulating tumor DNA is possible,
resulting in the identification of predictive biomarkers before
starting therapies. For example, plasma circulating free DNA levels
have been shown to accurately predict patients suitable for proton
vs. photon radiotherapy and may serve as a minimally invasive
predictive biomarker for acute and late gastrointestinal toxicity
reported by patients (142). In
the future, biomarker-based screening may further enhance precision
medicine, improving outcomes, multi-cancer detection by common
biomarkers, while reducing the financial and emotional burdens
associated with ineffective or excessive treatments. In addition,
integration with artificial intelligence will optimize the analysis
of complex biomarker datasets. Since biomarkers also change
following the treatment response, biomarkers other than PSMA should
also be evaluated at preclinical and clinical level to observe
response to therapy. Translating research findings into clinical
applications requires rigorous validation through clinical trials
to ensure biomarkers are robust and reproducible across diverse
populations.
Using biomarkers in healthcare also comes with
ethical, practical and technical challenges, particularly in areas
with limited resources. A key issue is the high cost of developing,
testing and using biomarker-based treatments, which can make these
difficult to access and increase health inequalities. Advanced
tools, such as liquid biopsies and next-generation sequencing
require special equipment and trained experts, which may not be
available in low-resource areas. Additionally, the need for regular
testing or real-time monitoring could overwhelm healthcare systems
that already face staff shortages or limited funding. Ethically,
focusing on costly personalized treatments could leave out people
who can't afford them, creating concerns about equitable care.
Additionally, technical challenges, such as the standardization of
biomarker assays and the interpretation of complex data, complicate
their widespread adoption. Addressing these barriers requires
innovative, cost-effective solutions and global collaborations to
ensure biomarker-driven therapies benefit all patients, regardless
of socioeconomic status. Making biomarker development more
efficient with affordable tests and ensuring they have practical
use in clinics can help connect research with real-world
applications. This will promote precision medicine while keeping
healthcare sustainable. Owing to the high cost of NGS, the majority
of biomarker-driven clinical trials are being conducted in
developed countries. More collaboration between developed and
developing countries should be explored to overcome this financial
issue. In summary, therapeutic biomarkers should be evaluated to
provide precision in personalized therapies for prostate
cancer.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
LG was involved in the conceptualization of the
study, as well as in the writing and preparation of the original
draft of the manuscript. DB and CJ prepared the tables. RG
supervised the study and edited the manuscript. All the authors
have read and approved the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ng KL: The Etiology of Prostate Cancer.
In: Prostate Cancer. Bott SRJ and Ng KL (eds). Exon Publications,
Brisbane, Queensland, 2021.
|
|
3
|
Sekhoacha M, Riet K, Motloung P, Gumenku
L, Adegoke A and Mashele S: Prostate cancer review: Genetics,
diagnosis, treatment options, and alternative approaches.
Molecules. 27(5730)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Adamaki M and Zoumpourlis V: Prostate
cancer biomarkers: From diagnosis to prognosis and precision-guided
therapeutics. Pharmacol Ther. 228(107932)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cai C, Chen S, Ng P, Bubley GJ, Nelson PS,
Mostaghel EA, Marck B, Matsumoto AM, Simon NI, Wang H, et al:
Intratumoral de novo steroid synthesis activates androgen receptor
in castration-resistant prostate cancer and is upregulated by
treatment with CYP17A1 inhibitors. Cancer Res. 71:6503–6513.
2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hellmis E, Schwentner C, Mandel P, Banek
S, Gleissner J and Bogemann M: Apalutamide in patients with
high-risk M0CRPC: Data from the pivotal SPARTAN study and initial
experience from a compassionate use program. Aktuelle Urol.
54:140–147. 2023.PubMed/NCBI View Article : Google Scholar : (In German).
|
|
7
|
Rajaram P, Rivera A, Muthima K, Olveda N,
Muchalski H and Chen QH: Second-generation androgen receptor
antagonists as hormonal therapeutics for three forms of prostate
cancer. Molecules. 25(2448)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Shen J, Chowdhury S, Agarwal N, Karsh LI,
Oudard S, Gartrell BA, Feyerabend S, Saad F, Pieczonka CM, Chi KN,
et al: Apalutamide efficacy, safety and wellbeing in older patients
with advanced prostate cancer from Phase 3 randomised clinical
studies TITAN and SPARTAN. Br J Cancer. 130:73–81. 2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ruiz de Porras V, Font A and Aytes A:
Chemotherapy in metastatic castration-resistant prostate cancer:
Current scenario and future perspectives. Cancer Lett. 523:162–169.
2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Scher HI, Lu D, Schreiber NA, Louw J, Graf
RP, Vargas HA, Johnson A, Jendrisak A, Bambury R, Danila D, et al:
Association of AR-V7 on circulating tumor cells as a
treatment-specific biomarker with outcomes and survival in
castration-resistant prostate cancer. JAMA Oncol. 2:1441–1449.
2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Deluce JE, Cardenas L, Lalani AK, Maleki
Vareki S and Fernandes R: Emerging Biomarker-guided therapies in
prostate cancer. Curr Oncol. 29:5054–5076. 2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Asif S and Teply BA: Biomarkers for
treatment response in advanced prostate cancer. Cancers (Basel).
13(5723)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Singh P, Patel M, Bhowmik D, Kumari N,
Prajapati KS and Gupta R: Identification of common biomarkers
affecting patient survival in cancers. World Acad Sci. 6:1–12.
2024.
|
|
14
|
Patel M, Singh P, Gandupalli L and Reeshu
G: Identification and evaluation of Survival-associated common
chemoresistant genes in cancer. Biomed Biotechnol Res J. 8:320–327.
2024.
|
|
15
|
Chen JY, Wang PY, Liu MZ, Lyu F, Ma MW,
Ren XY and Gao XS: Biomarkers for prostate cancer: From diagnosis
to treatment. Diagnostics (Basel). 13(3350)2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Pires FR, Sagarra R, Corrêa ME, Pereira
CM, Vargas PA and Lopes MA: Oral metastasis of a hepatocellular
carcinoma. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.
97:359–368. 2004.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Sobhani N, Neeli PK, D'Angelo A, Pittacolo
M, Sirico M, Galli IC, Roviello G and Nesi G: AR-V7 in metastatic
prostate cancer: A strategy beyond redemption. Int J Mol Sci.
22(5515)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hu R, Dunn TA, Wei S, Isharwal S, Veltri
RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al:
Ligand-independent androgen receptor variants derived from splicing
of cryptic exons signify hormone-refractory prostate cancer. Cancer
Res. 69:16–22. 2009.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Katleba KD, Ghosh PM and Mudryj M: Beyond
prostate cancer: An androgen receptor splice variant expression in
multiple malignancies, non-cancer pathologies, and development.
Biomedicines. 11(2215)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Ferguson DC, Mata DA, Tay TK, Traina TA,
Gucalp A, Chandarlapaty S, D'Alfonso TM, Brogi E, Mullaney K,
Ladanyi M, et al: Androgen receptor splice variant-7 in breast
cancer: Clinical and pathologic correlations. Mod Pathol.
35:396–402. 2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Lieberman AP and Robins DM: The androgen
receptor's CAG/glutamine tract in mouse models of neurological
disease and cancer. J Alzheimers Dis. 14:247–255. 2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Dauki AM, Blachly JS, Kautto EA, Ezzat S,
Abdel-Rahman MH and Coss CC: Transcriptionally active androgen
receptor splice variants promote hepatocellular carcinoma
progression. Cancer Res. 80:561–575. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sharp A, Coleman I, Yuan W, Sprenger C,
Dolling D, Rodrigues DN, Russo JW, Figueiredo I, Bertan C, Seed G,
et al: Androgen receptor splice variant-7 expression emerges with
castration resistance in prostate cancer. J Clin Invest.
129:192–208. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Watson PA, Chen YF, Balbas MD, Wongvipat
J, Socci ND, Viale A, Kim K and Sawyers CL: Constitutively active
androgen receptor splice variants expressed in castration-resistant
prostate cancer require full-length androgen receptor. Proc Natl
Acad Sci USA. 107:16759–16765. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wang Z, Shen H, Liang Z, Mao Y, Wang C and
Xie L: The characteristics of androgen receptor splice variant 7 in
the treatment of hormonal sensitive prostate cancer: A systematic
review and meta-analysis. Cancer Cell Int. 20(149)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
König P, Eckstein M, Jung R, Abdulrahman
A, Guzman J, Weigelt K, Serrero G, Hayashi J, Geppert C, Stöhr R,
et al: Expression of AR-V7 (androgen receptor variant 7) protein in
granular cytoplasmic structures is an independent prognostic factor
in prostate cancer patients. Cancers (Basel).
12(2639)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hu R, Lu C, Mostaghel EA, Yegnasubramanian
S, Gurel M, Tannahill C, Edwards J, Isaacs WB, Nelson PS, Bluemn E,
et al: Distinct transcriptional programs mediated by the
ligand-dependent full-length androgen receptor and its splice
variants in castration-resistant prostate cancer. Cancer Res.
72:3457–3462. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bevan CL, Hoare S, Claessens F, Heery DM
and Parker MG: The AF1 and AF2 domains of the androgen receptor
interact with distinct regions of SRC1. Mol Cell Biol.
19:8383–8392. 1999.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Chen Z, Wu D, Thomas-Ahner JM, Lu C, Zhao
P, Zhang Q, Geraghty C, Yan PS, Hankey W, Sunkel B, et al: Diverse
AR-V7 cistromes in castration-resistant prostate cancer are
governed by HoxB13. Proc Natl Acad Sci USA. 115:6810–6815.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Dai C, Dehm SM and Sharifi N: Targeting
the androgen signaling axis in prostate cancer. J Clin Oncol.
41:4267–4278. 2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Mostaghel EA: Alternative acts: Oncogenic
splicing of steroidogenic enzymes in prostate cancer. Clin Cancer
Res. 25:1139–1141. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Henzler C, Li Y, Yang R, McBride T, Ho Y,
Sprenger C, Liu G, Coleman I, Lakely B, Li R, et al: Truncation and
constitutive activation of the androgen receptor by diverse genomic
rearrangements in prostate cancer. Nat Commun.
7(13668)2016.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang T, Karsh LI, Nissenblatt MJ and
Canfield SE: Androgen receptor splice variant, AR-V7, as a
biomarker of resistance to androgen Axis-targeted therapies in
advanced prostate cancer. Clin Genitourin Cancer. 18:1–10.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu C, Armstrong CM, Lou W, Lombard AP,
Cucchiara V, Gu X, Yang JC, Nadiminty N, Pan CX, Evans CP, et al:
Niclosamide and bicalutamide combination treatment overcomes
Enzalutamide- and bicalutamide-resistant prostate cancer. Mol
Cancer Ther. 16:1521–1530. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Antonarakis ES, Lu C, Luber B, Wang H,
Chen Y, Nakazawa M, Nadal R, Paller CJ, Denmeade SR, Carducci MA,
et al: Androgen receptor splice variant 7 and efficacy of taxane
chemotherapy in patients with metastatic castration-resistant
prostate cancer. JAMA Oncol. 1:582–591. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Antonarakis ES, Lu C, Luber B, Wang H,
Chen Y, Zhu Y, ilberstein JL, Taylor MN, Maughan BL, Denmeade SR,
et al: Clinical significance of androgen receptor splice variant-7
mRNA detection in circulating tumor cells of men with metastatic
castration-resistant prostate cancer treated with First- and
Second-line abiraterone and enzalutamide. J Clin Oncol.
35:2149–2156. 2017.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kono M, Fujii T, Lim B, Karuturi MS,
Tripathy D and Ueno NT: Androgen receptor function and androgen
Receptor-targeted therapies in breast cancer: A review. JAMA Oncol.
3:1266–1273. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Gatalica Z, Hoag J, Hall DW, Alyaqoub FS,
Dombrowski S, Noel P, Szelinger S, Udhane S, Min W and Thakkar SG:
CGE23-072: The frequency of androgen receptor splice Variant 7
(AR-V7) in solid tumors. J Nat Comprehensive Nat Netw. 21:2023.
|
|
39
|
Armstrong AJ, Halabi S, Luo J, Nanus DM,
Giannakakou P, Szmulewitz RZ, Danila DC, Healy P, Anand M, Rothwell
CJ, et al: Prospective multicenter validation of androgen receptor
splice variant 7 and hormone therapy resistance in High-risk
Castration-resistant prostate cancer: The PROPHECY Study. J Clin
Oncol. 37:1120–1129. 2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Carles J, Alonso-Gordoa T, Mellado B,
Mendez-Vidal MJ, Vazquez S, Gonzalez-Del-Alba A, Piulats JM,
Borrega P, Gallardo E, Morales-Barrera R, et al: Radium-223 for
patients with metastatic castration-resistant prostate cancer with
asymptomatic bone metastases progressing on first-line abiraterone
acetate or enzalutamide: A single-arm phase II trial. Eur J Cancer.
173:317–3126. 2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Shenderov E, Boudadi K, Fu W, Wang H,
Sullivan R, Jordan A, Dowling D, Harb R, Schonhoft J, Jendrisak A,
et al: Nivolumab plus ipilimumab, with or without enzalutamide, in
AR-V7-expressing metastatic castration-resistant prostate cancer: A
phase-2 nonrandomized clinical trial. Prostate. 81:326–338.
2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Tewari A: AMG 160 aH-LE, PSMA-Targeted,
Bispecific T-cell Engager (BiTE®) immune Therapy for
mCRPC-Interim Results From a Phase I Study. [(Accessed on 30 April
2021)]; Available online: https://www.urotoday.com/conference-highlights/esmo-2020/prostate-cancer/124632-esmo-virtual-congress-2020-amg-160-a-half-life-extended-psma-targeted-bispecific-t-cell-engager-bite-immune-therapy-for-mcrpc-interim-results-from-a-phase-i-study.html.
|
|
43
|
Li W, Zhang B, Tang J, Cao Q, Wu Y, Wu C,
Guo J, Ling EA and Liang F: Sirtuin 2, a mammalian homolog of yeast
silent information regulator-2 longevity regulator, is an
oligodendroglial protein that decelerates cell differentiation
through deacetylating alpha-tubulin. J Neurosci. 27:2606–2616.
2007.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Rana Z, Diermeier S, Hanif M and Rosengren
RJ: Understanding failure and improving treatment using HDAC
inhibitors for prostate cancer. Biomedicines. 8(22)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Eckschlager T, Plch J, Stiborova M and
Hrabeta J: Histone deacetylase inhibitors as anticancer drugs. Int
J Mol Sci. 18(1414)2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Oehme I, Deubzer HE, Wegener D, Pickert D,
Linke JP, Hero B, Kopp-Schneider A, Westermann F, Ulrich SM, von
Deimling A, et al: Histone deacetylase 8 in neuroblastoma
tumorigenesis. Clin Cancer Res. 15:91–99. 2009.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang L, Zou X, Berger AD, Twiss C, Peng Y,
Li Y, Chiu J, Guo H, Satagopan J, Wilton A, et al: Increased
expression of histone deacetylaces (HDACs) and inhibition of
prostate cancer growth and invasion by HDAC inhibitor SAHA. Am J
Transl Res. 1:62–71. 2009.PubMed/NCBI
|
|
48
|
Weichert W, Roske A, Gekeler V, Beckers T,
Stephan C, Jung K, Fritzsche FR, Niesporek S, Denkert C, Dietel M,
et al: Histone deacetylases 1, 2 and 3 are highly expressed in
prostate cancer and HDAC2 expression is associated with shorter PSA
relapse time after radical prostatectomy. Br J Cancer. 98:604–610.
2008.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Shankar E, Pandey M, Verma S, Abbas A,
Candamo M, Kanwal R, Shukla S, MacLennan GT and Gupta S: Role of
class I histone deacetylases in the regulation of maspin expression
in prostate cancer. Mol Carcinog. 59:955–966. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Waltregny D, North B, Van Mellaert F, de
Leval J, Verdin E and Castronovo V: Screening of histone
deacetylases (HDAC) expression in human prostate cancer reveals
distinct class I HDAC profiles between epithelial and stromal
cells. Eur J Histochem. 48:273–290. 2004.PubMed/NCBI
|
|
51
|
Li Y and Seto E: HDACs and HDAC inhibitors
in cancer development and therapy. Cold Spring Harb Perspect Med.
6(a026831)2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zeng LS, Yang XZ, Wen YF, Mail SJ, Wang
MH, Zhang MY, Zheng XF and Wang HY: Overexpressed HDAC4 is
associated with poor survival and promotes tumor progression in
esophageal carcinoma. Aging (Albany NY). 8:1236–1249.
2016.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Beaver LM, Lӧhr CV, Clarke JD, Glasser ST,
Watson GW, Wong CP, Zhang Z, Williams DE, Dashwood RH, Shannon J,
et al: Broccoli sprouts delay prostate cancer formation and
decrease prostate cancer severity with a concurrent decrease in
HDAC3 protein expression in transgenic adenocarcinoma of the mouse
prostate (TRAMP) mice. Curr Dev Nutr. 2(nzy002)2018.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jang YG, Hwang KA and Choi KC: Rosmarinic
acid, a component of rosemary tea, induced the cell cycle arrest
and apoptosis through modulation of HDAC2 expression in prostate
cancer cell lines. Nutrients. 10(1784)2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Pandey M, Kaur P, Shukla S, Abbas A, Fu P
and Gupta S: Plant flavone apigenin inhibits HDAC and remodels
chromatin to induce growth arrest and apoptosis in human prostate
cancer cells: In vitro and in vivo study. Mol Carcinog. 51:952–962.
2012.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Zhang L, Zhang J, Jiang Q, Zhang L and
Song W: Zinc binding groups for histone deacetylase inhibitors. J
Enzyme Inhib Med Chem. 33:714–721. 2018.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Yue K, Qin M, Huang C, James Chou C, Jiang
Y and Li X: Comparison of three zinc binding groups for HDAC
inhibitors-A potency, selectivity and enzymatic kinetics study.
Bioorg Med Chem Lett. 70(128797)2022.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Moi D, Bonanni D, Belluti S, Linciano P,
Citarella A, Franchini S, Sorbi C, Imbriano C, Pinzi L and Rastelli
G: Discovery of potent pyrrolo-pyrimidine and purine HDAC
inhibitors for the treatment of advanced prostate cancer. Eur J Med
Chem. 260(115730)2023.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Eigl BJ, North S, Winquist E, Finch D,
Wood L, Sridhar SS, Powers J, Good J, Sharma M, Squire JA, et al: A
phase II study of the HDAC inhibitor SB939 in patients with
castration resistant prostate cancer: NCIC clinical trials group
study IND195. Invest New Drugs. 33:969–976. 2015.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Ferrari AC, Alumkal JJ, Stein MN, Taplin
ME, Babb J, Barnett ES, Gomez-Pinillos A, Liu X, Moore D, DiPaola R
and Beer TM: Epigenetic therapy with panobinostat combined with
bicalutamide rechallenge in castration-resistant prostate cancer.
Clin Cancer Res. 25:52–63. 2019.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Biersack B, Nitzsche B and Hopfner M: HDAC
inhibitors with potential to overcome drug resistance in
castration-resistant prostate cancer. Cancer Drug Resist. 5:64–79.
2022.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Chen Z, Wang X, Yang X, Xu Y, Yang Y, Wang
H, Li T, Bai P, Yuan G, Chen H, et al: Imaging assisted evaluation
of antitumor efficacy of a new histone deacetylase inhibitor in the
castration-resistant prostate cancer. Eur J Nucl Med Mol Imaging.
48:53–66. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Rosati R, Chen B, Patki M, McFall T, Ou S,
Heath E, Ratnam M and Qin Z: Hybrid Enzalutamide derivatives with
histone deacetylase inhibitor activity decrease heat shock protein
90 and androgen receptor levels and inhibit viability in
Enzalutamide-resistant C4-2 prostate cancer cells. Mol Pharmacol.
90:225–237. 2016.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Hu WY, Xu L, Chen B, Ou S, Muzzarelli KM,
Hu DP, Li Y, Yang Z, Vander Griend DJ, Prins GS and Qin Z:
Targeting prostate cancer cells with enzalutamide-HDAC inhibitor
hybrid drug 2-75. Prostate. 79:1166–1179. 2019.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Sun H, Mediwala SN, Szafran AT, Mancini MA
and Marcelli M: CUDC-101, a novel inhibitor of Full-length androgen
receptor (flAR) and androgen receptor variant 7 (AR-V7) activity:
Mechanism of action and in vivo efficacy. Horm Cancer. 7:196–210.
2016.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lai CJ, Bao R, Tao X, Wang J, Atoyan R, Qu
H, Wang DG, Yin L, Samson M, Forrester J, et al: CUDC-101, a
multitargeted inhibitor of histone deacetylase, epidermal growth
factor receptor, and human epidermal growth factor receptor 2,
exerts potent anticancer activity. Cancer Res. 70:3647–3656.
2010.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Li X, Kamenecka TM and Cameron MD:
Cytochrome P450-mediated bioactivation of the epidermal growth
factor receptor inhibitor erlotinib to a reactive electrophile.
Drug Metab Dispos. 38:1238–1245. 2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Goehringer N, Biersack B, Peng Y, Schobert
R, Herling M, Ma A, Nitzsche B and Höpfner M: Anticancer activity
and mechanisms of action of new chimeric EGFR/HDAC-inhibitors. Int
J Mol Sci. 22(8432)2021.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Hu C, Xia H, Bai S, Zhao J, Edwards H, Li
X, Yang Y, Lyu J, Wang G, Zhan Y, et al: CUDC-907, a novel dual
PI3K and HDAC inhibitor, in prostate cancer: Antitumour activity
and molecular mechanism of action. J Cell Mol Med. 24:7239–7253.
2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Wu CP, Hsieh YJ, Hsiao SH, Su CY, Li YQ,
Huang YH, Huang CW, Hsieh CH, Yu JS and Wu YS: Human ATP-binding
cassette transporter ABCG2 confers resistance to CUDC-907, a dual
inhibitor of histone deacetylase and phosphatidylinositol 3-Kinase.
Mol Pharm. 13:784–794. 2016.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Evans LW and Ferguson BS: Food bioactive
HDAC inhibitors in the epigenetic regulation of heart failure.
Nutrients. 10(1120)2018.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Lauri C, Chiurchioni L, Russo VM, Zannini
L and Signore A: PSMA expression in solid tumors beyond the
prostate gland: Ready for theranostic applications? J Clin Med.
11(6590)2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Uijen MJM, Derks YHW, Merkx RIJ, Schilham
MGM, Roosen J, Prive BM, van Lith SAM, van Herpen CML, Gotthardt M,
Heskamp S, et al: PSMA radioligand therapy for solid tumors other
than prostate cancer: Background, opportunities, challenges, and
first clinical reports. Eur J Nucl Med Mol Imaging. 48:4350–4368.
2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Kasperzyk JL, Finn SP, Flavin R,
Fiorentino M, Lis R, Hendrickson WK, Clinton SK, Sesso HD,
Giovannucci EL, Stampfer MJ, et al: Prostate-specific membrane
antigen protein expression in tumor tissue and risk of lethal
prostate cancer. Cancer Epidemiol Biomarkers Prev. 22:2354–2363.
2013.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Heskamp S, Hernandez R, Molkenboer-Kuenen
JDM, Essler M, Bruchertseifer F, Morgenstern A, Steenbergen EJ, Cai
W, Seidl C, McBride WJ, et al: α-versus β-emitting radionuclides
for pretargeted radioimmunotherapy of carcinoembryonic
antigen-expressing human colon cancer xenografts. J Nucl Med.
58:926–933. 2017.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Sheehan B, Guo C, Neeb A, Paschalis A,
Sandhu S and de Bono JS: Prostate-specific membrane antigen biology
in lethal prostate cancer and its therapeutic implications. Eur
Urol Focus. 8:1157–1168. 2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Zacherl MJ, Gildehaus FJ, Mittlmeier L,
Boning G, Gosewisch A, Wenter V, Unterrainer M, Schmidt-Hegemann N,
Belka C, Kretschmer A, et al: First clinical results for
PSMA-Targeted α-therapy using 225Ac-PSMA-I&T in Advanced-mCRPC
patients. J Nucl Med. 62:669–674. 2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Sathekge M, Bruchertseifer F, Knoesen O,
Reyneke F, Lawal I, Lengana T, Davis C, Mahapane J, Corbett C,
Vorster M and Morgenstern A: 225Ac-PSMA-617 in chemotherapy-naive
patients with advanced prostate cancer: A pilot study. Eur J Nucl
Med Mol Imaging. 46:129–138. 2019.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Tagawa ST, Thomas C, Sartor AO, Sun M,
Stangl-Kremser J, Bissassar M, Vallabhajosula S, Huicochea
Castellanos S, Nauseef JT, Sternberg CN, et al: Prostate-specific
membrane antigen-targeting alpha emitter via antibody delivery for
metastatic castration-resistant prostate cancer: A phase I
dose-escalation study of 225Ac-J591. J Clin Oncol. 42:842–581.
2024.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Baum RP, Kulkarni HR, Schuchardt C, Singh
A, Wirtz M, Wiessalla S, Schottelius M, Mueller D, Klette I and
Wester HJ: 177Lu-Labeled Prostate-specific membrane antigen
radioligand therapy of metastatic castration-resistant prostate
cancer: Safety and efficacy. J Nucl Med. 57:1006–1013.
2016.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Sandhu S, Joshua AM, Emmett L, Spain LA,
Horvath L, Crumbaker M, Anton A and Hofman MS: PRINCE: Phase I
trial of 177Lu-PSMA-617 in combination with pembrolizumab in
patients with metastatic castration-resistant prostate cancer
(mCRPC). J Clin Oncol. 40(5017)2022.
|
|
82
|
Dorff T, Horvath LG, Autio K,
Bernard-Tessier A, Rettig MB, Machiels JP, Bilen MA, Lolkema MP,
Adra N, Rottey S, et al: A phase I study of acapatamab, a Half-life
extended, PSMA-targeting bispecific T-cell engager for metastatic
castration-resistant prostate cancer. Clin Cancer Res.
30:1488–1500. 2024.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Hummel HD, Kufer P, Grullich C,
Seggewiss-Bernhardt R, Deschler-Baier B, Chatterjee M, Goebeler ME,
Miller K, de Santis M, Loidl W, et al: Pasotuxizumab, a
BiTE® immune therapy for castration-resistant prostate
cancer: Phase I, dose-escalation study findings. Immunotherapy.
13:125–141. 2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
De Bono JS, Fong L, Beer TM, Gao X,
Geynisman DM, Burris III HA, Strauss JF, Courtney KD, Quinn DI,
VanderWeele DJ, et al: Results of an ongoing phase 1/2a dose
escalation study of HPN424, a tri-specific half-life extended
PSMA-targeting T-cell engager, in patients with metastatic
castration-resistant prostate cancer (mCRPC). J Clin Oncol.
39(5013)2021.
|
|
85
|
Sandhu S, Joshua AM, Emmett L, Crumbaker
M, Bressel M, Huynh R, Banks PD, Wallace R, Hamid A, Inderjeeth AJ,
et al: LuPARP: Phase 1 trial of 177Lu-PSMA-617 and olaparib in
patients with metastatic castration resistant prostate cancer
(mCRPC). J Clin Oncol. 41(5005)2023.
|
|
86
|
Xiao X, Lao XM, Chen MM, Liu RX, Wei Y,
Ouyang FZ, Chen DP, Zhao XY, Zhao Q, Li XF, et al: PD-1hi
identifies a novel regulatory B-cell population in human hepatoma
that promotes disease progression. Cancer Discov. 6:546–559.
2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Salvi S, Fontana V, Boccardo S, Merlo DF,
Margallo E, Laurent S, Morabito A, Rijavec E, Dal Bello MG, Mora M,
et al: Evaluation of CTLA-4 expression and relevance as a novel
prognostic factor in patients with non-small cell lung cancer.
Cancer Immunol Immunother. 61:1463–1472. 2012.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Seidel JA, Otsuka A and Kabashima K:
Anti-PD-1 and Anti-CTLA-4 therapies in cancer: Mechanisms of
action, efficacy, and limitations. Front Oncol.
8(86)2018.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kwon ED, Drake CG, Scher HI, Fizazi K,
Bossi A, van den Eertwegh AJ, Krainer M, Houede N, Santos R,
Mahammedi H, et al: Ipilimumab versus placebo after radiotherapy in
patients with metastatic castration-resistant prostate cancer that
had progressed after docetaxel chemotherapy (CA184-043): A
multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol.
15:700–712. 2014.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Powles T, Yuen KC, Gillessen S, Kadel EE
III, Rathkopf D, Matsubara N, Drake CG, Fizazi K, Piulats JM,
Wysocki PJ, et al: Atezolizumab with enzalutamide versus
enzalutamide alone in metastatic castration-resistant prostate
cancer: A randomized phase 3 trial. Nat Med. 28:144–153.
2022.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Sharma P, Pachynski RK, Narayan V, Flechon
A, Gravis G, Galsky MD, Mahammedi H, Patnaik A, Subudhi SK,
Ciprotti M, et al: Initial results from a phase II study of
nivolumab (NIVO) plus ipilimumab (IPI) for the treatment of
metastatic castration-resistant prostate cancer (mCRPC; CheckMate
650). J Clin Oncol. 37(142)2019.
|
|
92
|
Beer TM, Kwon ED, Drake CG, Fizazi K,
Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, et
al: Randomized, double-blind, phase III trial of ipilimumab versus
placebo in asymptomatic or minimally symptomatic patients with
metastatic Chemotherapy-naive castration-resistant prostate cancer.
J Clin Oncol. 35:40–47. 2017.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Hansen AR, Massard C, Ott PA, Haas NB,
Lopez JS, Ejadi S, Wallmark JM, Keam B, Delord JP, Aggarwal R, et
al: Pembrolizumab for advanced prostate adenocarcinoma: Findings of
the KEYNOTE-028 study. Ann Oncol. 29:1807–1813. 2018.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Antonarakis ES, Piulats JM, Gross-Goupil
M, Goh JC, Vaishampayan UN, De Wit R, Alanko T, Fukasawa S, Tabata
K, Feyerabend S, et al: Update on KEYNOTE-199, cohorts 1-3:
Pembrolizumab (pembro) for docetaxel-pretreated metastatic
castration-resistant prostate cancer (mCRPC). J Clin Oncol.
38(104)2020.
|
|
95
|
Hoimes CJ, Graff JN, Tagawa ST, Hwang C,
Kilari D, Tije AJT, Omlin A, McDermott RS, Vaishampayan UN, Elliott
T, et al: KEYNOTE-199 cohorts (C) 4 and 5: Phase II study of
pembrolizumab (pembro) plus enzalutamide (enza) for enza-resistant
metastatic castration-resistant prostate cancer (mCRPC). J Clin
Oncol. 38(5543)2020.
|
|
96
|
Yu EY, Piulats JM, Gravis G, Laguerre B,
Arija JAA, Oudard S, Fong PCC, Kolinsky MP, Augustin M, Feyerabend
S, et al: KEYNOTE-365 cohort A updated results: Pembrolizumab
(pembro) plus olaparib in docetaxel-pretreated patients (pts) with
metastatic castration-resistant prostate cancer (mCRPC). J Clin
Oncol. 38(100)2020.
|
|
97
|
Berry WR, Fong PCC, Piulats JM, Appleman
LJ, Conter HJ, Feyerabend S, Shore ND, Gravis G, Laguerre B, Gurney
H, et al: KEYNOTE-365 cohort C updated results: Pembrolizumab
(pembro) plus enzalutamide (enza) in abiraterone (abi)-pretreated
patients (pts) with metastatic castrate-resistant prostate cancer
(mCRPC). J Clin Oncol. 38(102)2020.
|
|
98
|
Stein M, Fong L, Tutrone R, Mega A, Lam
ET, Vangala S, Dennie J, Petit R, Gutierrez A, Hayes S and Haas N:
KEYNOTE-046: Effects of ADXS-PSA with or without pembrolizumab on
survival and antigen spreading in metastatic, castration-resistant
prostate cancer patients. Cancer Res. 79(CT098)2019.
|
|
99
|
van den Eertwegh AJ, Versluis J, van den
Berg HP, Santegoets SJ, van Moorselaar RJ, van der Sluis TM, Gall
HE, Harding TC, Jooss K, Lowy I, et al: Combined immunotherapy with
granulocyte-macrophage colony-stimulating factor-transduced
allogeneic prostate cancer cells and ipilimumab in patients with
metastatic castration-resistant prostate cancer: A phase 1
dose-escalation trial. Lancet Oncol. 13:509–517. 2012.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Ross AE, Hurley PJ, Tran PT, Rowe SP,
Benzon B, Neal TO, Chapman C, Harb R, Milman Y, Trock BJ, et al: A
pilot trial of pembrolizumab plus prostatic cryotherapy for men
with newly diagnosed oligometastatic hormone-sensitive prostate
cancer. Prostate Cancer Prostatic Dis. 23:184–193. 2020.PubMed/NCBI View Article : Google Scholar
|
|
101
|
van Wilpe S, Kloots ISH, Slootbeek PHJ,
den Brok M, Westdorp H, Franken MD, Coskunturk M, Osinga T,
Bloemendal H, Adema G, et al: Ipilimumab with nivolumab in
molecularly selected patients with castration-resistant prostate
cancer: Primary analysis of the phase II INSPIRE trial. Ann Oncol.
35:1126–1137. 2024.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Wang XH, Wang ZQ, Mu ZY, Zhu LP, Zhong CF
and Guo S: The efficacy and safety of immune checkpoint inhibitors
in metastatic castration-resistant prostate cancer: A systematic
review and meta-analysis. Medicine (Baltimore).
101(e29715)2022.PubMed/NCBI View Article : Google Scholar
|
|
103
|
McNeel DG, Eickhoff JC, Wargowski E, Zahm
C, Staab MJ, Straus J and Liu G: Concurrent, but not sequential,
PD-1 blockade with a DNA vaccine elicits anti-tumor responses in
patients with metastatic, castration-resistant prostate cancer.
Oncotarget. 9:25586–25596. 2018.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Madan RA, Mohebtash M, Arlen PM, Vergati
M, Rauckhorst M, Steinberg SM, Tsang KY, Poole DJ, Parnes HL,
Wright JJ, et al: Ipilimumab and a poxviral vaccine targeting
prostate-specific antigen in metastatic castration-resistant
prostate cancer: A phase 1 dose-escalation trial. Lancet Oncol.
13:501–508. 2012.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Brahmer JR, Drake CG, Wollner I, Powderly
JD, Picus J, Sharfman WH, Stankevich E, Pons A, Salay TM, McMiller
TL, et al: Phase I study of single-agent anti-programmed death-1
(MDX-1106) in refractory solid tumors: Aafety, clinical activity,
pharmacodynamics, and immunologic correlates. J Clin Oncol.
28:3167–3175. 2010.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Aguiar PN Jr, Santoro IL, Tadokoro H, de
Lima Lopes G, Filardi BA, Oliveira P, Mountzios G and de Mello RA:
The role of PD-L1 expression as a predictive biomarker in advanced
non-small-cell lung cancer: A network meta-analysis. Immunotherapy.
8:479–488. 2016.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Karja V, Aaltomaa S, Lipponen P, Isotalo
T, Talja M and Mokka R: Tumour-infiltrating lymphocytes: A
prognostic factor of PSA-free survival in patients with local
prostate carcinoma treated by radical prostatectomy. Anticancer
Res. 25:4435–4438. 2005.PubMed/NCBI
|
|
108
|
Ness N, Andersen S, Valkov A, Nordby Y,
Donnem T, Al-Saad S, Busund LT, Bremnes RM and Richardsen E:
Infiltration of CD8+ lymphocytes is an independent prognostic
factor of biochemical failure-free survival in prostate cancer.
Prostate. 74:1452–1461. 2014.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Sorrentino C, Musiani P, Pompa P,
Cipollone G and Di Carlo E: Androgen deprivation boosts prostatic
infiltration of cytotoxic and regulatory T lymphocytes and has no
effect on disease-free survival in prostate cancer patients. Clin
Cancer Res. 17:1571–1581. 2011.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Jiao S, Subudhi SK, Aparicio A, Ge Z, Guan
B, Miura Y and Sharma P: Differences in tumor microenvironment
dictate T helper lineage polarization and response to immune
checkpoint therapy. Cell. 179:1177–1190.e13. 2019.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Claps M, Mennitto A, Guadalupi V, Sepe P,
Stellato M, Zattarin E, Gillessen SS, Sternberg CN, Berruti A, De
Braud FGM, et al: Immune-checkpoint inhibitors and metastatic
prostate cancer therapy: Learning by making mistakes. Cancer Treat
Rev. 88(102057)2020.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Valabrega G, Scotto G, Tuninetti V, Pani A
and Scaglione F: Differences in PARP inhibitors for the treatment
of ovarian cancer: Mechanisms of action, pharmacology, safety, and
efficacy. Int J Mol Sci. 22(4203)2021.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Chan CY, Tan KV and Cornelissen B: PARP
inhibitors in cancer diagnosis and therapy. Clin Cancer Res.
27:1585–1594. 2021.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Thiery-Vuillemin A, de Bono J, Hussain M,
Roubaud G, Procopio G, Shore N, Fizazi K, Dos Anjos G, Gravis G,
Joung JY, et al: Pain and health-related quality of life with
olaparib versus physician's choice of next-generation hormonal drug
in patients with metastatic castration-resistant prostate cancer
with homologous recombination repair gene alterations (PROfound):
An open-label, randomised, phase 3 trial. Lancet Oncol. 23:393–405.
2022.PubMed/NCBI View Article : Google Scholar
|
|
115
|
de Bono J, Mateo J, Fizazi K, Saad F,
Shore N, Sandhu S, Chi KN, Sartor O, Agarwal N, Olmos D, et al:
Olaparib for metastatic castration-resistant prostate cancer. N
Engl J Med. 382:2091–2102. 2020.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Thiery-Vuillemin A, Oya M, Procopio G, De
Menezes JJ, Colagiovanni Girotto G, Ghatalia P, Nole F, Din O,
Spiegelhalder P, Mincik I, et al: Olaparib plus abiraterone as
first-line therapy in men with metastatic castration-resistant
prostate cancer: Pharmacokinetics data from the PROpel trial. J
Clin Oncol. 40(5050)2022.
|
|
117
|
Thiery-Vuillemin ASF, Saad F, Armstrong
AJ, Oya M, Maia Vianna KC, Özgüroğlu M, Gedye C, Buchschacher GL,
Lee JY, Emmenegger U, et al: Tolerability of abiraterone (abi)
combined with olaparib (ola) in patients (pts) with metastatic
castration-resistant prostate cancer (mCRPC): Further results from
the phase III PROpel trial. J Clin Oncol. 40(5019)2022.
|
|
118
|
Chen X, Pan Y, Wang Q, Ren C, Li M, Hao X
and Liu X: Comparative efficacy of olaparib in combination with or
without novel antiandrogens for treating metastatic
castration-resistant prostate cancer. Front Endocrinol (Lausanne).
14(1225033)2023.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Abida W, Patnaik A, Campbell D, Shapiro J,
Bryce AH, McDermott R, Sautois B, Vogelzang NJ, Bambury RM, Voog E,
et al: Rucaparib in men with metastatic castration-resistant
prostate cancer harboring a BRCA1 or BRCA2 gene alteration. J Clin
Oncol. 38:3763–3772. 2020.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Abida W, Campbell D, Patnaik A, Shapiro
JD, Sautois B, Vogelzang NJ, Voog EG, Bryce AH, McDermott R, Ricci
F, et al: Non-BRCA DNA damage repair gene alterations and response
to the PARP inhibitor rucaparib in metastatic castration-resistant
prostate cancer: Analysis from the phase II TRITON2 study. Clin
Cancer Res. 26:2487–2496. 2020.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Rao A, Morris D, Assikis VJ, Gopalji Jha
G, Ryan CJ, Ablaza JA, Habeck J, Loehr A, Xiao J, Gangolli EA, et
al: Rucaparib plus enzalutamide in patients (pts) with metastatic
castration-resistant prostate cancer (mCRPC): Pharmacokinetics (PK)
and safety data from the phase Ib RAMP study. J Clin Oncol.
39(79)2021.
|
|
122
|
Smith MR, Scher HI, Sandhu S, Efstathiou
E, Lara PN Jr, Yu EY, George DJ, Chi KN, Saad F, Ståhl O, et al:
Niraparib in patients with metastatic castration-resistant prostate
cancer and DNA repair gene defects (GALAHAD): A multicentre,
open-label, phase 2 trial. Lancet Oncol. 23:362–373.
2022.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Chi KN, Rathkopf DE, Raymond Smith M,
Efstathiou E, Attard G, Olmos D, Lee JY, Small EJ, Juliana Gomes A,
Roubaud G, et al: Phase 3 MAGNITUDE study: First results of
niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as
first-line therapy in patients (pts) with metastatic
castration-resistant prostate cancer (mCRPC) with and without
homologous recombination repair (HRR) gene alterations. J Clin
Oncol. 40(12)2022.
|
|
124
|
Agarwal N, Azad A, Shore ND, Carles J, Fay
AP, Dunshee C, Karsh LI, Paccagnella ML, Santo ND, Elmeliegy M, et
al: Talazoparib plus enzalutamide in metastatic
castration-resistant prostate cancer: TALAPRO-2 phase III study
design. Future Oncol. 18:425–436. 2022.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Karzai F, VanderWeele D, Madan RA, Owens
H, Cordes LM, Hankin A, Couvillon A, Nichols E, Bilusic M, Beshiri
ML, et al: Activity of durvalumab plus olaparib in metastatic
castration-resistant prostate cancer in men with and without DNA
damage repair mutations. J Immunother Cancer. 6(141)2018.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Yu E, Piulats JM, Gravis G, Fong P,
Todenhöfer T, Laguerre B, Arranz J, Oudard S, Massard C, Stoeckle
M, et al: 73P Association between homologous recombination repair
mutations and response to pembrolizumab (pembro) plus olaparib
(ola) in metastatic castration-resistant prostate cancer (mCRPC):
KEYNOTE-365 Cohort A biomarker analysis. Ann Oncol. 32
(Suppl)(S387)2021.
|
|
127
|
Robinson D, Van Allen EM, Wu YM, Schultz
N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC,
Attard G, et al: Integrative clinical genomics of advanced prostate
cancer. Cell. 161:1215–1228. 2015.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Carver BS, Chapinski C, Wongvipat J,
Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J,
Scher H, et al: Reciprocal feedback regulation of PI3K and androgen
receptor signaling in PTEN-deficient prostate cancer. Cancer Cell.
19:575–586. 2011.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Sirico M, D'Angelo A, Gianni C, Casadei C,
Merloni F and De Giorgi U: Current state and future challenges for
PI3K inhibitors in cancer therapy. Cancers (Basel).
15(703)2023.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Sweeney C, Bracarda S, Sternberg CN, Chi
KN, Olmos D, Sandhu S, Massard C, Matsubara N, Alekseev B, Parnis
F, et al: Ipatasertib plus abiraterone and prednisolone in
metastatic castration-resistant prostate cancer (IPATential150): A
multicentre, randomised, double-blind, phase 3 trial. Lancet.
398:131–142. 2021.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Sarker D, Dawson NA, Aparicio AM, Dorff
TB, Pantuck AJ, Vaishampayan UN, Henson L, Vasist L, Roy-Ghanta S,
Gorczyca M, et al: A Phase I, Open-label, Dose-finding study of
GSK2636771, a PI3Kβ inhibitor, administered with enzalutamide in
patients with metastatic castration-resistant prostate cancer. Clin
Cancer Res. 27:5248–5257. 2021.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Pacey S, Shah N, Daviesbr BR, Bratt O,
Warren A, Davies RD, Gnanapragasam VJ, Ingle S, Stearn S, Machin A,
et al: A pharmacodynamic biomarker study of vistusertib (AZD2014),
an mTORC1/2 inhibitor, given prior to radical prostatectomy
(CANCAP02). J Clin Oncol. 36(5801)2018.
|
|
133
|
Graham L, Banda K, Torres A, Carver BS,
Chen Y, Pisano K, Shelkey G, Curley T, Scher HI, Lotan TL, et al: A
phase II study of the dual mTOR inhibitor MLN0128 in patients with
metastatic castration resistant prostate cancer. Invest New Drugs.
36:458–467. 2018.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Kemp Bohan PM, Cindass JL, Chick RC,
Vreeland TJ, Hale DF, Hickerson A, Clifton GT, Peoples GE and Liss
M: Results of a phase Ib trial of encapsulated rapamycin in
prostate cancer patients under active surveillance to prevent
progression. J Clin Oncol. 38(34)2020.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Narayan V, Vapiwala N, Subramanian P,
Christodouleas JP, Bekelman JE, Mick R, Walicki M, Ciconte J,
Rajendran RR and Haas NB: Phase I trial of everolimus plus
radiation therapy for salvage treatment of biochemical recurrence
in prostate cancer patients following prostatectomy. J Clin Oncol.
34(16617)2016.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Sweeney CJ, Percent IJ, Babu S, Cultrera
JL, Mehlhaff BA, Goodman OB, Morris DS, Schnadig ID, Albany C,
Shore ND, et al: Phase Ib/II study of enzalutamide with samotolisib
(LY3023414) or placebo in patients with metastatic
Castration-resistant prostate cancer. Clin Cancer Res. 28:2237–224.
2022.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Courtney KD, Manola JB, Elfiky AA, Ross R,
Oh WK, Yap JT, Van den Abbeele AD, Ryan CW, Beer TM, Loda M, et al:
A phase I study of everolimus and docetaxel in patients with
castration-resistant prostate cancer. Clin Genitourin Cancer.
13:113–123. 2015.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Chow H, Ghosh PM, deVere White R, Evans
CP, Dall'Era MA, Yap SA, Li Y, Beckett LA, Lara PN Jr and Pan CX: A
phase 2 clinical trial of everolimus plus bicalutamide for
castration-resistant prostate cancer. Cancer. 122:1897–1904.
2016.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Vaishampayan U, Shevrin D, Stein M,
Heilbrun L, Land S, Stark K, Li J, Dickow B, Heath E and Smith D:
Phase II trial of carboplatin, everolimus, and prednisone in
metastatic castration-resistant prostate cancer pretreated with
docetaxel chemotherapy: A prostate cancer clinical trial consortium
study. Urology. 86:1206–1211. 2015.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Rathkopf DE, Slovin SF, Morris MJ, Danila
DC, Delacruz A, Shelkey G, DeNunzio M, McLaughlin B and Scher H:
Targeting reciprocal feedback inhibition: Apalutamide and
everolimus in patients with metastatic castration-resistant
prostate cancer (mCRPC). J Clin Oncol. 15(204)2017.
|
|
141
|
Coleman N, Moyers JT, Harbery A, Vivanco I
and Yap TA: Clinical development of AKT inhibitors and associated
predictive biomarkers to guide patient treatment in cancer
medicine. Pharmgenomics Pers Med. 14:1517–1535. 2021.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Lockney NA, Henderson RH, Swarts SG, Zhang
Z, Zhang B, Li J, Zlotecki RA, Morris CG, Casey-Sawicki KA and
Okunieff PG: Measuring radiation toxicity using circulating
Cell-free DNA in prostate cancer patients. Int J Part Ther.
8:28–35. 2022.PubMed/NCBI View Article : Google Scholar
|