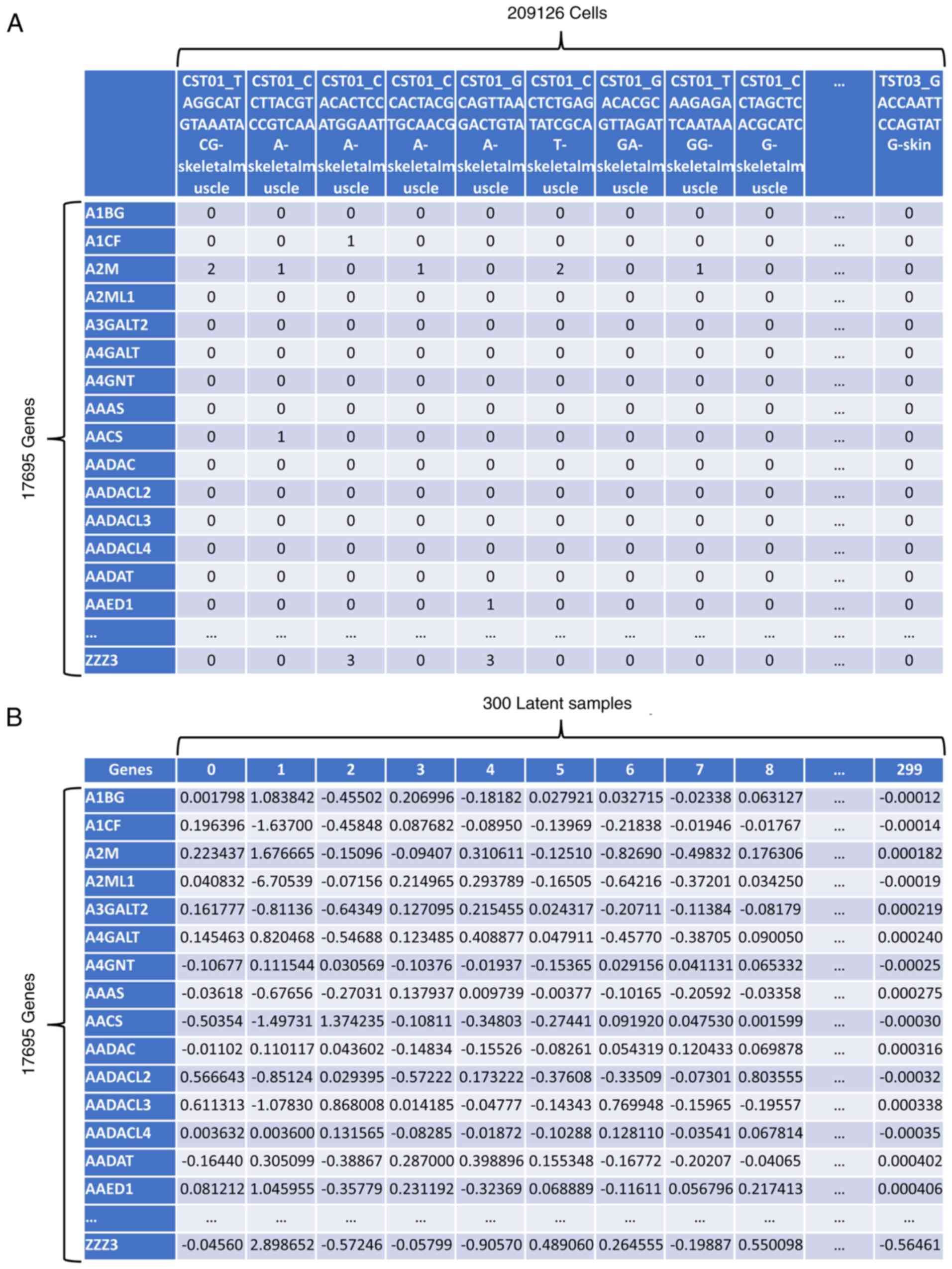
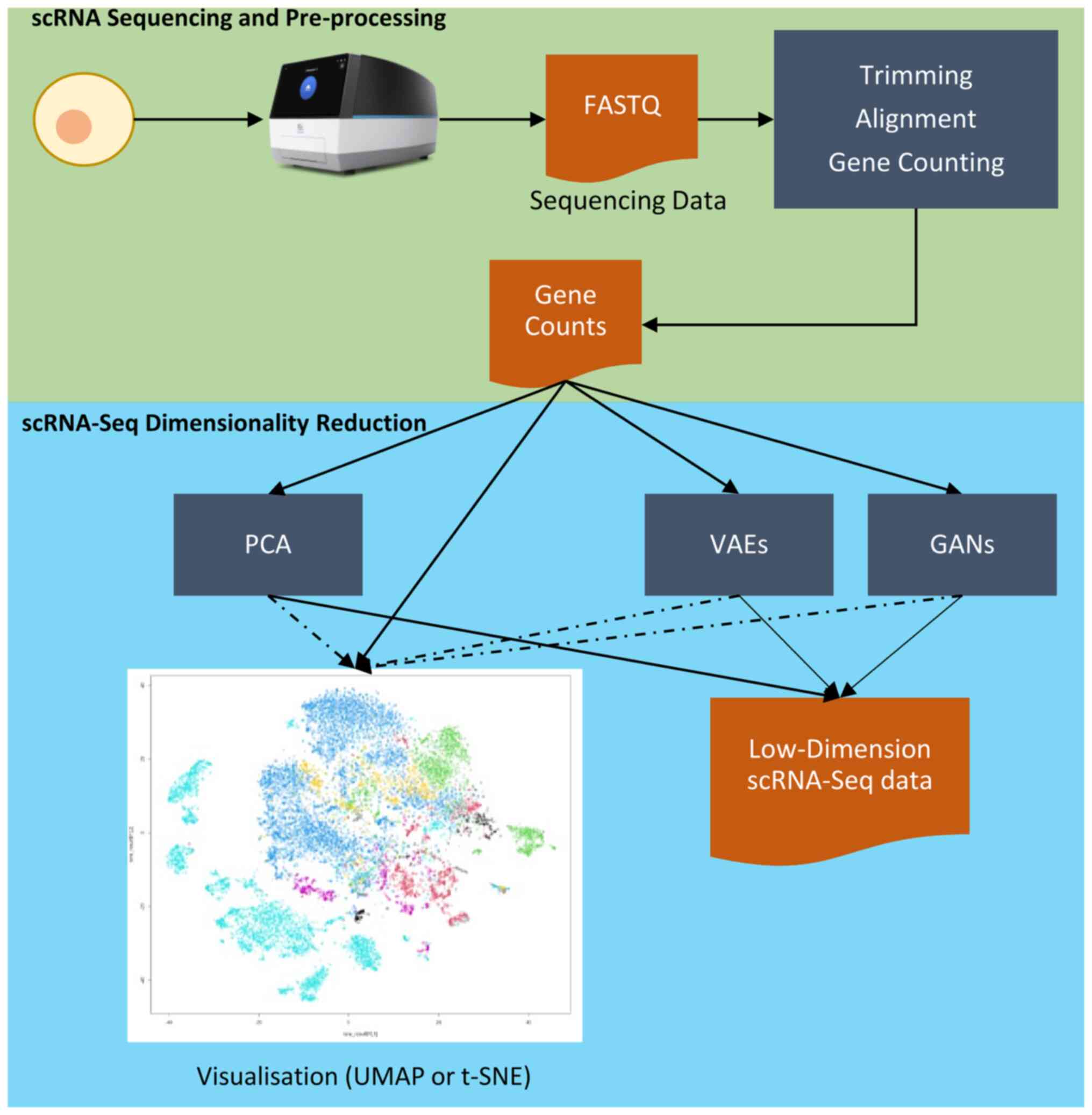
|
1
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Schena M, Shalon D, Davis RW and Brown PO:
Quantitative monitoring of gene expression patterns with a
complementary DNA microarray. Science. 270:467–470. 1995.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tang F, Barbacioru C, Wang Y, Nordman E,
Lee C, Xu N, Wang X, Bodeau J, Tuch BB, Siddiqui A, et al: mRNA-Seq
whole-transcriptome analysis of a single cell. Nat Methods.
6:377–382. 2009.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Haque A, Engel J, Teichmann SA and
Lonnberg T: A practical guide to single-cell RNA-sequencing for
biomedical research and clinical applications. Genome Med.
9(75)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cock PJ, Fields CJ, Goto N, Heuer ML and
Rice PM: The Sanger FASTQ file format for sequences with quality
scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res.
38:1767–1771. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kivioja T, Vaharautio A, Karlsson K, Bonke
M, Enge M, Linnarsson S and Taipale J: Counting absolute numbers of
molecules using unique molecular identifiers. Nat Methods. 9:72–74.
2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Satija R, Farrell JA, Gennert D, Schier AF
and Regev A: Spatial reconstruction of single-cell gene expression
data. Nat Biotechnol. 33:495–502. 2015.PubMed/NCBI View
Article : Google Scholar
|
|
8
|
Zogopoulos VL, Saxami G, Malatras A,
Papadopoulos K, Tsotra I, Iconomidou VA and Michalopoulos I:
Approaches in gene coexpression analysis in eukaryotes. Biology
(Basel). 11(1019)2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ilicic T, Kim JK, Kolodziejczyk AA, Bagger
FO, McCarthy DJ, Marioni JC and Teichmann SA: Classification of low
quality cells from single-cell RNA-seq data. Genome Biol.
17(29)2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zheng GX, Terry JM, Belgrader P, Ryvkin P,
Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et
al: Massively parallel digital transcriptional profiling of single
cells. Nat Commun. 8(14049)2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Wu Y and Zhang K: Tools for the analysis
of high-dimensional single-cell RNA sequencing data. Nat Rev
Nephrol. 16:408–421. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Qiu P: Embracing the dropouts in
single-cell RNA-seq analysis. Nat Commun. 11(1169)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Imoto Y, Nakamura T, Escolar EG, Yoshiwaki
M, Kojima Y, Yabuta Y, Katou Y, Yamamoto T, Hiraoka Y and Saitou M:
Resolution of the curse of dimensionality in single-cell RNA
sequencing data analysis. Life Sci Alliance.
5(e202201591)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Van der Maaten L and Hinton G: Visualizing
data using t-SNE. J Mach Learn Res. 9:2008.
|
|
15
|
Nanga S, Bawah AT, Acquaye BA, Billa MI,
Baeta FD, Odai NA, Obeng SK and Nsiah AD: Review of dimension
reduction methods. J Data Anal Inform Process. 09:189–231.
2021.
|
|
16
|
Sarker IH: Machine learning: Algorithms,
Real-world applications and research directions. SN Comput Sci.
2(160)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Alpaydin E: Introduction to Machine
Learning. MIT Press, Cambridge, Massachusetts, London, England,
2020.
|
|
18
|
Okada H, Chung UI and Hojo H: Practical
compass of Single-cell RNA-Seq Analysis. Curr Osteoporos Rep.
22:433–440. 2024.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Arora JK, Opasawatchai A, Poonpanichakul
T, Jiravejchakul N, Sungnak W, Thailand D, Matangkasombut O,
Teichmann SA, Matangkasombut P and Charoensawan V: Single-cell
temporal analysis of natural dengue infection reveals skin-homing
lymphocyte expansion one day before defervescence. iScience.
25(104034)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Linderman GC: Dimensionality reduction of
Single-cell RNA-Seq data. Methods Mol Biol. 2284:331–342.
2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Pearson K: LIII. On lines and planes of
closest fit to systems of points in space. Lond Edinb Dubl Phil
Mag. 2:559–572. 1901.
|
|
22
|
Jolliffe IT: Principal Component Analysis.
Springer, New York, NY, 2002.
|
|
23
|
Jolliffe IT and Cadima J: Principal
component analysis: A review and recent developments. Philos Trans
A Math Phys Eng Sci. 374(20150202)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Thorndike RL: Who belongs in the family?
Psychometrika. 18:267–276. 1953.
|
|
25
|
Tsuyuzaki K, Sato H, Sato K and Nikaido I:
Benchmarking principal component analysis for large-scale
single-cell RNA-sequencing. Genome Biol. 21(9)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ma S and Dai Y: Principal component
analysis based methods in bioinformatics studies. Brief Bioinform.
12:714–722. 2011.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Hinton GE and Roweis S: Stochastic
Neighbor Embedding. In: Advances in Neural Information Processing
Systems. Becker S, Thrun S and Obermayer K (eds.) MIT Press,
Cambridge, MA, pp857-864, 2003.
|
|
28
|
McInnes L, Healy J and Melville J: Umap:
Uniform manifold approximation and projection for dimension
reduction arXiv: 1802.03426, 2018.
|
|
29
|
Slovin S, Carissimo A, Panariello F,
Grimaldi A, Bouche V, Gambardella G and Cacchiarelli D: Single-cell
RNA sequencing analysis: A Step-by-Step overview. Methods Mol Biol.
2284:343–365. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Lachmann A, Torre D, Keenan AB, Jagodnik
KM, Lee HJ, Wang L, Silverstein MC and Ma'ayan A: Massive mining of
publicly available RNA-seq data from human and mouse. Nat Commun.
9(1366)2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Kobak D and Linderman GC: Initialization
is critical for preserving global data structure in both t-SNE and
UMAP. Nat Biotechnol. 39:156–157. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hao Y, Stuart T, Kowalski MH, Choudhary S,
Hoffman P, Hartman A, Srivastava A, Molla G, Madad S,
Fernandez-Granda C and Satija R: Dictionary learning for
integrative, multimodal and scalable single-cell analysis. Nat
Biotechnol. 42:293–304. 2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Goodfellow I, Bengio Y and Courville A:
Deep Learning. An MIT Press book. https://www.deeplearningbook.org/.
|
|
34
|
Ding J, Condon A and Shah SP:
Interpretable dimensionality reduction of single cell transcriptome
data with deep generative models. Nat Commun.
9(2002)2018.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kramer MA: Nonlinear principal component
analysis using autoassociative neural networks. AIChE J.
37:233–243. 1991.
|
|
36
|
Eraslan G, Simon LM, Mircea M, Mueller NS
and Theis FJ: Single-cell RNA-seq denoising using a deep count
autoencoder. Nat Commun. 10(390)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Agarwal D, Wang J and Zhang NR: Data
denoising and Post-denoising corrections in single cell RNA
sequencing. Statistical Science. 35:112–128. 2020.
|
|
38
|
Huang M, Wang J, Torre E, Dueck H, Shaffer
S, Bonasio R, Murray JI, Raj A, Li M and Zhang NR: SAVER: Gene
expression recovery for single-cell RNA sequencing. Nat Methods.
15:539–542. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Li WV and Li JJ: An accurate and robust
imputation method scImpute for single-cell RNA-seq data. Nat
Commun. 9(997)2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kingma DP and Welling M: Auto-encoding
variational bayes. arXiv, 2013.
|
|
41
|
Gronbech CH, Vording MF, Timshel PN,
Sonderby CK, Pers TH and Winther O: scVAE: Variational
auto-encoders for single-cell gene expression data. Bioinformatics.
36:4415–4422. 2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Pan W, Long F and Pan J: ScInfoVAE:
Interpretable dimensional reduction of single cell transcription
data with variational autoencoders and extended mutual information
regularization. BioData Min. 16(17)2023.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hinton GE and Salakhutdinov RR: Reducing
the dimensionality of data with neural networks. Science.
313:504–507. 2006.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Erfanian N, Heydari AA, Feriz AM, Ianez P,
Derakhshani A, Ghasemigol M, Farahpour M, Razavi SM, Nasseri S,
Safarpour H and Sahebkar A: Deep learning applications in
single-cell genomics and transcriptomics data analysis. Biomed
Pharmacother. 165(115077)2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Bica I, Andres-Terre H, Cvejic A and Lio
P: Unsupervised generative and graph representation learning for
modelling cell differentiation. Sci Rep. 10(9790)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Rahman MA, Tutul AA, Sharmin M and Bayzid
MS: BEENE: Deep learning-based nonlinear embedding improves batch
effect estimation. Bioinformatics. 39(btad479)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Chen RTQ, Li X, Grosse R and Duvenaud D:
Isolating sources of disentanglement in VAEs. In: Proceedings of
the 32nd International Conference on Neural Information Processing
Systems Curran Associates Inc., Montréal Canada, pp2615-2625,
2018.
|
|
48
|
Eraslan G, Drokhlyansky E, Anand S, Fiskin
E, Subramanian A, Slyper M, Wang J, Van Wittenberghe N, Rouhana JM,
Waldman J, et al: Single-nucleus cross-tissue molecular reference
maps toward understanding disease gene function. Science.
376(eabl4290)2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Koutrouli M, Nastou K, Piera Lindez P,
Bouwmeester R, Rasmussen S, Martens L and Jensen LJ: FAVA:
High-quality functional association networks inferred from
scRNA-seq and proteomics data. Bioinformatics.
40(btae010)2024.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Szklarczyk D, Kirsch R, Koutrouli M,
Nastou K, Mehryary F, Hachilif R, Gable AL, Fang T, Doncheva NT,
Pyysalo S, et al: The STRING database in 2023: Protein-protein
association networks and functional enrichment analyses for any
sequenced genome of interest. Nucleic Acids Res. 51:D638–D646.
2023.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Goodfellow IJ, Pouget-Abadie J, Mirza M,
Xu B, Warde-Farley D, Ozair S, Courville A and Bengio Y: Generative
adversarial nets. In: Proceedings of the 27th International
Conference on Neural Information Processing Systems-Volume 2 MIT
Press, Montreal, Canada, pp2672-2680, 2014.
|
|
52
|
Lan L, You L, Zhang Z, Fan Z, Zhao W, Zeng
N, Chen Y and Zhou X: Generative Adversarial Networks and Its
Applications in Biomedical Informatics. Front Public Health.
8(164)2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lacan A, Sebag M and Hanczar B: GAN-based
data augmentation for transcriptomics: Survey and comparative
assessment. Bioinformatics. 39:i111–i120. 2023.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Vinas R, Andres-Terre H, Lio P and Bryson
K: Adversarial generation of gene expression data. Bioinformatics.
38:730–737. 2022.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Marouf M, Machart P, Bansal V, Kilian C,
Magruder DS, Krebs CF and Bonn S: Realistic in silico generation
and augmentation of single-cell RNA-seq data using generative
adversarial networks. Nat Commun. 11(166)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Lall S, Ray S and Bandyopadhyay S: LSH-GAN
enables in-silico generation of cells for small sample high
dimensional scRNA-seq data. Commun Biol. 5(577)2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zhu X, Meng S, Li G, Wang J and Peng X:
AGImpute: Imputation of scRNA-seq data based on a hybrid GAN with
dropouts identification. Bioinformatics. 40(btae068)2024.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Chari T and Pachter L: The specious art of
single-cell genomics. PLoS Comput Biol. 19(e1011288)2023.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Chollet F: Keras. https://github.com/fchollet/keras; https://keras.io.
|
|
60
|
Abadi M, Agarwal A, Barham P, Brevdo E,
Chen Z, Citro C, Corrado GS, Davis A, Dean J, Devin M, et al:
TensorFlow: Large-scale machine learning on heterogeneous
distributed Systems. Distributed Parallel Cluster Computing: 16
Mar, 2016.
|
|
61
|
Mittal S and Vaishay S: A survey of
techniques for optimizing deep learning on GPUs. J Systems
Architecture. 99(101635)2019.
|
|
62
|
Kim J and Park H: Limited discriminator
GAN using explainable AI model for overfitting problem. ICT
Express. 9:241–246. 2023.
|