Introduction
Cancer is a major public health burden and is the
second leading cause of mortality following cardiovascular diseases
(1). Cancer is a genetic disease
characterized by uncontrolled cell proliferation that usually
invades and disrupts surrounding organs and tissues. This condition
poses a serious public health concern in both developed and
developing countries, in spite of current interventions (2). The regulation of growth stimulating
and inhibiting pathways is a dependent factor for the growth and
progression of healthy cells. Thus, alterations in the levels of
proto-oncogenes and tumor suppressor genes that code for proteins,
which regulate cell division, repair damaged DNA and initiate
apoptosis, are known to cause cancers. The outcome of these
alterations may be the production of cells that do not need
external signals for cell division and growth (3). Genetic instability, aided by
increased oxidative stress, results in the production of new tumor
phenotypes with a reduction in apoptosis and an increase in tumor
progression (4).
Cancer remains a main cause of morbidity and
mortality, despite the notable advancements made in clinical
interventions. Hepatocellular carcinoma (HCC) is known as primary
liver cancer and has been reported as one of the leading causes of
cancer-associated mortality that accounts for >80% of liver
cancer cases (5,6). HCC is a malignant tumor with a high
incidence rate that causes a dysregulation of metabolic enzymes
(7-9).
Among the risk factors of HCC are chronic hepatitis (hepatitis B
and C virus) infections, obesity, alcohol abuse, autoimmune
hepatitis, diabetes mellitus and metabolic diseases (10).
Conventionally, cancer management options include
surgery, radiotherapy, immunotherapy and chemotherapy. However, due
to the development of acquired or intrinsic chemo-resistance and
the decrease in the levels of apoptotic proteins, the majority of
the chemotherapeutic drugs used in the treatment of liver cancer,
such as cisplatin, adriamycin, 5-fluorouracil, paclitaxel and
doxorubicin have become ineffective (11). However, as regards liver cancer,
clinically, no satisfactory method is available to date for its
treatment. This situation renders the continuous search for novel
and suitable alternatives imperative.
Generally, plants are known as essential sources of
novel chemical entities suitable for anticancer drug discovery and
development, and a number of plant species are currently in use for
the treatment and prevention of cancer (12). Plants are known to contain
compounds, such as phenols, flavonoids, tannins, alkaloids,
lignans, terpenoids and quinones that contributes to therapeutic
value in disease management (13).
The bioactive components of plants have been used for the treatment
of inflammation, infections and tumors. This practice is very
common in poverty-stricken regions of the world and is based on
oral tradition and folklore. In Nigeria for example, bitter yam,
also known as Dioscorea dumetorum (D. dumetorum), is
a tuber from the family, Diosoreaceae that is used by traditional
medical practitioners for the treatment of diabetes, diarrhea,
gonorrhea, jaundice, malaria, cancers and pain management (14,15).
The bitter yam (doyar bisa in Hausa) is believed to contain
numerous bioactive agents that contribute to its potency in
traditional medicine. The Nigerian myth presumes that anything
bitter is medicinal and has therapeutic potential. However, even
though this myth has enabled the use of bitter yam for the local
management of a number of diseases including cancer, there is
limited scientific evidence available to support its anticancer
properties. Therefore, the present study was designed to
investigate the anticancer properties of D. dumetorum tubers
in the HepG2 liver cancer cell line.
Materials and methods
Reagents and chemicals
The reagents used in the present study included
trypsin-EDTA (MilliporeSigma), Roswell Park Memorial Institute
(RPMI) medium (Gibco; Thermo Fisher Scientific, Inc.) and
3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT)
reagent (Sigma-Aldrich; Merck KGaA). Phosphate-buffered saline
(PBS), trypan blue, dimethyl sulphoxide (DMSO), molecular grade
water and analytical grade solvents (methanol, hexane, chloroform
and ethyl-acetate) were obtained from MilliporeSigma.
Plant material
Fresh tubers of D. dumetorum (Kunth) Pax were
purchased from the local morning market along Ahmadu Bello Way,
Kaduna-North Local Government Area of Kaduna State, Nigeria. The
tubers were identified and authenticated at the Herbarium, in the
Department of Biological Sciences, Kaduna State University, Kaduna,
Nigeria. The voucher specimen number assigned to the sample was
KASU/BCH/0778.
Preparation of plant material
The yam tubers were washed, peeled and chopped into
smaller sections that were dried under the shade. The dried chips
were grinded using a domestic warring blender and maintained in an
air-tight container prior to extraction.
Extraction of sample
For the extraction of the sample, ~1 kg of the
powdered D. dumetorum tuber was soaked successively in
hexane and methanol for 48 h each time at room temperature. The
extracts were collected separately by filtration using a muslin
cloth. Thereafter, the extracts were dried in a rotary evaporator
(Heidolph Instruments GmbH & Co. KG) set at 40˚C.
The extracts were weighed and stored at 4˚C for further
use.
Bioassay guided fractionation of D.
dumetorum
The crude hexane and methanolic extracts of D.
dumetorum were subjected to cytotoxicity assay to identify the
more active extract. The methanolic extract was more cytotoxic to
the liver cancer cell line and was subjected to further
fractionation using the modified solvent-solvent fractionation
method as previously described (16). The methanolic extract (5 g) was
mixed with a four-solvent system that included hexane, chloroform,
ethyl acetate and water in a separating funnel at room temperature.
The mixture was allowed to stand for 2 h for fractionation to
occur. The solvent mixture fractionated based on their densities,
each carrying different components of the bioactive methanolic
extract. The fractions were collected separately and subjected to
cytotoxicity assay.
Thin layer chromatography (TLC)
profiling of the extracts
The methanolic extract of D. dumetorum and
its fractions were subjected to TLC to evaluate the purity of the
fractions. The procedure was performed using TLC plates (5x1 cm)
(MilliporeSigma) pre-coated with silica gel. The mixtures to be
resolved were spotted on the baseline of plates and placed in a
beaker containing the mobile phase. The mobile phase was allowed to
move through the plates by capillary action until it reached the
solvent front. Thereafter, the plates were visualized under UV
light and the retention factor (RF) values of spots (compounds)
were calculated.
Gas chromatography-mass spectrometry
(GC-MS) analysis
The most bioactive fraction (chloroform) of the
methanolic extract of D. dumetorum was subjected to GC-MS
analysis which was performed using the Mass Hunter GCMS system
(Agilent Technologies, Inc.) with a 5975C Mass Spectrometer fitted
with a HP5-MS capillary column. The interpretation of the mass
spectral data was performed using the database of the National
Institute Standard and Technology (NIST), which has >62,000
patterns. The mass spectra of the unknown components were compared
with the spectra of the known components in the NIST library. The
components of the test materials were identified by name, molecular
weight and structure.
Cell line and culture
The HepG2 (liver cancer) cancer cell line (cat. no.
HB-8065; American Type Culture Collection) and maintained at
37˚C in an incubator supplemented with 5%
CO2. The HepG2 cells were grown in RPMI-1640 medium
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (FBS; Sigma-Aldrich, USA) and 1%
penicillin-streptomycin (Thermo Fisher Scientific, Inc.). At 70 to
80% cell culture confluence, sub-culturing was routinely performed
to maintain the cells.
Cytotoxicity assay (MTT assay)
The cytotoxicity study was performed using MTT
assay, as previously described by Waziri et al (17). The HepG2 cells were seeded at a
density of 2x103 cells per well for 24 h and treated
with either 12.5, 25, 50, 100 or 200 µg/ml of the extract or
fraction for 48 h, while 0.1% DMSO and doxorubicin (MilliporeSigma)
in same treatment concentrations as the extract/fraction were used
as negative and positive controls, respectively. Following 48 h of
treatment, 20 µl MTT solution (5 mg/ml) were added to each well and
the plate was re-incubated at 37˚C for 2 h. The reaction
was terminated by the addition of 150 µl DMSO to solubilize the
MTT-formazan crystals formed by metabolically viable cells. The
optical density was measured at 570 nm using an xMark microplate
spectrophotometer (Bio-Rad Laboratories, Inc.). Each experiment was
repeated three times, and each dilution had at least three
replicates. The percentage cytotoxicity was calculated using the
following formula:
Cell morphology assay
The effects of the chloroform fraction of D.
dumetorum on HepG2 cells were monitored using phase contrast
microscopy. The cells were seeded in a 6-well plate at a density of
5x104 cells/well overnight and treated with various
concentrations (20, 40, 60 80 and 100 µg/ml) of the chloroform
fraction of the methanol extract of D. dumetorum for 24 h.
Thereafter, the cells were viewed under a phase contrast microscope
(AmScope).
Caspase-3, -8 and -9 assays
Using the Caspase-3 colorimetric assay kit (cat. no.
K106-100, BioVision), the effects of the chloroform fraction of
D. dumetorum on the apoptosis of HepG2-cells were
investigated. Briefly, the cells were seeded overnight in a 96-well
plate at a density of 1x104 cells per well and treated
with 20, 40, 60, 80 and 100 µg/ml of the chloroform fraction for 24
h. Thereafter, the cells were harvested using trypsin and
centrifuged at 20.12 x g for 5 min at 4˚C to obtain
pellets. The cell pellets were washed with PBS and re-suspended in
50 µl chilled (4˚C) Cell Lysis Buffer in the assay kit
before incubating on ice for 10 min for lysis to occur. Following
incubation on ice, the cells were centrifuged at 20,124 x g for 1
min at 4˚C to collect supernatant for protein
quantification to ensure even protein concentrations in all
samples. Approximately 50 µl of each supernatant was mixed with 50
µl 2X Reaction Buffer in the assay kit (containing 10 mM DTT) in a
96-well plate. This was followed by the addition of 5 µl of 4 mM
DEVD-pNA in the assay kit (200 µM, final concentration) and
incubation for 2 h at 37˚C. The same procedure was
repeated for the caspase-8 (cat. no. 113-100) and -9 (cat. no.
K119-100) assays using their respective assay kits. The optical
density (OD) of each sample was measured at 405 nm using the xMark
microtiter spectrophotometer (Bio-Rad Laboratories, Inc.) and the
fold change was calculated relative to the negative control.
Gene expression assay. RNA
isolation
RNA was isolated from the chloroform
fraction-treated HepG2 cells using the GF-1 total RNA extraction
kit (Vivantis Technologies Sdn Bhd). Following treatment with
increasing concentrations of the chloroform fraction, the cells
were harvested and centrifuged at 20.12 x g for 5 min at
4˚C to collect the pellets. Subsequently, ~350 µl Buffer
TR in the extraction kit was added to the pellets (suspended in 50
µl PBS) and thoroughly mixed by vortexing to produce cell lysate,
which was transferred into a homogenization column assembled in a
collection tube and centrifuged at 46,887 x g for 2 min at
4˚C. The flow through was saved and equal volume of 80%
ethanol was added and mixed gently. Subsequently, ~650 µl of the
mixture were transferred into an RNA Binding Column provided along
with the kit, fitted to a collection tube and spun at 20,124 x g
for 1 min at 4˚C. The flow through was discarded and 500
µl Wash Buffer in the assay kit was added to the Binding Column and
centrifuged at maximum speed (46,887 x g) for 1 min at
4˚C prior to the addition of 70 µl DNase I Digestion Mix
and the mixture was incubated for 15 min at room temperature.
Following incubation, 500 µl Inhibition Removal Buffer was added
and centrifuged at maximum speed (46,887 x g) for 1 min at
4˚C to discard the flow through. The pellets in the
Binding column were washed twice each with 500 µl Wash Buffer, in
each case the flow through was discarded following centrifugation
at 20,124 x g for 1 min at 4˚C. RNA was finally
collected by the addition of 60 µl RNase-free Water directly on the
membrane of the RNA Binding Column fitted into a new Eppendorf tube
(1.5 ml; Eppendorf AG, Hamburg, Germany) and centrifuged for 1 min
at 20,124 x g at 4˚C to collect the pure RNA. The RNA
was quantified at 260 nm using a NanoDrop®
spectrophotometer (Thermo Fisher Scientific, Inc.) and stored at
-20˚C.
cDNA synthesis. The RNA extracted was
transcribed into cDNA using ReverTra Ace™ qPCR RT Master
Mix with gDNA Remover (Toyobo Co., Ltd.). For genomic DNA removal,
the DNase I reaction solution was prepared comprising of 2 µl of 4X
DN master mix, 6 µl of RNA template (0.5 µg) and 2 µl of nuclease
free water and incubated for 5 min at 37˚C. For the
reverse transcription, 2 µl of 5X RT master mix II was added and
incubated first at 37˚C for 15 min, and 50˚C
for 5 min and finally heated at 98˚C for 5 min to
terminate the reverse transcription. The cDNA synthesized was
stored at -20˚C for further analysis.
Primer design. The primers for the apoptosis
related genes were designed on the NCBI website using Primer-BLAST
software (Primer3, version 2.5.0). The primers used are listed in
Table I. They were synthesized by
Integrated DNA Technologies (IDT). Each primer was provided in a
lyophilized form and reconstituted to a stock concentration of 100
µM using nuclease-free water.
 | Table IPrimers of genes used in the present
study. |
Table I
Primers of genes used in the present
study.
| Gene | Primer
sequence |
|---|
| β-actin | F:
5'-ACCTAACTTGCGCAGAAAACAAGA-3' |
| | R:
5'-ACTGCTGTCACCTTCACCGT-3' |
| Bax | F:
5'-GAGTGTCTCAAGCGCATCGG-3' |
| | R:
5'-AGTAGAAAAGGGCGACAACCC-3' |
| p53 | F:
5'-CCTGGATTGGCAGCCAGACT-3' |
| | R:
5'-CCATTGCTTGGGACGGCAAG-3' |
| Bcl-2 | F:
5'-ATCGCCCTGTGGATGACTGAG-3' |
| | R:
5'-AGGGCCAAACTGAGCAGAGTC-3' |
| MDM2 | F:
5'-GCGTGCCAAGCTTCTCTGTG-3' |
| | R:
5'-CCTGAGTCCGATGATTCCTGCT-3' |
Reverse transcription-quantitative PCR
(RT-qPCR). The expression of the apoptotic genes was evaluated
by RT-qPCR using SYBR-Green master mix (Toyobo Co., Ltd.) and the
synthesized primers (Table I),
while β-actin was used as the reference gene. PCR was performed as
follows: 95˚C for 60 sec of initial denaturation,
followed by 40 cycles of 95˚C for 15 sec and
60˚C for 30 sec. The relative expression of each gene
was performed using the 2-ΔΔCq
method (18).
Statistical analysis
All experiments were performed in triplicate. The
statistical software package SPSS (version 27, IBM Corp.) was used
to analyze the data. One-way analysis of variance (ANOVA) was
conducted to compare the mean values between control and treatment
groups at the 95% confidence level, followed by Tukey's post hoc
test to identify specific group differences. Data obtained from the
study are expressed as the mean ± standard deviation (SD). A value
of P<0.05 was considered to indicate a statistically significant
difference.
Results
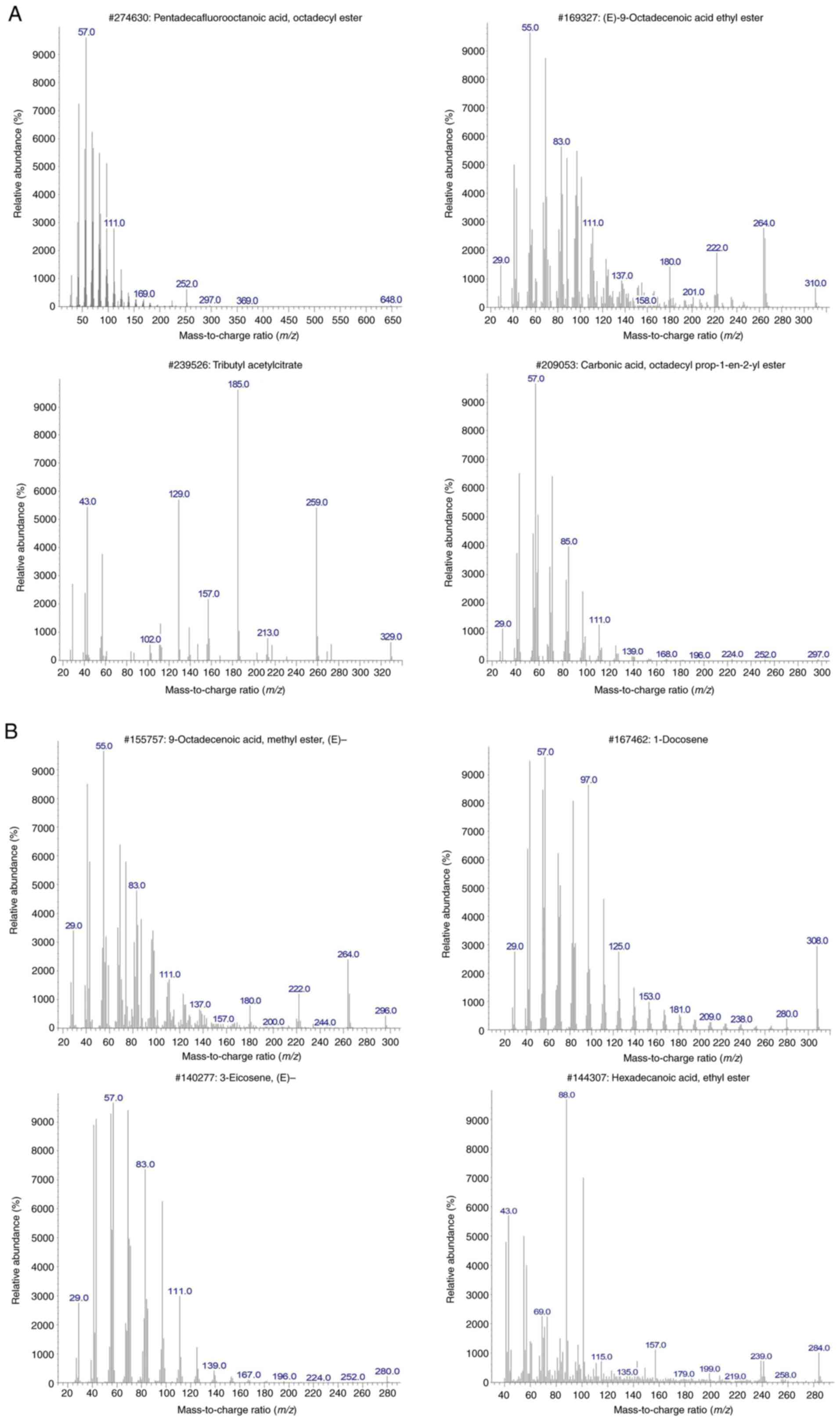
Results of GC-MS analysis
The GC-MS analysis revealed that the chloroform
fraction was composed mainly of pentadecafluorooctanoic acid,
(E)-9-octadecenoic acid ethyl ester (elaidic acid), tributyl acetyl
citrate, octadecyl prop-1-en-2-yl ester, 9-octadecenoic acid (oleic
acid), 1-docosene, 3-eicosene, and hexadecanoic acid (palmitic
acid) (Table II and Fig. 1A and B).
| Table IIGas chromatography-mass
spectrophotometer analysis of CFMEDD. |
Table II
Gas chromatography-mass
spectrophotometer analysis of CFMEDD.
| Serial no. | Names of
compounds | Molecular
formula | Molecular weight
(g/mol) | Retention time
(min) | Peak area (%) |
|---|
| 1 |
Pentadecafluorooctanoic acid |
C8HF15O2 | 414.07 | 26.42 | 11.78 |
| 2 | (E)-9-Octadecenoic
acid ethyl ester |
C20H38O2 | 310.50 | 24.24 | 11.37 |
| 3 | Tributyl
acetylcitrate |
C20H34O8 | 402.50 | 25.64 | 9.88 |
| 4 | Octadecyl
prop-1-en-2-yl ester |
C22H42O3 | 354.6 | 25.15 | 6.16 |
| 5 | 9-Octadecenoic
acid |
C18H34O2 | 282.5 | 23.00 | 4.86 |
| 6 | 1-Docosene |
C22H44 | 308.6 | 26.24 | 4.72 |
| 7 | 3-Eicosene |
C20H40 | 280.5 | 25.42 | 4.16 |
| 8 | Hexadecanoic
acid |
C16H32O2 | 256.42 | 19.81 | 4.06 |
Cytotoxic effects of the extract on
HepG2 cells
The methanolic extract had a lower IC50
value (29.84±0.004 µg/ml) than the hexane extract (50.54±0.004
µg/ml) of D. dumetorum, while that of doxorubicin (standard
drug) was the lowest, as shown in Table III. The IC50 value of
the methanol extract was significantly (P<0.05) lower than that
of the hexane extract and was selected for further fractionation.
Of the fractions of the methanolic extract of D. dumetorum
screened, the chloroform fraction was the most active with the
lowest IC50 value (28.5±0.00 µg/ml), followed by the
ethyl acetate fraction (31.69±0.03 µg/ml), hexane fraction
(52.02±0.03 µg/ml) and aqueous fraction (86.28±0.05 µg/ml). For
this reason, the chloroform fraction was selected as the fraction
used for the treatment of the HepG2 cancer cell line.
| Table IIIIn vitro cytotoxic effects of
the crude extracts and methanol sub-fractions of Dioscorea
dumetorum tuber on HepG2 (liver cancer) cells. |
Table III
In vitro cytotoxic effects of
the crude extracts and methanol sub-fractions of Dioscorea
dumetorum tuber on HepG2 (liver cancer) cells.
| Tuber extract
(Dioscorea dumetorum) | IC50
(µg/ml) |
|---|
| Crude
extracts | |
|
Hexane
extract |
50.54±0.004c |
|
Methanol
extract |
29.84±0.004b |
|
Doxorubicin
(positive control) |
11.0±0.01a |
| Sub-fractions of
methanol extract | |
|
Aqueous
fraction |
86.28±0.050d |
|
Hexane
fraction |
52.02±0.030e |
|
Chloroform
fraction |
28.5±0.00d |
|
Ethyl
acetate fraction |
31.69±0.030e |
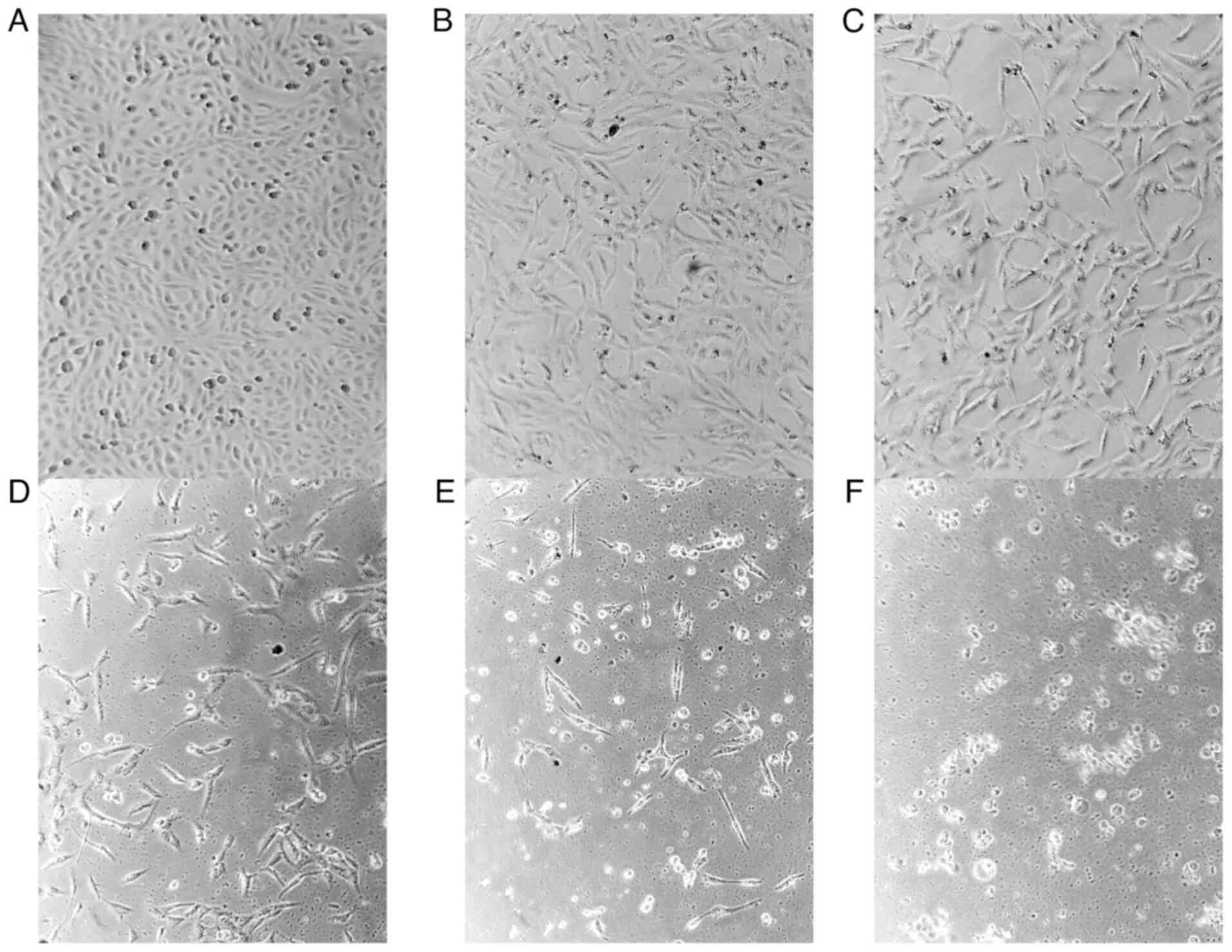
Effect of the chloroform fraction of
the methanolic extract of D. dumetorum (CFMEDD) on HepG2 cell
morphology
Treatment with the chloroform fraction caused
increased HepG2 cell death and distorted cell morphology (Fig. 2B-F). The formation of round-shaped
and floating cells is an evidence of cell death, and this feature
was more prominent in the cells treated with the highest
concentration of the extract (Fig.
2F). In addition, the chloroform fraction-treated cells
exhibited a distorted cell morphology and signs of chromatin
condensation, which is a feature of apoptosis. The phase contrast
micrograph revealed an intact cell morphology in the negative
control-treated cells (Fig.
2A).
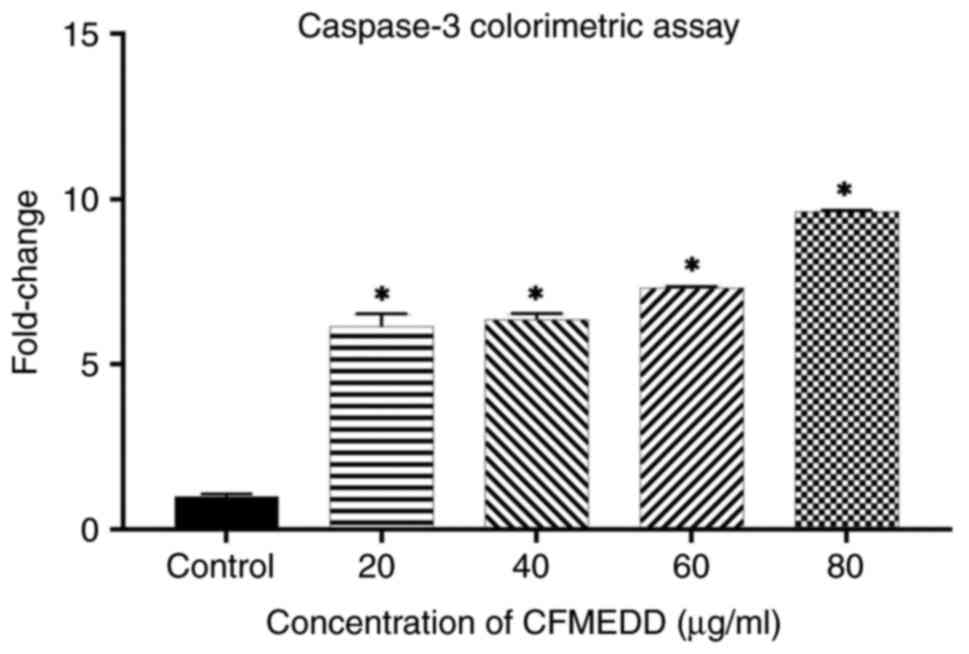
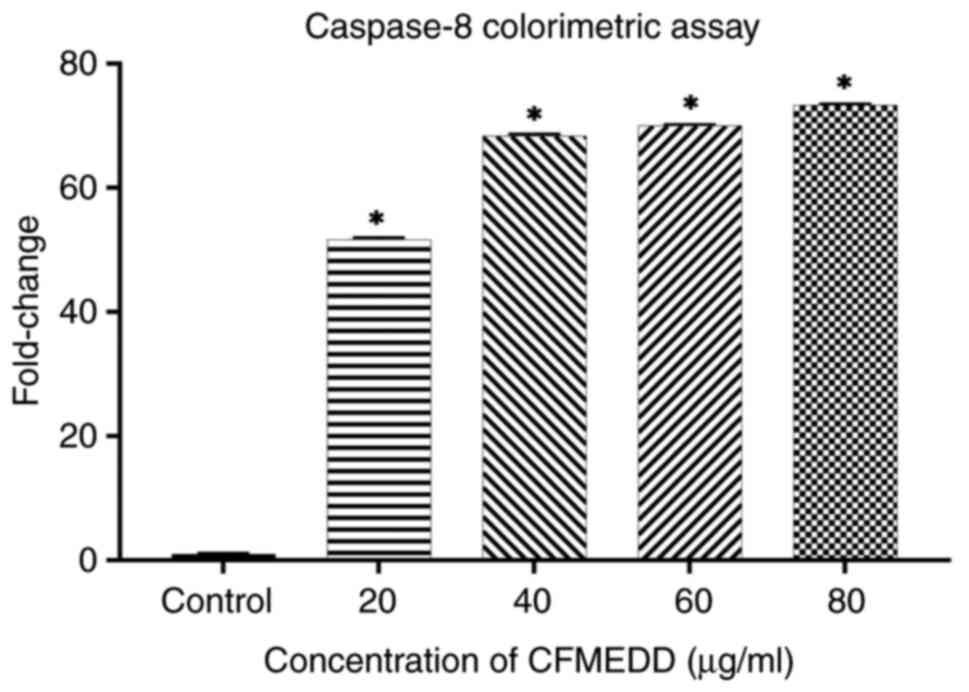
The chloroform fraction increases the
expression of caspase-3, -8 and -9 in HepG2 cells
Treatment with the chloroform fraction significantly
increased (P<0.05) the protein expression levels of caspase-3
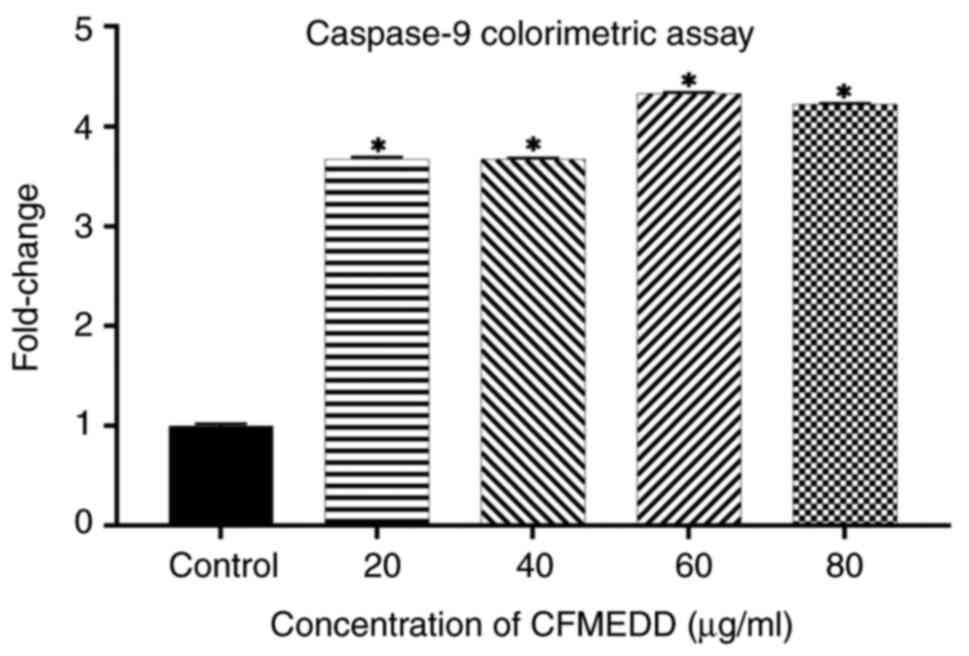
and -8 in a concentration-dependent manner (Figs. 3 and 4). As regards caspase-9, its expression
significantly increased as the treatment concentration increased
from 20 to 60 µg/ml, and decreased slightly at the concentration of
80 µg/ml (Fig. 5). The expression
of the caspases in the treated cells was significantly (P<0.05)
higher than that of the untreated cells (negative control).
Caspase-8 is main caspase of the death receptor pathway, while
caspase-9 is the main caspase of the mitochondrial pathway.
Effects of chloroform fraction on mRNA
expression in HepG2 cells
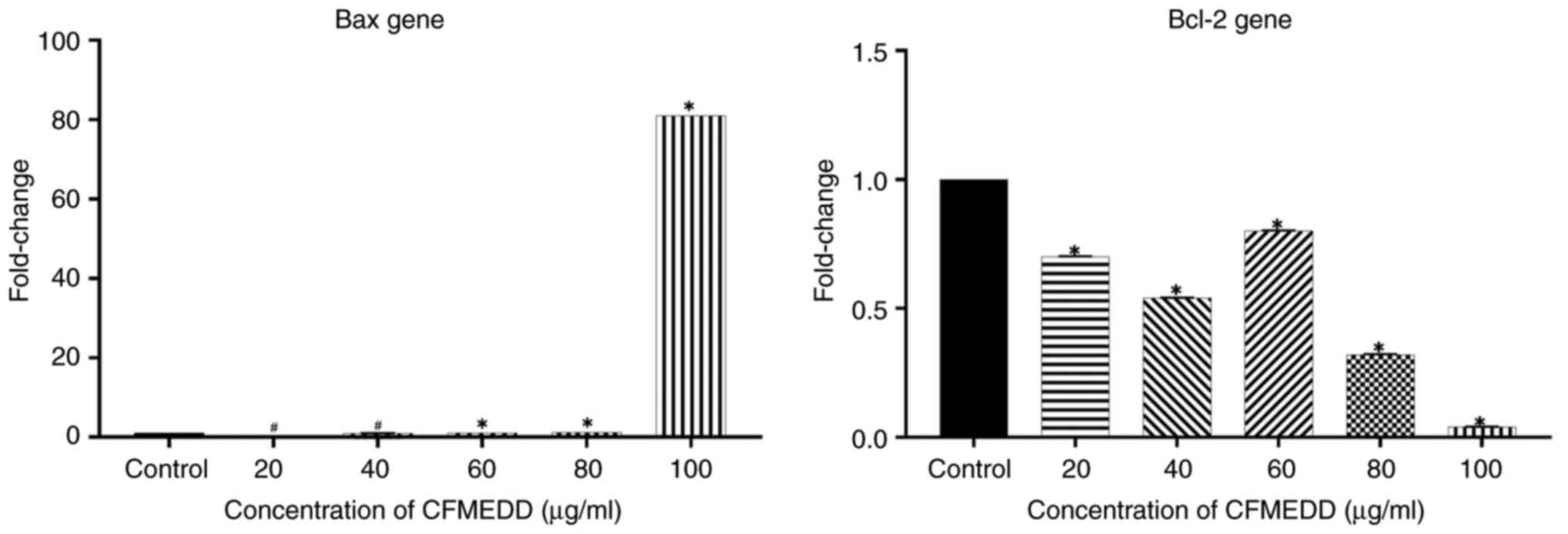
As shown in Fig. 6,
the treatment caused a significant (P<0.05) increase in the mRNA
expression of Bax at a dose of 100 µg/ml and a significant
(P<0.05) decrease in Bcl-2 mRNA corresponding to the treatment
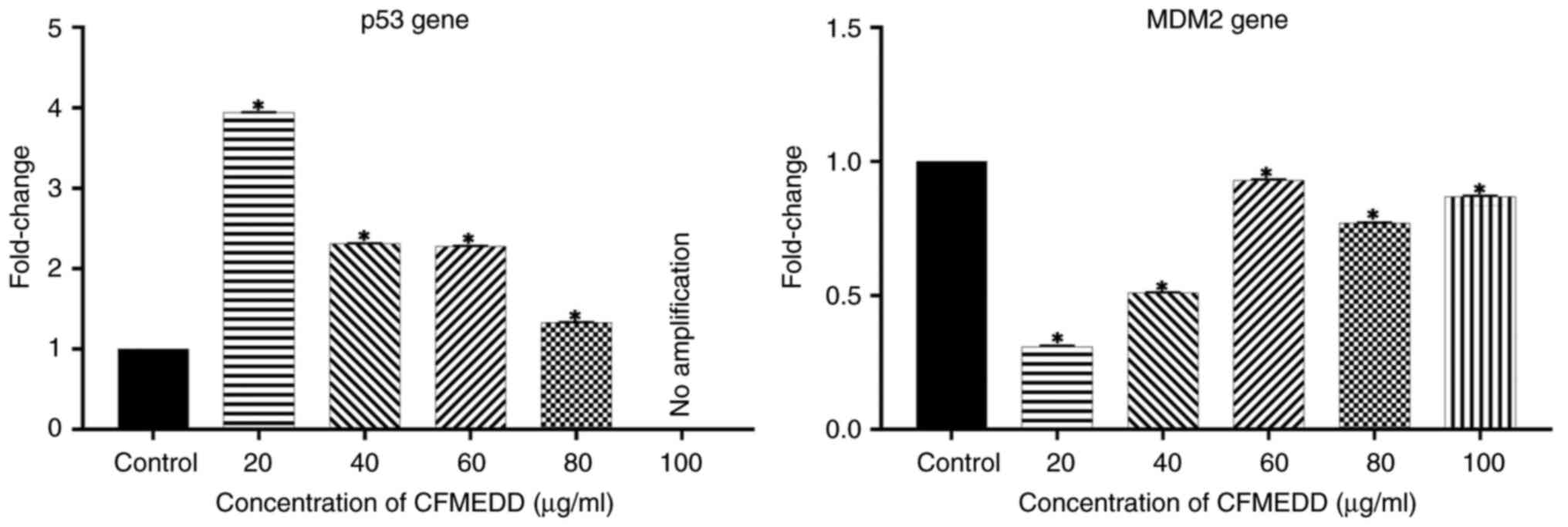
concentration. In a similar manner, the chloroform fraction
treatment caused a significant (P<0.05) increase in the mRNA
expression of p53 compared with the control cells (Fig. 7). Conversely, the mRNA expression
of the oncogene, murine double minute 2 (MDM2) significantly
(P<0.05) decreased compared with the negative control cells
(Fig. 7).
Discussion
The present study was performed to evaluate the
mechanisms of action of the CFMEDD on HepG2 (liver cancer) cells.
The results revealed that CFMEDD induced greater cytotoxic effects
than all fractions in HepG2 cells with an IC50 value of
28.45±0.00 µg/ml, while the IC50 value of the standard
drug, doxorubicin, was 11.0±0.01 µg/ml. Of the eight major
compounds identified by the GC-MS analysis of the chloroform
fraction, octadecenoic and hexadecenoic acids have been reported to
demonstrate in vitro anticancer properties. Specifically,
octadecenoic acid was found to demonstrate significant
anti-proliferative effect on human gastric (SGC-7901),
hepatocellular carcinoma (BEL-7402), and leukaemia (HL-60) tumour
cell strains, while hexadecenoic acid was previously used in the
treatment of breast, colon and liver cancers (19-21).
Therefore, it was suspected that both octadecenoic and hexadecenoic
acids may have contributed to the cytotoxicity of the chloroform
fraction of HepG2 cancer cell lines in the present study.
The examination of HepG2 cell morphology following
treatment with CFMEDD provides valuable insight into the cytotoxic
effects at a cellular level. Herein, the phase contrast micrographs
(Fig. 2) demonstrated a
dose-dependent alteration in HepG2 cell morphology, including
certain features of apoptosis, such as chromatin condensation and
cell membrane disruption. Chromatin condensation, cell shrinking
and membrane blebbing are events that precede the formation of
apoptotic bodies and eventual apoptosis (22-25).
In the present study, the protein expression of caspase-3, -8 and
-9 in HepG2 cells significantly increased following treatment with
the CFMEDD. Caspase-9 is an initiator of apoptosis via the
mitochondrial pathway, while caspase-8 initiates apoptosis via the
extrinsic death receptor pathway. The findings of the present study
suggest that CFMEDD induces the apoptosis of liver cancer cells via
both the mitochondrial and extrinsic pathways of apoptosis. Both
the extrinsic and intrinsic pathways are closely linked (26). Caspase-8 is negatively controlled
by anti-apoptotic proteins. The loss of caspase-9 promoter or gene
functions that lead to a reduced caspase-9 activity has been
implicated as one of the causes of cancer development (27). Caspase-3 is an executioner of cell
apoptosis whose activation is triggered by both caspase-8 and -9.
It was hypothesized that both caspase-8 and -9 may have triggered
the increased expression of caspase-3 in the present study.
Caspase-3 executes apoptosis via the selective destruction of
subcellular structures, organelles and the genome (28). The increased expression of
caspase-3 and -9 has also been reported in liver cancer cells
treated with clausenidin isolated from Clausena excavata
(17). In addition, activated
caspases cleave at least 100 different proteins that are
responsible for DNA replication, transcription, translation,
phosphorylation and dephosphorylation. The caspases can also cleave
the inhibitor of caspase activated DNase complex to release active
DNase in certain tissues. Activated DNases are responsible for the
internucleosomal cleavage of genomic DNA to produce smaller
fragments (29). The activity of
DNases also produces morphological alterations in cells. In the
present study, phase contrast micrographs revealed features typical
of apoptosis that could have been triggered by DNase activity.
In the present study, the results of RT-qPCR
provided further evidence of the involvement of the intrinsic
mitochondrial pathway in the apoptosis of liver cancer cells in
vitro. The treatment of HepG2 cancer cell lines with the CFMEDD
resulted in a significant decrease in the levels of Bcl-2 and MDM2
genes in a concentration-dependent manner. These two genes function
to prevent the apoptosis of cancer cells. Specifically, the Bcl-2
family genes play a key role in intrinsic mitochondrial apoptosis
(30). Bcl-2 protein is an
anti-apoptotic protein that interacts with pro-apoptotic proteins,
such as Bax, limiting pore development and the release of
cytochrome c (31). On the
other hand, an increase in Bax expression causes tumor cells to
die, resulting in cell death, while an increase in Bcl-2 expression
prevents cell death (32). In the
present study, it was hypothesized that the apoptosis of HepG2
cells may have been triggered via the decreased expression of the
Bcl-2 gene in the treated cells.
Furthermore, the gene expression analysis performed
herein revealed the upregulation of the tumor suppressor gene, p53,
and the downregulation of the oncogene, MDM2. Cancer is a product
of mutations conferred by oncogenes with dominant survival
characteristics and the downregulation of tumor suppressor genes
(33). Gene mutations do not occur
with great efficiency due to the existence of DNA surveillance and
the repair system under the control of tumor suppressors, such as
the p53 gene. The majority of cancers are the result of damage to
the p53 gene. However, the present study revealed a significant
increase in p53 gene expression in the treated cells. p53 generates
numerous signals that lead to the apoptosis of cancer cells. On the
other hand, MDM2 is a negative regulator of p53 and other proteins
involved in DNA repair and apoptosis (34). In fact, MDM2 leads to the
destruction of p53 and to the consequent development of cancers.
The interaction between MDM2 and p53 is detrimental to cells, as it
decreases p53 activity and enables cells to escape apoptosis.
Therefore, a number of anticancer agents target the inhibition of
the MDM2-p53 interaction (10,35).
The lower the expression of the MDM2 gene, the higher the
expression of the p53 gene, and its consequent activity. In the
present study, treatment with CFMEDD led to the decreased
expression of MDM2, which may have enabled the p53-mediated
apoptosis of the HepG2 cancer cells. Therefore, it was hypothesized
that this may be one of the mechanisms of action of the CFMEDD in
HepG2 cells.
In addition, the expression of Bax mRNA began to
increase at concentrations >60 µg/ml and reached its peak at the
concentration of 100 µg/ml. At this highest concentration (100
µg/ml), a >80-fold increase in the expression of the Bax gene
was observed in the CFMEDD-treated HepG2 cells. Conversely, Bcl-2
expression significantly decreased (P<0.05) in a
concentration-dependent manner. Bcl-2 is an anti-apoptotic gene
that enables tumor cells to survive, while Bax is a pro-apoptotic
gene that enables tumors to undergo apoptosis. The decision on
whether a cell should undergo apoptosis depends on the ratio of Bax
to Bcl-2. Apoptosis is lost when there is an overexpression of
Bcl-2, as reported in a number of cancers (36). In the present study, treatment of
the HepG2 cells with CFMEDD significantly downregulated the
expression of the Bcl-2 gene, and this may have triggered the onset
of the apoptosis of the HepG2 cells. p53 protein is known to
activate Bax protein for apoptosis to occur in cells (37). The present study observed a
significant increase in the expression of Bax mRNA, whose
translation leads to the production of Bax protein.
In conclusion, the present study demonstrates that,
amongst other mechanisms, the Bax gene induces the apoptosis of
CFMEDD-treated HepG2 cells, triggered by p53 activation. From the
findings presented herein, it can be concluded that CFMEDD holds
promise as a potential therapeutic agent for the treatment of liver
cancer due to its ability to induce the apoptosis of HepG2 cells
via the extrinsic and intrinsic pathways.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
All authors (CVP, PMW, RA, DT, MIT and AEA) were
involved in the conception and design of the study. CVP and PMW
provided the materials (reagents and chemicals used). CVP, DT, MIT
and AEA were involved in the in vitro assays. DT performed
the statistical analysis of the data. CVP and DT were involved in
the interpretation of data. PMW and RA were involved in the
reviewing and editing of the manuscript. PMW and DT confirm the
authenticity of all the raw data and all authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organization: Cancer: Key
facts. WHO, Geneva, 2018. https://www.who.int/news-room/fact-sheets/detail/cancer.
Accessed February 7, 2019.
|
|
2
|
Tariq A, Sadia S, Pan K, Ullah I, Mussarat
S, Sun F, Abiodun OO, Batbaatar A, Li Z, Song D, et al: A
systematic review on ethnomedicines of anti-cancer plants.
Phytother Res. 31:202–264. 2017.PubMed/NCBI View
Article : Google Scholar
|
|
3
|
Aliyu-Amoo H, Isa HI, Njoya EM and McGaw
LJ: Antiproliferative effect of extracts and fractions of the root
of Terminalia avicennioides (Combretaceae) Guill and Perr.
On HepG2 and Vero Cell Lines. Int J Phytomed Phytother.
7(71)2021.
|
|
4
|
Kadan S, Rayan M and Rayan A: Anticancer
activity of anise (Pimpinella anisum L.) seed extract. Open
Nutraceuticals J. 6:1–5. 2013.
|
|
5
|
Wang Z, Li Z, Ye Y, Xie L and Li W:
Oxidative stress and liver cancer: Etiology and therapeutic
targets. Oxid Med Cell Longev. 2016(7891574)2016.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Maluccio M and Covey A: Recent progress in
understanding, diagnosing, and treating hepatocellular carcinoma.
CA Cancer J Clin. 62:394–399. 2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sun Y, Ma W, Yang Y, He M, Li A, Bai L, Yu
B and Yu Z: Cancer nanotechnology: Enhancing tumor cell response to
chemotherapy for hepatocellular carcinoma therapy. Asian J Pharm
Sci. 14:581–594. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Nekvindova J, Mrkvicova A, Zubanova V,
Hyrslova Vaculova A, Anzenbacher P, Soucek P, Radova L, Slaby O,
Kiss I, Vondracek J, et al: Hepatocellular carcinoma: Gene
expression profiling and regulation of xenobiotic-metabolizing
cytochromes P450. Biochem Pharmacol. 177(113912)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Florio AA, Campbell PT, Zhang X,
Zeleniuch-Jacquotte A, Wactawski-Wende J, Smith-Warner SA, Sinha R,
Simon TG, Sesso HD, Schairer C, et al: Abdominal and gluteofemoral
size and risk of liver cancer: The liver cancer pooling project.
Int J Cancer. 147:675–685. 2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Manosroi A, Akazawa H, Kitdamrongtham W,
Akihisa T, Manosroi W and Manosroi J: Potent antiproliferative
effect on liver cancer of medicinal plants selected from the
Thai/Lanna medicinal plant recipe database ‘MANOSROI III’. Evid
Based Complement Alternat Med. 2015(397181)2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Khalil R, Ali Q, Hafeez M and Malik A:
Phytochemical activities of Conocarpus erectus: An overview.
Biol Clin Sci Res J. 2020:1–6. 2020.
|
|
13
|
Kaur R, Kapoor K and Kaur H: Plants as a
source of anticancer agents. J Nat Prod Plant Resour. 1:19–124.
2011.
|
|
14
|
Jesus M, Martins AP, Gallardo E and
Silvestre S: Diosgenin: Recent highlights on pharmacology and
analytical methodology. J Anal Methods Chem.
2016(4156293)2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Salehi B, Sener B, Kilic M, Sharifi-Rad J,
Naz R, Yousaf Z, Mudau FN, Fokou PVT, Ezzat SM, El Bishbishy MH, et
al: Dioscorea plants: A genus rich in vital
nutra-pharmaceuticals-A review. Iran J Pharm Res. 18(Suppl):68–89.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Keshava R, Muniyappa N and Gope R:
Bioactivity guided fractionation and elucidation of anti-cancer
properties of Imperata cylindrica leaf extracts. Asian Pac J Cancer
Prev. 21:707–714. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Waziri PM, Abdullah R, Yeap SK, Omar AR,
Kassim NK, Malami I, How CW, Etti IC and Abu ML: Clausenidin
induces caspase-dependent apoptosis in colon cancer. BMC Complement
Altern Med. 16(256)2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Win DT: Oleic acid-The anti-breast cancer
component in olive oil. AU J.T. 9:75–78. 2005.
|
|
20
|
Yu F, Lu S, Yu F, Shi J, McGuire PM and
Wang R: Cytotoxic activity of an octadecenoic acid extract from
Euphorbia kansui (Euphorbiaceae) on human tumour cell
strains. J Pharm Pharmacol. 60:253–259. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Sangpairoj K, Settacomkul R, Siangcham T,
Meemon K, Niamnont N, Sornkaew N, Tamtin M, Sobhon P and
Vivithanaporn P: Hexadecanoic acid-enriched extract of Halymenia
durvillei induces apoptotic and autophagic death of human
triple-negative breast cancer cells by upregulating ER stress.
Asian Pac J Trop Biomed. 12:132–140. 2022.
|
|
22
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Häcker G: The morphology of apoptosis.
Cell Tissue Res. 301:5–17. 2000.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Pfeffer CM and Singh ATK: Apoptosis: A
target for anticancer therapy. Int J Mol Sci.
19(448)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Obeng E: Apoptosis (programmed cell death)
and its signals-A review. Braz J Biol. 81:1133–1143.
2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lossi L: The concept of intrinsic versus
extrinsic apoptosis. Biochem J. 479:357–384. 2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Olsson M and Zhivotovsky B: Caspases and
cancer. Cell Death Differ. 18:1441–1449. 2011.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Nagata S: Apoptotic DNA fragmentation. Exp
Cell Res. 256:12–18. 2000.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Cheng EH, Wei MC, Weiler S, Flavell RA,
Mak TW, Lindsten T and Korsmeyer SJ: BCL-2, BCL-XL sequester BH3
domain-only molecules preventing BAX-and BAK-mediated mitochondrial
apoptosis. Mol Cell. 8:705–711. 2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wei MC, Zong WX, Cheng EH, Lindsten T,
Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB and
Korsmeyer SJ: Proapoptotic BAX and BAK: A requisite gateway to
mitochondrial dysfunction and death. Science. 292:727–730.
2001.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Weinberg RA: How cancer arises. Sci Am.
275:62–70. 1996.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Eischen CM: Role of Mdm2 and Mdmx in DNA
repair. J Mol Cell Biol. 9:69–73. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chène P: Inhibiting the p53-MDM2
interaction: An important target for cancer therapy. Nat Rev
Cancer. 3:102–109. 2003.PubMed/NCBI View
Article : Google Scholar
|
|
36
|
Campbell KJ and Tait SWG: Targeting BCL-2
regulated apoptosis in cancer. Open Biol. 8(180002)2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Jain AK and Barton MC: p53: Emerging roles
in stem cells, development and beyond. Development.
145(dev158360)2018.PubMed/NCBI View Article : Google Scholar
|