Introduction
Nonalcoholic fatty liver disease (NAFLD) is defined
as a clinicopathological syndrome with the primary characteristic
being hepatocellular macrovesicular steatosis, where pathology and
other explicit factors give rise to hepatic injury, ranging from
simple steatosis to nonalcoholic steatohepatitis (NASH), which has
the potential to develop into cirrhosis, hepatocellular carcinoma
(HCC) and end-stage liver disease (1).
NAFLD is proven to be related to obesity, type 2 diabetes (T2DM),
insulin resistance (IR), hyperlipidemia and gender. The onset and
persistence of steatohepatitis results from the early predisposing
factors of accumulating visceral adiposity and free fatty acids
(FFAs). According to the ‘two hit hypothesis’ (2), the degree of obesity and presence of IR
being the first hit promote the accumulation of fatty acids in
liver, and increase the susceptibility of hepatic injury caused by
the second hit. Active factors of the ‘two hit hypothesis’, for
instance, inflammation, dysfunction of Kupffer cells, oxidative
stress, mitochondrial dysfunction and a regulative disorder of
adipocytokines, lead to a greater risk of serious liver disease.
The primary factor in causing NAFLD is the presence of IR that is
associated with overexpression of hypoadiponectinemia,
hyperleptinemia and cytokines, all of which can intensify IR and
promote the accumulation of fatty acids in liver. Excessive FFAs in
hepatocytes are said to induce the occurrence of oxidative stress,
and the release of inflammatory factors and adipogenesis-related
factors result in the formation of necroinflammation,
hepatofibrogenesis and cirrhosis.
Currently, NAFLD is gradually becoming more
prevalent and affects ~20–30% of the general population worldwide.
Most estimates indicate that 70% obese patients may have hepatic
steatosis, among which, the risk of developing NASH accounts for
20%, and patients with NASH that progresses to cirrhosis is
estimated to be ~20–25% (3). NASH is
now accepted as the major underlying cause of cryptogenic
cirrhosis, which suggests that genetic factors are involved in the
development of NAFLD. As of yet, the genetic mechanism of NAFLD is
not able to be comprehensively and thoroughly understood, but
research on genetic variation influencing IR, lipid metabolism and
oxidative stress are important issues of scientific interest to
improve patients' management. The aims of the present review is to
concentrate on the available knowledge of gene polymorphisms
implicated in the development of NAFLD induced by a disorder in
glucose metabolism and fatty acid metabolism, oxidative stress and
related cytokines.
Pathogeneisis
The molecular events lead to the occurrence of
intrahepatic lipid accumulation followed by increased FFAs in the
liver, reduced fatty acid oxidation, or elevated de novo
synthesis of triglycerides (TG), whereby the very low-density
lipoprotein (VLDL) synthetic rate is unable to keep up. FFAs derive
from dietary fatty acids and lipolysis takes place in the
peripheral, especially visceral, subcutaneous adipose tissue.
Hepatic glucose and FFA uptake occurs via an insulin-independent
manner. Insulin serves a crucial role in the pathogenesis of NAFLD
by upregulating colony-stimulating factor expression. Almost all
patients with NAFLD suffer from hepatic IR with a failure to
suppress lipolysis as a result from a deficiency of sensitivity to
insulin in adipose tissue. IR leads to lipid storage in hepatocytes
for two major routes: One is hyperlipidemia and the other is
hyperinsulinemia. The storage and release of FFAs from adipose
tissue is mediated by hormones and cytokines, such as estrogen,
cortisol, growth hormone, glucagon, insulin and insulin-like
growing factor, which augment levels of fatty acids through
altering energy metabolism in turn to enhance FFA uptake and
synthesis in liver, ultimately resulting in lipid accumulation. FFA
is an amphipathic molecule with high toxicity, and the
reinforcement of peroxidation engendered by unsaturated fatty acids
finally impairs hepatocytes. Lipid peroxidation free radicals and
end-products like malondialdehyde are able to bring about abnormal
fluidity and permeability of cell membrane, causing cell
dysfunction, apoptosis or death. Excess FFAs conduce to
hepatotoxicity in NAFLD/NASH, since FFA oxidation in hepatic
mitochondria leads to oxidative stress. Following this, elevated
levels of peroxides and free radicals, as a consequence of fatty
acid oxidation, causes oxidative damage, endoplasmic reticulum
stress and apoptosis (4). Once the
absorption of FFAs increases, this is followed by a compensatory
increase in mitochondrial β-oxidation, which will further enhance
the generation of reactive oxygen species (ROS) that can tightly
bind to membrane phospholipids to block the respiratory chain of
electron transfer in mitochondria (5).
Another location where ROS is generated is in microsomes. FFA is
the inductive agent of P450-2E1, which occupies an important
position in the cascade of ROS and lipid peroxidation generation
(6). In microsomes, FFAs are easily
oxidized and degraded into acyl-CoA. As the ligand combines with
some enzymes of the fatty acid oxidation system in the liver,
acyl-CoA controls gene-induction and promoting the synthesis of
unwinding protein, which could prevent hepatocyte apoptosis.
Patients carrying variant alleles of acyl-CoA dehydrogenases may
result in the progression of steatohepatitis, because of aberrant
fatty acid oxidation (7). In addition,
the following undoubtedly contribute to lipid storage within the
liver (5,8–10):
Insufficient synthesis and inhibition of apolipoproteins;
Inhibition of newly synthesized VLDL transportation from the
endoplasmic reticulum to the Golgi complex; Dysfunction of the
Golgi-endoplasmic reticulum-lysosome complex; Reduction of
saccharification or secretory vesicles of newly synthesized VLDL;
Interference with exocytosis of newly synthesized VLDL as a result
from malfunction of secretory vesicles migrating to hepatocyte
serosa; Chronic hunger, poor digestion and absorption, low protein
diet leading to the deficiency of ‘anti-fatty liver factors’
(including methionine, choline and phosphatidylcholine), bringing
about insufficient synthesis of apolipoproteins, in particular apoB
and VLDL. It has been demonstrated that lipid metabolism
disturbance is regulated insufficiently by vital transcription
factors that are indispensable for lipogenesis, such as the
activation of AMP-activated protein kinase (AMPK), Jun N-terminal
kinase (JNK), protein kinase C and nuclear factor (NF)-κB (11). Other adipokines, such as resistin and
leptin also serve as prospectively latent markers of NAFLD. These
events cooperatively promote the susceptibility to NAFLD and its
progression (Fig. 1).
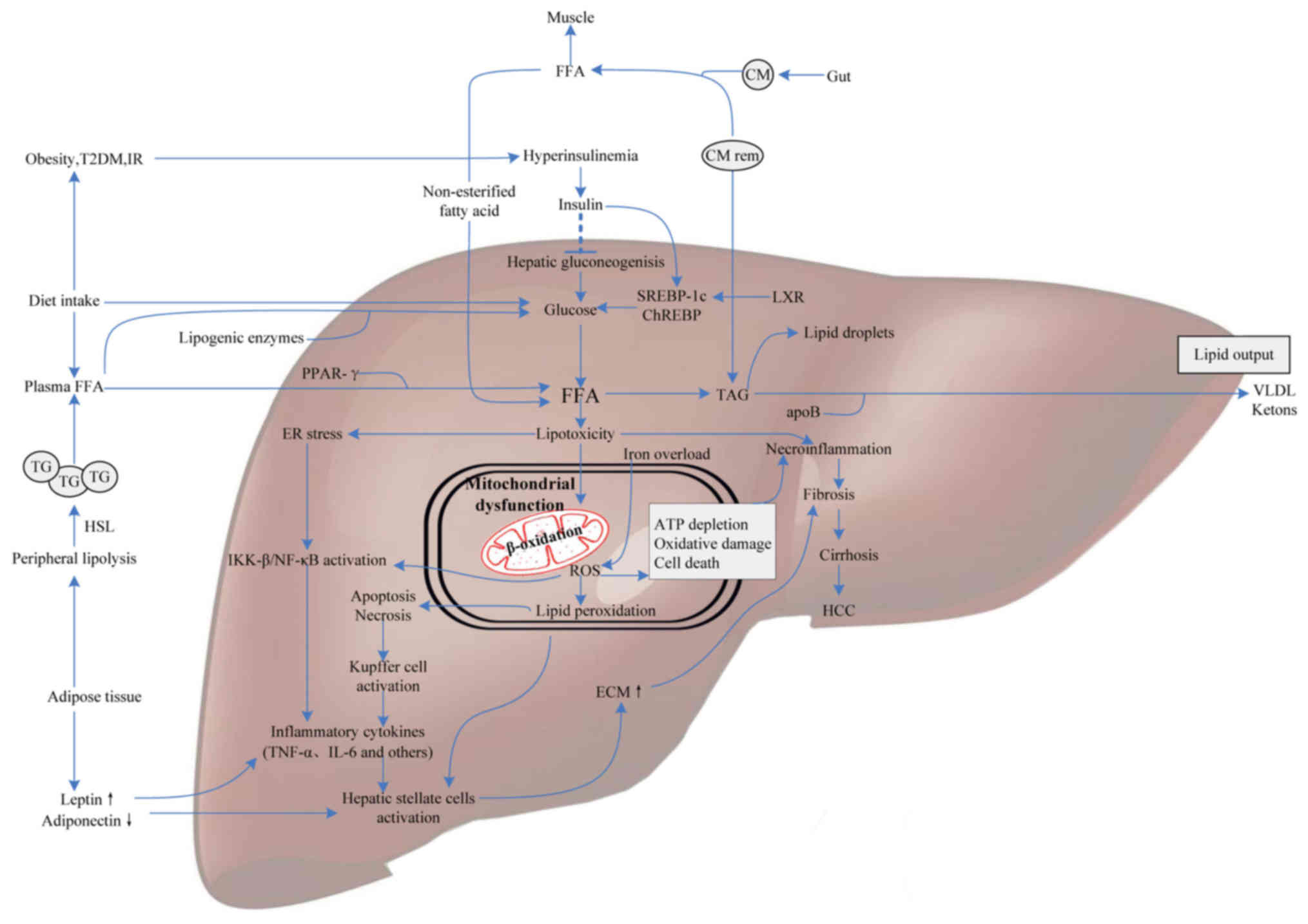 | Figure 1.Molecular mediators involved in NAFLD
pathogenesis. FFAs derive from dietary fatty acids and lipolysis in
the periphery, and then are delivered to the liver by CM via the
portal vein. Meanwhile, individuals with obesity, IR or T2DM
influence hepatic gluconeogenesis and transcriptional activity of
SREBP-1c and ChREBP by increasing the generation of glucose,
ultimately leading to intrahepatic FFA accumulation. Excess FFAs
present lipotoxicity with the ability to impair hepatocytes
triggering necroinflammation and mitochondrial dysfunction. Some
FFAs in the liver are converted into TAG and then exported out of
liver in the form of VLDL, others are oxidized in hepatic
mitochondria, generating a mass of ROS. Additionally, iron overload
generating hydroxyl radicals through the Fenton reaction also
contributes to oxidative stress. ROS can cause oxidative damage,
endoplasmic reticulum stress and apoptosis leading to
necroinflammation via IKK-β activation, NF-κB activation and
Kupffer cell activation pathways. Subsequently, the release of
inflammatory cytokines, like TNF-α and IL-6, which give rise to
hepatofibrogenesis, and finally progress to cirrhosis and HCC.
Therefore, molecular mediators involved in glucose metabolism,
fatty acid metabolism and oxidative stress may contribute to the
progression of NAFLD. NAFLD, nonalcoholic fatty liver disease;
FFAs, free fatty acids; IR, insulin resistance; CM, chylomicron;
T2DM, type 2 diabetes; SREBP-1c, sterol regulatory element binding
protein-1c; ChREBP, carbohydrate-responsive element-binding
protein; TAG, triacylglycerol; VLDL, very low-density lipoprotein;
ROS, reactive oxygen species; IKK-β, IkB kinase-β; NF-κB, nuclear
factor-κB; HCC, hepatocellular carcinoma; PPAR, peroxisome
proliferator-activated receptor; ApoB, apolipoprotein B; TG,
triglyceride; ECM, extracellular matrix; TNF-α, tumor necrosis
factor-α; LXR, liver X receptor; HSL, hormone-sensitive lipase. |
Gene polymorphisms in mediators of glucose
metabolism
IR and aberration of glucose metabolism are a
central part of the onset and development of NAFLD. The formation
of IR in the liver is caused by some factors that do the same to
the progression of NAFLD/NASH, and will be discussed below.
Insulin receptor substrate (IRS) and
ectoenzyme nucleotide pyrophosphatase phosphodiesterase 1
(ENPP1)
Among the genes concerned with regulating IR, IRS-1
and ENPP1 directly interact with insulin receptor (INSR) signaling.
Mechanically, insulin binds to the INSR with a high affinity for
hepatocytes, resulting in INSR phosphorylation. The activation of
IRS-1 and the downstream kinase AKT phosphorylates the
transcription factor FOXO1 in turn to decrease glucose production
and cell apoptosis. FOXO1 expression is observed to be increased
and resistant to the inhibition of hyperinsulinemia in patients
with NASH. ENPP1 is a membrane glycoprotein, and overexpression of
ENPP1 induces the occurrence of IR and the disturbance of glucose
metabolism (12). IR, as determined by
the Lys121Gln single nucleotide polymorphism (SNP), is seen to
function through different mechanisms, for instance, the alteration
of appetite controlled by hypothalamus, and the reduced levels of
high-density lipoprotein (HDL) (13).
The IRS protein functions as a regulator in the insulin-like growth
factor-1-activated signaling. The Gly972Arg SNP, as previously
reported, is also associated with IR, which brings about more
serious liver fibrosis and progression of NAFLD (14). Presumably, NAFLD patients carrying both
ENPP1 and IRS-1 SNPs possess higher possibility of fibrogenesis
than those with unitary ENPP1 or IRS-1 SNP (13,14).
Thereby, the coincident presence of ENPP1 and IRS-1 SNPs in NAFLD
patients synergistically triggers hepatic injury.
Adiponectin
Adiponectin is a specific plasma protein derived
from brown adipose tissue that serves a crucial role in regulating
glucose homeostasis. In liver tissue, adiponectin plays a part in
reducing the generation of glucose and FFAs, the concentration of
which circulating in plasma negatively associates with TG, and is
downregulated in obese populations. In addition, adiponectin has
capable of exerting insulin sensitivity effects and acts as an
anti-inflammatory molecule. However, inflammation is able to impede
the release of adiponectin, thus adipose tissue inflammation is
taken for one of the prime mechanisms involved in decreasing plasma
levels of adiponectin in obesity (15). Adiponectin predominantly activates AMPK
and peroxisome proliferator-activated receptor (PPAR)-γ to improve
insulin sensitivity, reduce the synthesis of fatty acids and
enhance fatty acids oxidation for combating steatosis. Therefore,
the levels of adiponectin attribute to the development of
NAFLD/NASH, and the adiponectin gene has the ability of modulating
individual differences. In a Japanese population, adiponectin gene
variants, +45GT and +276GT locus of alleles were correlated to the
risk of NAFLD. The frequency of the +45GG genotype was
significantly more pervasive among NAFLD females, whose
hepatofibrosis were more serious than that in control group
(16). However, no association was
observed between NAFLD and the genetic polymorphism of adiponectin
at positions 11391, −11377, +45 and +276 in a Chinese population.
Nonetheless, subjects carrying −11377 and +45 SNPs were associated
with higher TG levels, body mass index (BMI) and lower plasma
adiponectin levels (17). Another
study suggested that +45TG and +276GT genetic variations were
closely related to the predisposition to NAFLD in Chinese and
Indian populations (18). The
adiponectin rs266729 (−11377G/C) polymorphism may be a candidate
gene associated with the susceptibility to NAFLD in a southeast
Iranian population, but has not yet been confirmed in other
ethnicities (19). Collectively,
adiponectin gene polymorphisms have an intimate association with
BMI, blood lipid levels, T2DM, obesity and hyperlipemia, which add
to the risk of NAFLD.
Resistin
Resistin is a hormone secreted by fat cells, and is
regarded as the link of adiposity to diabetes. The major target of
resistin is the liver in vivo, and reduced or increased
resistin levels respectively gives rise to improvement or worsening
of hepatic IR through interfering with glycometabolism (20). The reduction of hepatic glucose in the
absence of resistin is integral to the activation of the AMPK
pathway. It was demonstrated that hepatic steatosis and VLDL
secretion were decreased in resistin-deficient mice treated with
high-fat diet, which suggested a function of resistin on steatosis
induced (21). Resistin suppresses the
effect of insulin on glucose absorption and glucose tolerance
impairment, which was regulated by CCAAT/enhancer binding protein α
and PPAR-γ in the phosphatidyl inositol 3-kinase and
mitogen-activated protein kinase pathways (22). Therefore, SNPs may influence resistin
gene expression. For adult women, the −420C/G SNP of the resistin
gene was related with abdominal obesity and metabolic risk factors;
this polymorphism in the promoter region captures only a small part
of the total genetic contribution to the variation of circulating
resistin (23). Another genotype of
the resistin +229AA polymorphism was confirmed to be associated
with higher risk of obesity and NAFLD incidence in T2DM patients
(24). This evidence is consistent
with the research in obese Egyptian patients (25). However, how resistin gene polymorphisms
influence the signaling pathway mechanism of NAFLD has not been
figured out clearly yet. Obviously, resistin as a proinflammatory
factor is involved in the inflammatory cascade reaction and
triggering ‘the second hit’.
Leptin and leptin receptor
(LepRb)
Leptin is protein-related product encoded by an
obesity gene and is involved in the modulation of body fat, insulin
signaling and the immune system. Leptin acts through leptin
receptors (LepRb), a member of the class-1 cytokine receptor
family, originally demonstrated in hypothalamic neurons, to control
satiety and maintain the body's constant energy balance. Leptin is
a contributing factor of elevating intracellular fatty acids via
promoting IR and altering insulin signaling in hepatocytes for
hepatic steatosis. At a later stage, hepatic steatosis caused by
leptin acting on proinflammatory response amplification evolves
into steatohepatitis. However, both roles of leptin and LepRb in
NAFLD are not completely clear. The LepRb gene polymorphism in
humans has been reported to be in association with IR, T2DM,
obesity and lipid metabolism, as well as the distribution of local
body mass. The LepRb SNP (rs6700896) increased the risk for NAFLD
as well as T2DM in an Egyptian population, and this enhanced leptin
level was accompanied by a decrease of LepRb concentration
(26). Furthermore, by mediating lipid
metabolism and insulin sensitivity, the LepRb G2057A SNP is
conducive to the onset of NAFLD (27).
However, the LepRb Arg233Gln polymorphism has been indicated in
relation to lower cholesterol, low-density lipoprotein (LDL) levels
and fibrosis score in NAFLD (28).
Patients carrying the SNPs of LepRb (rs1137100) with patatin-like
phospholipase domain containing 3 (rs738409) have ~three-fold risk
of suffering from NAFLD than those carrying either SNP, both genes
are correlatively upregulated in the absence of excess lipids
(28). Leptin inhibits steatosis via
downregulating stearoyl-CoA desaturase-1 and sterol regulatory
element binding protein (SREBP) 1c. Gene polymorphisms that code
leptin or regulate leptin secretion and tissue sensitivity proteins
may be potential genes in the development of NAFLD.
Gene polymorphisms in mediators of lipid
metabolism
The increased release of FFAs is closely relevant to
the onset and development of NAFLD. Lipotoxicity have an impact on
insulin signaling cascade, and then results in the accumulation of
lipids, ultimately leading up to the unbalance of gene-mediated
lipid metabolism.
Uncoupling protein 3 (UPC3)
UPC3 is a family of mitochondrial transporters known
for uncoupling oxidative phosphorylation via proton leakage from
the inner mitochondrial membrane, with primarily selective
expression in skeletal muscle, as well as brown adipose tissue, and
is implicated in modulating thermoregulation and energy metabolism
(29). Thereby, the expression of UPC3
mRNA undeniably has a bearing on an increase in metabolic rate and
a lower BMI. A number of polymorphisms have been identified in the
UPC3 gene. Particularly, the −55C/T polymorphism is responsible for
UPC3 mRNA levels, T2DM and weight gain (29). In obese Indonesian children, subjects
with T/T genotypes of UPC3 were demonstrated to present a lower
total energy expenditure than those with other genotypes (30). The level of serum LDL-cholesterol at
baseline was substantiated in relation to the rs3781907, rs1726745,
rs11235972 and rs1800849 variants of the UCP3 gene. Xu et al
(31) demonstrated that the
rs11235972GG genotype appears at a higher frequency in NAFLD, but
no increased risk associating the rs1800849 variant with the
development of NAFLD was confirmed. However, this remains
controversial with respect to the impact of these polymorphisms on
NAFLD susceptibility. Further additional larger studies should also
be conducted concerned with allowing stratification for ethnicity
and gene-gene interactions in order to distinctly elucidate the
possible roles of UCP3 polymorphisms in NAFLD.
Microsomal triglyceride transfer
protein (MTTP)
MTTP, as a pivotal enzyme involved in the
incorporation of TG into VLDL for lipid export out of the liver, is
localized to the endoplasmic reticulum of hepatocytes and
enterocytes. The human MTTP gene is located on chromosome 4q24 and
made up of 18 exons and 17 introns (32). Low hepatic expression of MTTP induced
by MTTP gene polymorphisms may be related to the pathogenesis of
NAFLD. Functional polymorphisms of the MTTP gene (promoter −493G/T,
−164 T/C and Ile128Thr) are involved in lower LDL levels and are
protected against other traits of metabolic syndrome, some studies
indicated (33,34). Among these, lower transcription of MTTP
was exhibited in a common SNP at position −493G/T, which
predisposed to NASH by modulating lipoprotein metabolism (35). The MTTP-493G>T resulting from a
G>T substitution in the intron region of NM_000253.2, which may
reduce the expression of MTTP and lead to an inadequate formation
of VLDL and chylomicron, resulting in the dysregulation of hepatic
lipid metabolism. In a study involving women treated with a western
type diet, higher fasting levels for plasma cholesterol (but not
synthesis) and high-absorption status were demonstrated in TT
homozygote women compared with G carriers (36). The presence of the G allele of MTTP
−493G/T has an association with lower hepatic MTTP expression,
which protects against steatosis in chronic hepatitis (37). Now, it is universally acknowledged that
the MTTP −493G>T polymorphism is correlated with the risk of
NAFLD. Thus, the MTTP −493G>T polymorphism in the future may be
used as a valuable and practical biomarker for early diagnosis of
NAFLD.
PPAR-α and PPAR-γ signaling
PPAR-α belongs to the nuclear hormone receptor
superfamily and acts as a primary regulatory role on lipid
metabolism. PPAR-α is able to activate fatty acid oxidation and
lipid hydrolysis, as well normalize glucose and insulin levels to
prevent hepatic lipid accumulation (38). Downregulation of PPAR-α is involved in
NASH pathogenesis by lessening FFA catabolism (39). PPAR-α-agonist fibrates and fenofibrates
are used to improve dyslipidemia and insulin sensitivity in
patients. The state of PPAR-α deficiency is involved in the
steatohepatitis pathogenesis. Several studies have demonstrated a
correlation between the Val227Ala of PPAR-α and low serum lipids
(40,41). It may have a protective role against
the development of obesity and be implicated in the pathogenesis of
NAFLD. The carriers of the Leu162Val SNP, which develops IR by
attenuating oxidative stress, had a association with decreased
hepatic lipolysis, dyslipidemia and T2DM, but not with liver damage
in NAFLD (41).
PPAR-γ, as another transcription factor, also
belongs to the nuclear hormone receptor superfamily, and is
primarily known to mediate adipocyte differentiation. A number of
genetic variants at several nucleotide loci of the PPAR-γ gene
bring about conformational changes in protein structures and gene
location. For disparate ethnic groups, a missense Pro12Ala
substation in the PPARγ2 gene (rs1801282) was demonstrated to have
a bearing on higher NAFLD risk, but was neither associated with
liver damage, nor insulin sensitivity (42). The decreasing DNA-binding affinity
caused by rs1801282 variant weakens transcriptional activation that
reduces PPAR-γ activity and activates lipogenic enzymes, resulting
in the exacerbation of disease (38,42).
Nevertheless, the association between rs1801282 variants and NAFLD
risk was thrown into doubt in a meta-analysis study (43). In a middle-aged and older Chinese
population, smokers with the C/C genotype of the PPARγ2 gene
polymorphism synergistically demonstrated a 3.75-fold higher risk
of NAFLD than non-smokers with the C/G genotype by aggravating
oxidative stress (44). Another
evidence indicated that a higher susceptibility to NAFLD was
associated with G/G genotype at rs10865710 loci, whereas no
influence of rs7649970 (45). Gawrieh
et al (46) first provided the
evidence that populations were at risk-reduction for NAFLD with a
haplotype containing both minor alleles of Pro12Ala and C1431T that
histological features manifested as lobular inflammation and
fibrosis, which were associated with NASH. It may be a promising
for subjects with NASH carrying this haplotype for treating with
PPARG agonists.
Notwithstanding genetic variants of PPARα/β may
influence disease progression of these studies make
recommendations, the implications of these SNPs on the risk of
NAFLD remain ill-defined, and further elucidation is required for
the functional role of these two PPAR genes on mechanism of NAFLD
pathogenesis.
SREBPs and SREBP cleavage activating
protein (SCAP)
SREBP serves a modulatory role in lipid metabolism.
Three informs of SREBP has been identified that contain SREBP-1a,
SREBP-1c and SREBP-2 in the human SREBP family, which are
correlated with lipogenesis, cholesterol homeostasis and adipocyte
development (47). SREBP-1a is capable
of activating the genes relating to fatty acid and cholesterol
synthesis. Compared with SREBP-1a, the SREBP-1c is a weaker
transcriptional activator, the expression of SREBP-1c is negatively
related to tumor necrosis factor (TNF)-α and IL-1β secretion that
is involved in liver fibrosis progression. In response to oxidative
stress, the activation of AMPK pathway inhibits de novo
lipogenesis via restraining the transcriptional activity of
SREBP-1c (48). Evidently, the
development of NASH may be associatively dependent on the
downregulation of SREBP-1c. Both SREBP-1a and SREBP-1c originate
from a single gene referred to as SREBF-1. Other than SREBF-1
acting on fatty acid synthesis, TG synthesis and phospholipid
synthesis, the activation of the SREBP-2 gene directly functions on
maintaining cholesterol homeostasis. Recently, a potential
biomarker for predicting the development of NAFLD in a Han Chinese
population was recommended by determining the genotypes of the
SREBP-2 rs2228314 G>C polymorphism, the variant gene was
indicated to carry a risk of NAFLD (49). SCAP is implicated in delivering SREBPs
from the endoplasmic reticulum to the Golgi complex. The activation
of SREBPs is subsequently translocated into the nucleus to
stimulate the expression of target genes for synthesis. In
liver-specific SCAP knockout mice, the synthesis rate of fatty
acids in liver was decreased by 70–80% as a result of the
significant reduction of expression levels of SREBP-1, SREBP-2 and
SREBP target genes. Interestingly, the SCAP rs2101247 A allele (AA
and GA genotypes) suffered from the risk of NAFLD may decrease,
which has been reported in a female metabolic syndrome population
(50), but it is unclear how, and the
potential mechanisms, for how the gender-specific distribution in
genetic susceptibility to NAFLD development are caused by
particular genotypes.
Gene polymorphisms in mediators of oxidative
stress
The occurrence of oxidative stress induced by
hepatonecrosis, inflammation and fibrosis, may be a factor of ‘the
second hit hypothesis’ in the onset and development of NAFLD.
Excessive oxidative stress derives from mitochondrial dysfunction
and iron overload. The unceasingly increased ROS, and the product
of oxidative stress, results in the rapid depletion of ATP, DNA
injury, instability of protein and release of inflammatory factors,
sequentially break cellular completeness of structure and function,
which promotes the development of NAFLD (6,10).
Manganese superoxide dismutase
(MnSOD)
MnSOD is nuclear encoded mitochondrial protein for
scavenging ROS. The MnSOD activity protects against ROS by
detoxifying superoxides into oxygen and hydrogen peroxide, the
impairment of which contributes to the higher oxidative stress for
NAFLD. In a Japanese population with NASH, a common variant of the
MnSOD gene 1183T/T results in an amino acid substitution in the
signaling sequence targeting the enzyme to the mitochondria, which
weakens the capability of MnSOD through mitochondria, leading to
the reduction of MnSOD and augmentation of oxygen radicals in
mitochondria, ultimately resulting in hepatocellular damage
(51). Another MnSOD polymorphism
(C47T, rs4880) has been investigated as a possible susceptible
factor in NASH and fibrosis, due to protein-import diminished into
mitochondria (52). More
interestingly, the concentration of MnSOD reduced in males with
NAFLD, instead of in females, possibly because due to the differing
sex hormone levels in different sexes (53). Thereby, the low level of MoSOD in males
makes them more susceptible to increase oxidative stress and
disease progression. In addition, the presence of the
Ala16Vla-MnSOD allele with the G-463A myeloperoxidase variant may
be attribute to an excess of hepatic iron accumulation and HCC
development, along with the increase of ROS-related oxidative
stress and DNA damage (54).
Cytochrome P4502E1 (CYP4502E1)
CYP4502E1 is an endoplasmic mono-oxygenase and it
serves a role in NAFLD. The elicitors such as ethanol, diet and
endogenously produced fatty acids, affect CYP4502E1 activation. As
a result of incomplete transfer of electrons to molecular oxygen,
CYP2450E1 releases superabundant ROS generation that leads to
raised oxidative stress in the pathophysiology of NAFLD. With
excessive CYP4502E1 activation in the perivenular hepatocytes, the
area of hepatocyte injury was the greatest (55). There are six kinds of restriction
fragment length polymorphism in CYP2450E1, and two point mutations
of PstI/RsaI located in 5′-flanking region affect the expression of
CYP2450E1 at the level of transcription. It was indicated that the
variant of c2 allele makes the CYP2450E1 level increase, which
functions by blocking tyrosine phosphorylation of IRS-1 and IRS-2,
ultimately resulting in IR and an increased risk of HCC (56). The CYP4502E1*5B (RsaI, −1053C>T)
variant is relevant to the improvement of its transcription and
enzyme activity, which increase the generation of ROS in alcoholic
cirrhotic patients, but not in NAFDL patients (57). Consequently, larger cohort studies
should be explored in future research to verify and evaluate the
effect of the CYP2E1 Pst I/Rsa I polymorphism-associated NAFLD
risk.
Iron overload and hemochromatosis
(HFE)
Among the candidate factors of oxidative stress,
iron is a notable pro-oxidant and acts through the Fenton reaction
to generate hydroxyl radicals (58).
Excessive hepatic iron accumulation causes oxidative stress and
promotes chronic liver damage in patients with genetic
hemochromatosis associated with HFE variants, especially C282Y
(rs1800562) and H63D (rs1799945). In mouse, iron-induced
inflammatory activation may contribute to the necroinflammation,
while the iron-mediated downregulation M2 pathway in macrophages
has an impact on fibrogenesis in NASH (58), which supports for a mechanistic role
for iron in NASH. HFE is characterized by the enhanced absorption
of dietary iron that results in progressive deposition of metal in
the liver. The HFE-knockout mice presented a deficiency of
hepatic-intestinal iron and lipid signaling, which enabled them to
be subjected to an accelerated progression of injury to fibrosis
with diet-induced hepatic lipotoxicity. The mechanism is relevant
to reduced hepcidin release leading to elevated iron absorption and
deposition, which promotes hepatic IR by disability of glucose
clearance and the facilitation of fibrogenesis by ROS generation of
hepatic stellate cells (HSCs) (59).
In general, HFE variants are common in white populations: The
frequency of the C282Y homozygote, H63D homozygote, H63D/wild-type
heterozygote in white populations respectively accounts for 0.44,
2.4 and 24%, while in Asians, this was ~0, 0–1 and 2.6–11%,
respectively (60). In American
patients with NAFLD, carriers of the C282Y variant, instead of H63D
HFE variant, were associated with hepatocellular iron deposition
and lower serum ALT and AST levels than wt/wt subjects, implying
that NASH pathogenesis may be independent of mild hepatocellular
iron deposition (61). However, the
iron deposition, rather than the HFE mutation, is the key factor in
the development of NAFLD, NASH and HCC (62). Also, β-globin variants are associated
with both hepatic iron and fibrosis, and arising rates of iron
accumulation are comparatively common in subjects with C282Y HFE
hemochromatosis and the beta thalassemia trait. The presence of
β-globin variants seems to be related to more severe liver disease.
Therefore, routine blood analysis may be conducive to identify the
risk of NASH progression in the carriers of the beta thalassemia
trait.
Gene polymorphisms in mediators of liver
fibrosis
There are countless factors associated with liver
fibrosis, including the activation of HSCs and the synthesis and
degradation of collagen. Fibrosis is a dynamic process of
continuous extracellular matrix (ECM) remodeling. The genes
encoding hepatofibrotic and fibrinolytic proteins are candidates
for fibrotic progression of NAFLD.
The leakage of endotoxins from the gut, bacterial
overgrowth and endotoxemia has been identified as a possible
stimulus of NASH. Toll-like receptor 4 (TLR4), a lipopolysaccharide
receptor, interacting with endotoxins, which leads to the release
of a mass of proinflammatory mediators that induce hepatic injury
and fibrogenesis (63,64). The importance of TLR4 signaling with
Kupffer cells in the pathogenesis of steatohepatitis was verified
in mice (65). TLR4 in Kupffer cells
serve a crucial role in the genesis of NASH via ROS-dependent
induction and X-box binding protein 1 activation. Dysregulation of
TLR4 signaling as a result of SNPs, alters the ligand binding and
balance between pro- and anti-inflammatory cytokines. It was
reported that the rs4986790 (D299G) and rs4986791 (T399I) were
related to IR, metabolic syndrome and T2DM (66). Nevertheless, a meta-analysis of 15,059
subjects drew a conclusion that Asp299Gly and Thr399Ile
polymorphisms of the TLR4 gene makes no difference with increased
risk of T2DM (67). In a small portion
of the general population, Kiziltas et al (68) first revealed in humans that TLR4
signaling is pivotal for the pathogenesis of NASH, and the TLR4
heterozygous gene mutation (Asp299Gly) may have a preventive role
against the genesis of NAFLD, which strongly suggests that TLR4
genotypes will be used as probable biomarkers in future clinical
trials for developing antagonists and agonists of TLR4 to better
manage patients with NAFLD.
Transforming growth factor (TGF)-β1 is another
potent inducer of hepatic fibrogenesis. TGF-β1 mediates hepatic
fibrosis via activating HSCs with the generation of ECM proteins.
TGF-β1 also indirectly act through increasing connective tissue
growth factors leading to liver fibrogenesis (69). In the human TGF-β1 locus, several SNPs
have been described previously. Two commonly studied polymorphisms
of the TGF-β1 gene are C-509T (rs1800469) and T-869C (rs1800470).
The level of TGF-β1 expression resulted from allelic variants are
significantly different (70). In the
Egyptian population, the serum TGF-β1 level in hepatocirrhosis
patients with the TGF-β1 −509TT genotype were remarkably increased
compared with those with CT/CC genotypes, and the −509 T allele
carriers were seven-fold more prone to develop liver cirrhosis than
−509 C allele carriers (71), which is
in agreement with the study in Italian populations (72). Additionally, the frequency of T allele
is remarkably high in HCV patients, more frequently leading to
progression of fibrosis (73).
However, with respect to the T-869C polymorphism associated with
HCC, there are some controversial results. The reasons for this
large difference in risk in different studies is unknown.
Gene polymorphisms in mediators of related
cytokines
TNF-α
TNF-α is a cytokine induced by macrophages and
others cells (such as adipocytes and hepatocytes) with
comprehensive functionality in metabolic regulation, inflammation
and tumor apoptosis. Now, enhanced TNF-α expression has been
demonstrated in NASH/NAFLD, and is therefore regarded as a risk
biomarker of disease progression. TNF-α blocks IRS signaling and
induces IR by triggering IkB kinase-β (IKK-β), the upstream
activator of NF-κB, and other critical intracellular kinases, such
as JNK activation. In animal models of genetic and diet-induced
obesity, an improvement of NASH and IR were attained by various
strategies targeting blockade of TNF-α activity. The TNF-α −238
promoter polymorphism was identified a higher prevalence in
patients with NAFLD, and the −863 variant was in relation to the
decrease of TNF-α concentration (74).
Furthermore, lower LDL-cholesterol, BMI and susceptibility of NASH
in Mexican and Chinese cohorts were observed in association with
the −238 polymorphism. However, frequencies of variants −1031C and
−863A were more prevalent in subjects with NASH in a different
study (75). A meta-analysis
evaluating polymorphisms in TNF-α confirmed the relationship of the
−238 polymorphism with NAFLD, whereas the −308 polymorphism was not
determined as a susceptible factor (76). Additionally, TNF-α G-308A, G-238A and
C-863A polymorphisms are also related to HCC in Asians populations
(77). It is still unclear whether,
and certainly which, TNF-α polymorphism is relevant to the
development of NASH. Further studies are in progress to focus on
replicating results larger cohorts and different populations.
Interleukin (IL)-6
IL-6 is a cytokine involved controlling the balance
between proinflammatory and anti-inflammatory pathways. The IL-6
gene is located on chromosome 7p21, and the −174 polymorphism of
the IL-6 gene promoter is described to be in linkage disequilibrium
with other commonly studied makers of inflammation. The G-174C of
IL-6 gene polymorphism has been reported to relate to its
transcription rate and the development of T2DM. In children with
obesity, BMI, middle upper arm circumference, tricipital skin-fold
thickness and serum albumin levels were demonstrated to have a
correlation with the CC allele carriers of IL-6 gene (78). Moreover, the IL-6 −174C variant is more
frequent in NASH individuals than that in cohorts with NAFLD.
However, its possible role in NAFLD has not been fully studied.
Currently, the presence of the GG genotype was observed to be in
high frequency in HCV-related chronic hepatitis, liver cirrhosis
and HCC (79). The variant predisposes
to produce higher levels of IL-6 that contributes to the
progression of HCC. In addition, the IL-6 receptor SNP (rs6684439)
in Chinese patients with HBV is related to a lower risk of HCC
(80), because of decreased levels of
circulating sIL-6R.
Conclusions
NAFLD is the most common chronic liver disease that
influences mortality. It is becoming a public health problem,
especially the common progressive types of NAFLD, which ultimately
progress to HCC. Patients with NAFLD frequently suffer from
metabolic syndrome (81). The
occurrence of NAFLD caused by many factors involving an
overwhelmingly complex pathophysiological process and the joint
action of multiple mechanisms. Ethnic background, gender, age and
geographic location influence the results varied widely. The
function of gene polymorphisms, which implicate IR, fatty acid
metabolism, oxidative stress and hepatofibrogenesis, are reflected
in every part of the pathogenesis of NAFLD. Therefore, obesity,
impairment of glucose homeostasis and genetic influences are deemed
as important pathogenic contributors in the advancing of NAFLD and
NASH. The molecular mechanisms of oxidative stress, lipotoxicity
and abnormal adipocytokine release caused by gene polymorphisms in
NAFLD/NASH, have been gradually elucidated. It is of great
significance for developing effective strategies in the future.
However, due to the complexity and limitation of gene polymorphism
research on NAFLD, most studies meet with the absence of
statistical power, replication in various cohorts, well-matched
samples and the presence of heterogeneities in disease stages,
thus, researchers still do not completely understand the
physiopathological mechanism of NAFLD caused by gene polymorphisms.
This makes the research on NAFLD gene polymorphisms be filled with
unknown and challenges. With the technological development and
applications of disease gene location, the authors believe that a
further breakthrough through studying NAFLD gene polymorphisms will
be achieved to help identify an effective treatment for NAFLD.
Glossary
Key words
Abbreviations:
|
NAFLD
|
nonalcoholic fatty liver disease
|
|
NASH
|
nonalcoholic steatohepatitis
|
|
HCC
|
hepatocellular carcinoma
|
|
T2DM
|
type 2 diabetes
|
|
IR
|
insulin resistance
|
|
FFAs
|
free fatty acids
|
|
TNF-α
|
tumor necrosis factor-α
|
|
TG
|
triglyceride
|
|
VLDL
|
very low-density lipoprotein
|
|
ROS
|
reactive oxygen species
|
|
AMPK
|
AMP protein kinase
|
|
JNK
|
Jun N-terminal kinase
|
|
NF-κB
|
nuclear factor-κB
|
|
IRS
|
insulin receptor substrate
|
|
ENPP1
|
ectoenzyme nucleotide pyrophosphate
phosphodiesterase 1
|
|
SNP
|
nucleotide polymorphism
|
|
HDL
|
high-density lipoprotein
|
|
SCD-1
|
stearoyl-CoA desaturase-1
|
|
IGF-1
|
insulin-like growth factor-1
|
|
PPAR-α/γ
|
peroxisome proliferator-activated
receptor-α/γ
|
|
BMI
|
body mass index
|
|
ALT
|
alanine transaminase
|
|
AST
|
aspartate aminotransferase
|
|
PI3-K
|
phosphatidyl inositol 3-kinase
|
|
MAPK
|
mitogen-activated protein kinase
|
|
LepRb
|
leptin receptor
|
|
LDL
|
low-density lipoprotein
|
|
SREBP
|
sterol regulatory element binding
protein
|
|
UPC3
|
uncoupling protein 3
|
|
MTTP
|
microsomal triglyceride transfer
protein
|
|
CM
|
chylomicron
|
|
SCAP
|
SREBP cleavage activating protein
|
|
MnSOD
|
manganese superoxide dismutase
|
|
CYP4502E1
|
cytochrome P4502E1
|
|
HFE
|
hemochromatosis
|
|
TLR4
|
Toll-like receptor 4
|
|
XBP-1
|
X-box binding protein 1
|
|
TGF-β1
|
transforming growth factor-β1
|
|
ECM
|
extracellular matrix
|
|
IKK-β
|
IkB kinase-β
|
|
IL-6
|
interleukin-6
|
References
|
1
|
Zhang X, Harmsen WS, Mettler TA, Kim WR,
Roberts RO, Therneau TM, Roberts LR and Chaiteerakij R:
Continuation of metformin use after a diagnosis of cirrhosis
significantly improves survival of patients with diabetes.
Hepatology. 60:2008–2016. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Day CP and James OF: Steatohepatitis: A
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Anstee QM, Daly AK and Day CP: Genetics of
alcoholic and nonalcoholic fatty liver disease. Semin Liver Dis.
31:128–146. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lake AD, Novak P, Hardwick RN,
Flores-Keown B, Zhao F, Klimecki WT and Cherrington NJ: The
adaptive endoplasmic reticulum stress response to lipotoxicity in
progressive human nonalcoholic fatty liver disease. Toxicol Sci.
137:26–35. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Anderson N and Borlak J: Molecular
mechanisms and therapeutic targets in steatosis and
steatohepatitis. Pharmacol Rev. 60:311–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Naik A, Belič A, Zanger UM and Rozman D:
Molecular interactions between NAFLD and xenobiotic metabolism.
Front Genet. 4:22013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang D, Liu ZX, Choi CS, Tian L, Kibbey
R, Dong J, Cline GW, Wood PA and Shulman GI: Mitochondrial
dysfunction due to long-chain Acyl-CoA dehydrogenase deficiency
causes hepatic steatosis and hepatic insulin resistance. Proc Natl
Acad Sci USA. 104:17075–17080. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Duvnjak M, Lerotić I, Barsić N, Tomasić V,
Jukić L Virović and Velagić V: Pathogenesis and management issues
for non-alcoholic fatty liver disease. World J Gastroenterol.
13:4539–4550. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bradbury MW and Berk PD: Lipid metabolism
in hepatic steatosis. Clin Liver Dis. 8639–671. (xi)2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
de Almeida IT, Cortez-Pinto H, Fidalgo G,
Rodrigues D and Camilo ME: Plasma total and free fatty acids
composition in human non-alcoholic steatohepatitis. Clin Nutr.
21:219–223. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Malaguarnera L, Di Rosa M, Zambito AM,
dell'Ombra N, Nicoletti F and Malaguarnera M: Chitotriosidase gene
expression in Kupffer cells from patients with non-alcoholic fatty
liver disease. Gut. 55:1313–1320. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kadowaki T, Yamauchi T, Kubota N, Hara K,
Ueki K and Tobe K: Adiponectin and adiponectin receptors in insulin
resistance, diabetes, and the metabolic syndrome. J Clin Invest.
116:1784–1792. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Müssig K, Heni M, Thamer C, Kantartzis K,
Machicao F, Stefan N, Fritsche A, Häring HU and Staiger H: The
ENPP1 K121Q polymorphism determines individual susceptibility to
the insulin-sensitising effect of lifestyle intervention.
Diabetologia. 53:504–509. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dongiovanni P, Valenti L, Rametta R, Daly
AK, Nobili V, Mozzi E, Leathart JB, Pietrobattista A, Burt AD,
Maggioni M, et al: Genetic variants regulating insulin receptor
signalling are associated with the severity of liver damage in
patients with non-alcoholic fatty liver disease. Gut. 59:267–273.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Parola M and Marra F: Adipokines and redox
signaling: Impact on fatty liver disease. Antioxid Redox Signal.
15:461–483. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tokushige K, Hashimoto E, Noto H, Yatsuji
S, Taniai M, Torii N and Shiratori K: Influence of adiponectin gene
polymorphisms in Japanese patients with non-alcoholic fatty liver
disease. J Gastroenterol. 44:976–982. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wong VW, Wong GL, Tsang SW, Hui AY, Chan
AW, Choi PC, So WY, Tse AM, Chan FK, Sung JJ, et al: Genetic
polymorphisms of adiponectin and tumor necrosis factor-alpha and
nonalcoholic fatty liver disease in Chinese people. J Gastroenterol
Hepatol. 23:914–921. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang BF, Wang Y, Ao R, Tong J and Wang BY:
AdipoQ T45 G and G276 T polymorphisms and susceptibility to
nonalcoholic fatty liver disease among Asian populations: A
meta-analysis and meta-regression. J Clin Lab Anal. 30:47–57. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hashemi M, Bojd H Hanafi, Nasab E
Eskandari, Bahari A, Hashemzehi NA, Shafieipour S, Narouie B,
Taheri M and Ghavami S: Association of adiponectin rs1501299 and
rs266729 gene polymorphisms with nonalcoholic fatty liver disease.
Hepat Mon. 13:e95272013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Muse ED, Obici S, Bhanot S, Monia BP,
McKay RA, Rajala MW, Scherer PE and Rossetti L: Role of resistin in
diet-induced hepatic insulin resistance. J Clin Invest.
114:232–239. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singhal NS, Patel RT, Qi Y, Lee YS and
Ahima RS: Loss of resistin ameliorates hyperlipidemia and hepatic
steatosis in leptin-deficient mice. Am J Physiol Endocrinol Metab.
295:E331–E338. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tomaru T, Steger DJ, Lefterova MI, Schupp
M and Lazar MA: Adipocyte-specific expression of murine resistin is
mediated by synergism between peroxisome proliferator-activated
receptor gamma and CCAAT/enhancer-binding proteins. J Biol Chem.
284:6116–6125. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kumar S, Gupta V, Srivastava N, Gupta V,
Mishra S, Mishra S, Shankar M Natu, Roy U, Chandra A, Negi MP, et
al: Resistin 420C/G gene polymorphism on circulating resistin,
metabolic risk factors and insulin resistance in adult women.
Immunol Lett. 162(2 Pt B): 287–291. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang LY, Jin YJ, Jin QS, Lin LY, Zhang DD
and Kong LL: Association between resistin +299A/A genotype and
nonalcoholic fatty liver disease in Chinese patients with type 2
diabetes mellitus. Gene. 529:340–344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
El-Shal AS, Pasha HF and Rashad NM:
Association of resistin gene polymorphisms with insulin resistance
in Egyptian obese patients. Gene. 515:233–238. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lu H, Sun J, Sun L, Shu X, Xu Y and Xie D:
Polymorphism of human leptin receptor gene is associated with type
2 diabetic patients complicated with non-alcoholic fatty liver
disease in China. J Gastroenterol Hepatol. 24:228–232. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Swellam M and Hamdy N: Association of
nonalcoholic fatty liver disease with a single nucleotide
polymorphism on the gene encoding leptin receptor. IUBMB Life.
64:180–186. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zain SM, Mohamed Z, Mahadeva S, Cheah PL,
Rampal S, Chin KF, Mahfudz AS, Basu RC, Tan HL and Mohamed R:
Impact of leptin receptor gene variants on risk of non-alcoholic
fatty liver disease and its interaction with adiponutrin gene. J
Gastroenterol Hepatol. 28:873–879. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brondani LA, Assmann TS, de Souza BM,
Bouças AP, Canani LH and Crispim D: Meta-analysis reveals the
association of common variants in the uncoupling protein (UCP) 1–3
genes with body mass index variability. PLoS One. 9:e964112014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mexitalia M, Yamauchi T, Utari A, Sjarif
DR, Subagio HW, Soemantri A and Ishida T: The role of uncoupling
protein 2 and 3 genes polymorphism and energy expenditure in obese
Indonesian children. J Pediatr Endocrinol Metab. 26:441–447. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xu YP, Liang L, Wang CL, Fu JF, Liu PN, Lv
LQ and Zhu YM: Association between UCP3 gene polymorphisms and
nonalcoholic fatty liver disease in Chinese children. World J
Gastroenterol. 19:5897–5903. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hashemi M, Hoseini H, Yaghmaei P,
Moazeni-Roodi A, Bahari A, Hashemzehi N and Shafieipour S:
Association of polymorphisms in glutamate-cysteine ligase catalytic
subunit and microsomal triglyceride transfer protein genes with
nonalcoholic fatty liver disease. DNA Cell Biol. 30:569–575. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian Y, Li H, Wang S, Yan J, Chen Z, Li Z,
Feng H, Zhou H and Ouyang D: Association of L-FABP T94A and MTP
I128T polymorphisms with hyperlipidemia in Chinese subjects.
Lipids. 50:275–282. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Peng XE, Wu YL, Lu QQ, Hu ZJ and Lin X:
MTTP polymorphisms and susceptibility to non-alcoholic fatty liver
disease in a Han Chinese population. Liver Int. 34:118–128. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gambino R, Cassader M, Pagano G, Durazzo M
and Musso G: Polymorphism in microsomal triglyceride transfer
protein: A link between liver disease and atherogenic postprandial
lipid profile in NASH? Hepatology. 45:1097–1107. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wolff E, Vergnes MF, Defoort C, Planells
R, Portugal H, Nicolay A and Lairon D: Cholesterol absorption
status and fasting plasma cholesterol are modulated by the
microsomal triacylglycerol transfer protein −493 G/T polymorphism
and the usual diet in women. Genes Nutr. 6:71–79. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Siqueira ER, Oliveira CP, Correa-Giannella
ML, Stefano JT, Cavaleiro AM, Fortes MA, Muniz MT, Silva FS,
Pereira LM and Carrilho FJ: MTP −493G/T gene polymorphism is
associated with steatosis in hepatitis C-infected patients. Braz J
Med Biol Res. 45:72–77. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Dongiovanni P, Rametta R, Fracanzani AL,
Benedan L, Borroni V, Maggioni P, Maggioni M, Fargion S and Valenti
L: Lack of association between peroxisome proliferator-activated
receptors alpha and gamma2 polymorphisms and progressive liver
damage in patients with non-alcoholic fatty liver disease: Α case
control study. BMC Gastroenterol. 10:1022010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Stienstra R, Saudale F, Duval C, Keshtkar
S, Groener JE, van Rooijen N, Staels B, Kersten S and Müller M:
Kupffer cells promote hepatic steatosis via
interleukin-1beta-dependent suppression of peroxisome
proliferator-activated receptor alpha activity. Hepatology.
51:511–522. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen S, Li Y, Li S and Yu C: A Val227Ala
substitution in the peroxisome proliferator activated receptor
alpha (PPAR alpha) gene associated with non-alcoholic fatty liver
disease and decreased waist circumference and waist-to-hip ratio. J
Gastroenterol Hepatol. 23:1415–1418. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Naito H, Yamanoshita O, Kamijima M, Katoh
T, Matsunaga T, Lee CH, Kim H, Aoyama T, Gonzalez FJ and Nakajima
T: Association of V227A PPARalpha polymorphism with altered serum
biochemistry and alcohol drinking in Japanese men. Pharmacogenet
Genomics. 16:569–577. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Domenici FA, Brochado MJ, Martinelli AL,
Zucoloto S, da Cunha SF and Vannucchi H: Peroxisome
proliferator-activated receptors alpha and gamma2 polymorphisms in
nonalcoholic fatty liver disease: A study in Brazilian patients.
Gene. 529:326–331. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sahebkar A: Does PPARγ2 gene Pro12Ala
polymorphism affect nonalcoholic fatty liver disease risk? Evidence
from a meta-analysis. DNA Cell Biol. 32:188–198. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yang Z, Wen J, Li Q, Tao X, Ye Z, He M,
Zhang W, Huang Y, Chen L, Ling C, et al: PPARG gene Pro12Ala
variant contributes to the development of non-alcoholic fatty liver
in middle-aged and older Chinese population. Mol Cell Endocrinol.
348:255–259. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cao CY, Li YY, Zhou YJ, Nie YQ and Wan YJ:
The C-681G polymorphism of the PPAR-γ gene is associated with
susceptibility to non-alcoholic fatty liver disease. Tohoku J Exp
Med. 227:253–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gawrieh S, Marion MC, Komorowski R,
Wallace J, Charlton M, Kissebah A, Langefeld CD and Olivier M:
Genetic variation in the peroxisome proliferator activated
receptor-gamma gene is associated with histologically advanced
NAFLD. Dig Dis Sci. 57:952–957. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bien CM and Espenshade PJ: Sterol
regulatory element binding proteins in fungi: Hypoxic transcription
factors linked to pathogenesis. Eukaryot Cell. 9:352–359. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nagaya T, Tanaka N, Suzuki T, Sano K,
Horiuchi A, Komatsu M, Nakajima T, Nishizawa T, Joshita S, Umemura
T, et al: Down-regulation of SREBP-1c is associated with the
development of burned-out NASH. J Hepatol. 53:724–731. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wang Y, Tong J, Chang B, Wang BF, Zhang D
and Wang BY: Relationship of SREBP-2 rs2228314 G>C polymorphism
with nonalcoholic fatty liver disease in a Han Chinese population.
Genet Test Mol Biomarkers. 18:653–657. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Sun S, Wang M, Song H, Wu T, Wei H, He S,
Ding Z and Ji G: SCAP gene polymorphisms decrease the risk of
nonalcoholic fatty liver disease in females with metabolic
syndrome. J Genet. 92:565–570. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Younossi ZM, Gorreta F, Ong JP, Schlauch
K, Del Giacco L, Elariny H, van Meter A, Younoszai A, Goodman Z,
Baranova A, et al: Hepatic gene expression in patients with
obesity-related non-alcoholic steatohepatitis. Liver Int.
25:760–771. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Al-Serri A, Anstee QM, Valenti L, Nobili
V, Leathart JB, Dongiovanni P, Patch J, Fracanzani A, Fargion S,
Day CP, et al: The SOD2 C47T polymorphism influences NAFLD fibrosis
severity: Evidence from case-control and intra-familial allele
association studies. J Hepatol. 56:448–454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Krautbauer S, Eisinger K, Lupke M,
Wanninger J, Ruemmele P, Hader Y, Weiss TS and Buechler C:
Manganese superoxide dismutase is reduced in the liver of male but
not female humans and rodents with non-alcoholic fatty liver
disease. Exp Mol Pathol. 95:330–335. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Nahon P, Sutton A, Rufat P, Ziol M,
Akouche H, Laguillier C, Charnaux N, Ganne-Carrié N, Grando-Lemaire
V, N'Kontchou G, et al: Myeloperoxidase and superoxide dismutase 2
polymorphisms comodulate the risk of hepatocellular carcinoma and
death in alcoholic cirrhosis. Hepatology. 50:1484–1493. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kathirvel E, Chen P, Morgan K, French SW
and Morgan TR: Oxidative stress and regulation of anti-oxidant
enzymes in cytochrome P4502E1 transgenic mouse model of
non-alcoholic fatty liver. J Gastroenterol Hepatol. 25:1136–1143.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Liu W, Tian F, Dai L and Chai Y:
Cytochrome P450 2E1 gene polymorphism and alcohol drinking on the
risk of hepatocellular carcinoma: A meta-analysis. Mol Biol Rep.
41:7645–7650. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Khan AJ, Ruwali M, Choudhuri G, Mathur N,
Husain Q and Parmar D: Polymorphism in cytochrome P450 2E1 and
interaction with other genetic risk factors and susceptibility to
alcoholic liver cirrhosis. Mutat Res. 664:55–63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Handa P, Morgan-Stevenson V, Maliken BD,
Nelson JE, Washington S, Westerman M, Yeh MM and Kowdley KV: Iron
overload results in hepatic oxidative stress, immune cell
activation, and hepatocellular ballooning injury, leading to
nonalcoholic steatohepatitis in genetically obese mice. Am J
Physiol Gastrointest Liver Physiol. 310:G117–G127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Nelson JE, Brunt EM and Kowdley KV:
Nonalcoholic Steatohepatitis Clinical Research Network: Lower serum
hepcidin and greater parenchymal iron in nonalcoholic fatty liver
disease patients with C282Y HFE mutations. Hepatology.
56:1730–1740. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lee SH, Jeong SH, Lee D, Lee JH, Hwang SH,
Cho YA, Park YS, Hwang JH, Kim JW, Kim N, et al: An epidemiologic
study on the incidence and significance of HFE mutations in a
Korean cohort with nonalcoholic fatty liver disease. J Clin
Gastroenterol. 44:e154–e161. 2010.PubMed/NCBI
|
|
61
|
Nelson JE, Yeh MM and Kowdley KV:
Relationship of C282Y HFE mutations, hepatic iron deposition and
histologic features in patients with nonalcoholic fatty liver
disease. Gastroenterology. 140:S938–S939. 2011.
|
|
62
|
Valenti L, Fracanzani AL, Bugianesi E,
Dongiovanni P, Galmozzi E, Vanni E, Canavesi E, Lattuada E, Roviaro
G, Marchesini G, et al: HFE genotype, parenchymal iron
accumulation, and liver fibrosis in patients with nonalcoholic
fatty liver disease. Gastroenterology. 138:905–912. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Rivera CA, Adegboyega P, van Rooijen N,
Tagalicud A, Allman M and Wallace M: Toll-like receptor-4 signaling
and Kupffer cells play pivotal roles in the pathogenesis of
non-alcoholic steatohepatitis. J Hepatol. 47:571–579. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Nagaya T, Tanaka N, Kimura T, Komatsu M
and Tanaka E: Enhanced expression of toll-like receptor 4 and MyD88
is associated with disease progression in human nonalcoholic fatty
liver disease. Gastroenterology. 140:S–978. 2011.
|
|
65
|
Thuy S, Ladurner R, Volynets V, Wagner S,
Strahl S, Königsrainer A, Maier KP, Bischoff SC and Bergheim I:
Nonalcoholic fatty liver disease in humans is associated with
increased plasma endotoxin and plasminogen activator inhibitor 1
concentrations and with fructose intake. J Nutr. 138:1452–1455.
2008.PubMed/NCBI
|
|
66
|
Peng D, Jiang F, Zhang R, Tang S, Chen M,
Yan J, Sun X, Luo Y, Hu C and Jia W: Association of Toll-like
Receptor 4 Gene polymorphisms with susceptibility to type 2
diabetes mellitus in the Chinese population. J Diabetes. 7:485–492.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yin YW, Wang Q, Sun QQ, Hu AM and Liu HL:
Toll-like receptor 4 gene Asp299Gly and Thr399Ile polymorphisms in
type 2 diabetes mellitus: A meta-analysis of 15,059 subjects.
Diabetes Res Clin Pract. 107:338–347. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Kiziltas S, Ata P, Colak Y, Mesçi B,
Senates E, Enc F, Ulasoglu C, Tuncer I and Oguz A: TLR4 gene
polymorphism in patients with nonalcoholic fatty liver disease in
comparison to healthy controls. Metab Syndr Relat Disord.
12:165–170. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Tache D, Bogdan F, Pisoschi C, Baniţă M,
Stănciulescu C, Fusaru AM and Comănescu V: Evidence for the
involvement of TGF-β1-CTGF axis in liver fibrogenesis secondary to
hepatic viral infection. Rom J Morphol Embryol. 52:409–412.
2011.PubMed/NCBI
|
|
70
|
Xiang TX, Cheng N, Li XN and Wu XP:
Association between transforming growth factor-β1 polymorphisms and
hepatocellular cancer risk: A meta-analysis. Hepatol Res.
42:583–590. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mohy A and Fouad A: Role of transforming
growth factor-β1 in serum and − 509 C>T promoter gene
polymorphism in development of liver cirrhosis in Egyptian
patients. Meta Gene. 2:631–637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Falleti E, Fabris C, Toniutto P, Fontanini
E, Cussigh A, Bitetto D, Fornasiere E, Avellini C, Minisini R and
Pirisi M: TGF-β1 genotypes in cirrhosis: Relationship with the
occurrence of liver cancer. Cytokine. 44:256–261. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Dai CY, Chuang WL, Lee LP, Pan WC, Huang
JF, Hsieh MY, Hou NJ, Lin ZY, Chen SC, Hsieh MY, et al: Association
between transforming growth factor-beta 1 polymorphism and
virologic characteristics of chronic hepatitis C. Transl Res.
152:151–156. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Valenti L, Fracanzani AL, Dongiovanni P,
Santorelli G, Branchi A, Taioli E, Fiorelli G and Fargion S: Tumor
necrosis factor alpha promoter polymorphisms and insulin resistance
in nonalcoholic fatty liver disease. Gastroenterology. 122:274–280.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Tokushige K, Takakura M,
Tsuchiya-Matsushita N, Taniai M, Hashimoto E and Shiratori K:
Influence of TNF gene polymorphisms in Japanese patients with NASH
and simple steatosis. J Hepatol. 46:1104–1110. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wang JK, Feng ZW, Li YC, Li QY and Tao XY:
Association of tumor necrosis factor-α gene promoter polymorphism
at sites −308 and −238 with non-alcoholic fatty liver disease: A
meta-analysis. J Gastroenterol Hepatol. 27:670–676. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Wei Y, Liu F, Li B, Chen X, Ma Y, Yan L,
Wen T, Xu M, Wang W and Yang J: Polymorphisms of tumor necrosis
factor-alpha and hepatocellular carcinoma risk: A HuGE systematic
review and meta-analysis. Dig Dis Sci. 56:2227–2236. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Marginean O, Banescu C, Duicu C, Marginean
M Oana and Marginean C: The role of interleukin-6 gene 572G/C
polymorphism in child obesity. Clin Nutr. 33:S1172014. View Article : Google Scholar
|
|
79
|
Giannitrapani L, Soresi M, Balasus D,
Licata A and Montalto G: Genetic association of interleukin-6
polymorphism (−174 G/C) with chronic liver diseases and
hepatocellular carcinoma. World J Gastroenterol. 19:2449–2455.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Deng Y, Li M, Wang J, Xie L, Li T, He Y,
Lu Q, Li R, Tan A, Qin X, et al: Susceptibility to hepatocellular
carcinoma in the Chinese population-associations with interleukin-6
receptor polymorphism. Tumour Biol. 35:6383–6388. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bedogni G, Miglioli L, Masutti F,
Tiribelli C, Marchesini G and Bellentani S: Prevalence of and risk
factors for nonalcoholic fatty liver disease: The Dionysos
nutrition and liver study. Hepatology. 42:44–52. 2005. View Article : Google Scholar : PubMed/NCBI
|














