Introduction
Lung cancer is the leading cause of cancer-related
mortality worldwide, with a 5-year overall survival rate of <15%
(1). Non-small cell lung cancer
(NSCLC) is the most common histological type of lung cancer,
representing ~85% of cases, and its incidence has risen steadily
over the past few decades, particularly in women (2). Although target-directed therapy has been
demonstrated to improve chemotherapy response among NSCLC patients
(3), for the numerous NSCLC patients
with an unknown mutation status or with no apparent gene mutations,
platinum-based regimens are the standard of care recommended by the
European Society for Medical Oncology Clinical Practise Guidelines
(4); however, chemotherapy for
advanced inoperable NSCLC is generally palliative. The major factor
contributing to failure of chemotherapy in the treatment of lung
cancer is development of drug resistance (5).
The lung is continuously exposed to chemicals and
carcinogens that cause oxidative stress to lipids, proteins and
DNA. In response, lung cells must activate cytoprotective
mechanisms to defend and protect themselves against these insults
(6). Lung cancer tumors seem to
inherit this ability, which affords them protection against insults
that generate oxidative stress or against the toxicity of
xenobiotics produced in the microenvironmental conditions of tumour
growth (7). The kelch-like
ECH-associated protein 1 (Keap1)-nuclear factor erythroid-2-related
factor-2 (Nrf2) pathway is a key determinant for cells in coping
with oxidative stress (8). In
response to environmental or endogenous insults, the Keap1-Nrf2
pathway increases the expression of a series of
cytoprotective/defensive proteins that protect cells against
oxidative stress and promote cell survival (9).
Previous data indicate that abnormal states of the
Keap1-Nrf2 pathway exist in lung cancer (10–12).
Elevated Nrf2 levels and Keap1 dysfunction have been frequently
identified in lung cancer, and it is possible that they are
associated with tumour progression, cytoprotection, resistance to
chemotherapeutic drugs and poor prognosis (13).
The current review summarizes recent advancements in
our understanding of alterations in the Keap1-Nrf2 pathway in
NSCLC, its role in drug resistance, and discusses its usefulness as
a potential biomarker. An improved understanding of the role served
by the Keap1-Nrf2 pathway in the regulation of cytoprotective
mechanisms may aid the search for novel anticancer targets that
afford decreased tumour defense systems and increased sensitivity
to treatments.
Keap1-Nrf2 pathway
The Keap1-Nrf2 pathway serves a central role in
protecting cells from oxidative and/or electrophilic stress. Nrf2,
also known as NFE2L2, belonging to the cap'n'collar subfamily of
the basic leucine zipper transcription factors, is the primary
regulator of the inducible cell defense system, which mediates the
expression of >200 oxidative stress-related genes (14), including those transcribing
antioxidant proteins, proteasome subunits, chaperones, growth
factors and their receptors, certain transcription factors, phase I
and II detoxification enzymes, and drug efflux pumps that can
accelerate the metabolic inactivation of antitumor agents and
decrease intracellular drug concentrations (8,15,16).
Nrf2 activity is tightly regulated by Keap1, which
is a cytoplasmic adaptor protein of the Cullin3 (Cul3)-based
E3-ligase (17). Under normal
physiological conditions, Keap1 constitutively targets Nrf2 for
polyubiquitination and degradation by the 26S proteasome (18). However, upon oxidative stress, Keap1
is inactivated and the ubiquitination of Nrf2 halted, which leads
to the accumulation of Nrf2 in the cytoplasm. Consequently, Nrf2 is
translocated into the nucleus via the importin-α5/importin-β1
import pathway, where it induces the transcription of a series of
antioxidant responsive element (ARE)-responsive genes and
ultimately leads to the activation of the defensive system as well
as mechanisms of chemoradiation resistance (19) (Fig.
1).
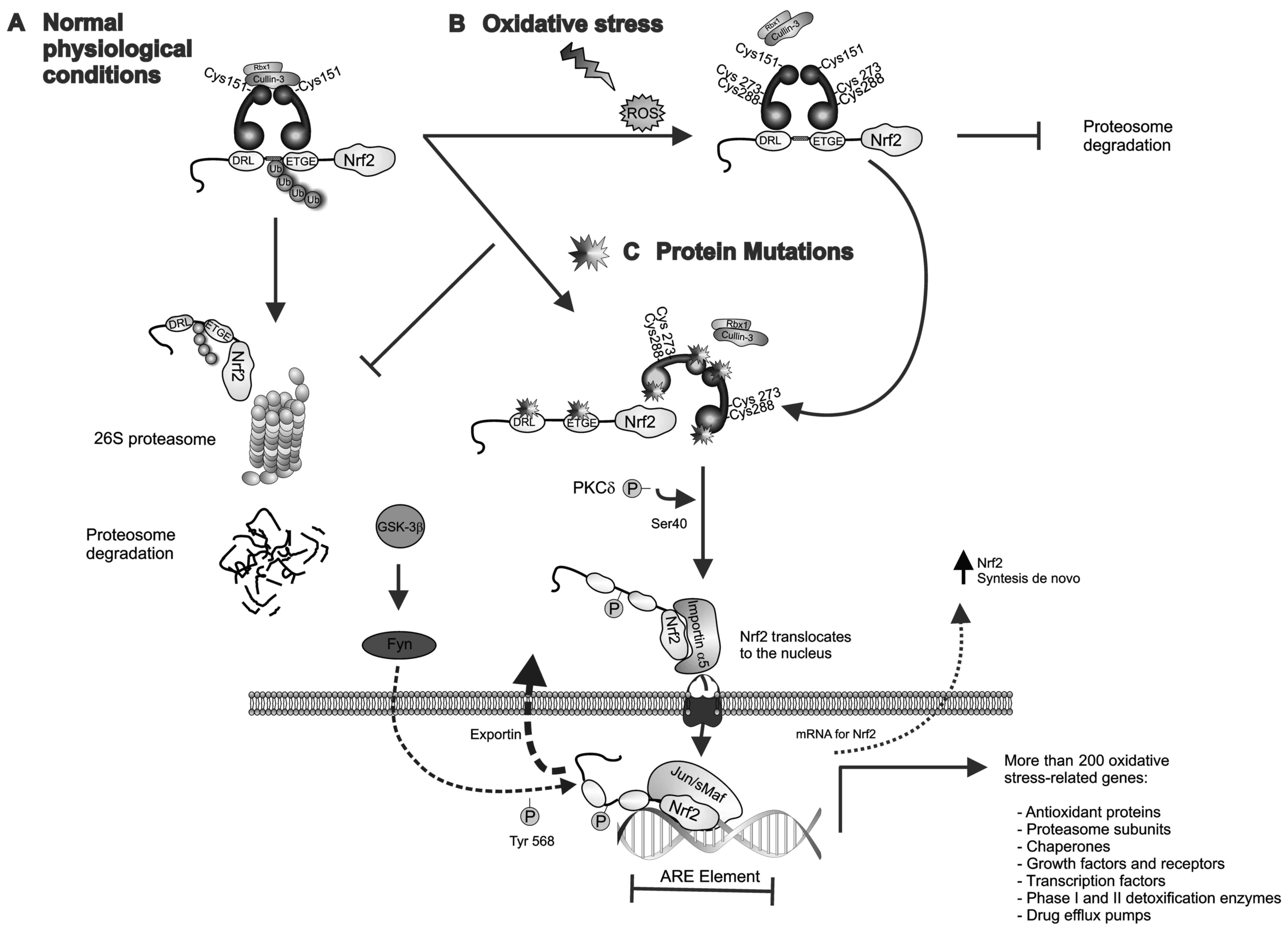 | Figure 1.Keap1-Nrf2 pathway. (A) Under
physiological/basal conditions, the Keap1-Cullin3 complex promotes
ubiquitin E3-ligase activity and proteasomal degradation of Nrf2.
(B) Upon exposure to chemicals or ROS, reactive cysteine residues
of Keap1 are modified leading to a disruption of the Keap1-Nrf2
complex and the release of Nrf2, which also allows de novo
synthesized Nrf2 to accumulate in the cytoplasm. The
phosphorylation of Ser-40 in the Neh2 domain of Nrf2 by PKCδ
results in Nrf2 activation and translocation to the nucleus, where
Nrf2-mediated transcriptional activity of ARE-responsive genes is
initiated. GSK-3β phosphorylates the tyrosine kinase Fyn and
induces its nuclear accumulation. Fyn phosphorylates Nrf2 at
tyrosine-568, facilitating its nuclear export and degradation. (C)
Loss-of-function mutations in Keap1 or Nrf2 lead to constitutive
activation of Nrf2 by disrupting the Keap1-Nrf2 interaction. In the
nucleus, Nrf2 heterodimerizes with sMaf proteins and regulates the
expression of >200 oxidative stress-related genes. ARE,
antioxidant response element; Fyn, proto-oncogene Src-family
tyrosine kinase; GSK-3β, glycogen synthase kinase 3; Keap1,
kelch-like ECH-associated protein 1; sMaf, musculoaponeurotic
fibrosarcoma oncogene; Nrf2, nuclear factor erythroid-2-related
factor-2; PKCδ, protein kinase C-δ; ROS, reactive oxygen
species. |
Several KEAP1 and NRF2 gene mutations which disrupt
the Keap1-Nrf2 interaction and result in Nrf2 overexpression have
been identified in several cancer tissues, including lung, breast,
bladder, ovarian and liver, and numerous types of cancer cell line
(10,20–23).
Mechanisms of Keap1-Nrf2 pathway
deregulation
Distinct mechanisms have been described for the
activation of Nrf2 in NSCLC, which include:
i) Somatic mutations: Gain-of-function mutations in
NRF2 and loss-of-function mutations in KEAP1 (8). Initially, KEAP1 gene mutations (G430C
and G364C) were identified in a human lung adenocarcinoma cell line
(NCI-H1648), each involving a glycine to cysteine substitution in
the Kelch-repeat domain of Keap1 (20). These adenocarcinoma cells exhibited
reduced affinity of Keap1 to Nrf2 and in consequence a constitutive
activation of Nrf2 was observed (18). Subsequent to this, other somatic
mutations have also been identified in the Kelch or intervening
linker (IVR) domains of the Keap1 protein in NSCLC cell lines
(NL20, A549, H460, H1435, H23, H358, H1993, H1395, H838, H1299 and
H292) and in tissues from NSCLC patients (10,11).
ii) KEAP1 hypermethylation: DNA methylation by DNA
methyltransferases in the promoter region of KEAP1 may affect its
expression and hinder its ability to bind to Nrf2, resulting in
Nrf2 activation (12,24). Conversely, CpG methylation of the NRF2
promoter appears to downregulate NRF2 expression indirectly
(25,26).
iii) Accumulation of p21Cip1/WAF1 and
p62lck may disrupt the Keap1-Nrf2 complex (27): In response to injuries producing
reactive oxygen species (ROS), cells generate an Nrf2-dependent
anti-oxidant response in an attempt to repair the ROS- and/or
electrophile-induced damages. However, if the ROS provoke DNA
damage, the transcription factor p53 may be activated, which
induces cell cycle arrest to allow time for the DNA repair
(28). It is this point where the
cyclin-dependent kinase inhibitor p21Cip1/WAF1 (a direct
downstream target of p53) may associate with the DLG motif of Nrf2,
inhibiting Keap1-dependent Nrf2 ubiquitination, resulting in
stabilization of the Nrf2 protein and, ultimately, leading to the
enhanced expression of oxidative stress-related genes (29). However, if DNA is unrepaired, a second
response of the cell may occur and p53-induced apoptosis is
promoted. Under this condition, p53 appears to suppress the
transcription of target genes of Nrf2 (30). However, impairment of autophagy in
cancer cells is usually accompanied by accumulation of
p62/sequestosome 1 protein. This protein, targeted directly onto
the Kelch-repeat domain of Keap1 via its STGE motif, may thereby
disrupt the Keap1-Nrf2 complex (31).
Again, this interaction may cause a decrease in the ubiquitination
of Nrf2 and elicit as a result an increase in Nrf2 stability
(29,32).
iv) Transcriptional upregulation of NRF2 by
oncogenes: Oncogenes including C-MYCERT12,
K-RASG12D and BRAFV619E may increase the
transcriptional level of NRF2 and NRF2-regulated genes, resulting
in a marked increase of cytoprotection of tumour cells (33).
v) Metabolic activation of Nrf2 by Kreb's cycle
intermediates: In the Kreb's cycle, fumarate modifies cysteine
residues within Keap1, which disrupts the ability to ubiquitinate
Nrf2 and in consequence prolongs activation of Nrf2 (34). In addition, defects in fumarate
hydratase may stimulate an importin-mediated nuclear transport of
Nrf2 and the transcription of antioxidant enzymes through the
succination of Keap1 (35).
Furthermore, it has been documented that metabolic reprogramming of
both catabolic and anabolic pathways appear to be driven by
Keap1-Nrf2 aberrations. For example, DeNicola et al
(36) demonstrated that activation of
Nrf2 regulated serine and glycine metabolism and was linked with
clinical aggressiveness in NSCLC, and Mitsuishi et al
(37) showed that Nrf2 redirected
glucose and glutamine into anabolic pathways, particularly under
the sustained activation of phosphatidylinositide 3-kinase
(PI3K)-protein kinase B (PKB)/Akt signaling, which was advantageous
for proliferation and survival in A549 cells.
vi) Loss of exon 2 in the NRF2 gene: Aberrant NRF2
transcripts may result from exon 2 skipping, which translates an
Nrf2 protein isoform missing either the DLG or ETGE motifs of the
regulatory Nrf2-ECH homology (Neh)2 domain, resulting in persistent
Nrf2 localization in the nucleus (38,39);
vii) In recent years, there has been recognition
that the repression of Nrf2 by Cul1-β-transducin repeat-containing
protein (β-TrCP) E3 ubiquitin ligase is augmented by glycogen
synthase kinase-3β (GSK-3β). Thus, Nrf2 may be regulated by GSK-3β
through the creation of a DSGIS motif-containing phosphodegron
present in the Neh6 domain of Nrf2 that is recognized by
Cul13-β-TrCP (40). Conversely, the
phosphorylation of Nrf2 by GSK-3β may be inhibited by growth factor
signaling through the PI3K-Akt pathway (41).
Keap1-Nrf2 pathway disruption and
prognosis
There is increasing data to suggest that KEAP1-NRF2
mutational status is associated with poor prognosis and
chemotherapeutic resistance in NSCLC (10). For example, Inoue et al
(42) examined the expression of Nrf2
by immunohistochemical analyses, in clinical tissue samples from
109 NSCLC cases, and observed that higher nuclear accumulation of
Nrf2 correlated with worse lung cancer-specific survival. By
immunohistochemistry, Yang et al (43) analyzed the Nrf2 expression status of
60 patients with stage IIIB or IV NSCLC and compared the response
to platinum-based treatments in both groups. Although positive
staining for Nrf2 was found in nearly all cases to varying degrees,
patients with stage IV disease exhibited higher Nrf2 expression
than patients with stage IIIB disease (P=0.017). Interestingly,
patients with <75% positive staining achieved a higher response
rate than those with 75–100% positive staining (P=0.003; r=0.447),
suggesting that Nrf2 expression may be a useful index to predict
the efficacy of platinum-based treatments (43).
Interestingly, the results of several studies of
NSCLC tumour samples have also indicated that the occurrence of
NRF2 or KEAP1 mutation is mutually exclusive and associated with
different histologies. Solis et al (44) studied 304 tumour tissue samples [190
adenocarcinomas (ADCs) and 114 squamous cell carcinomas (SqCCs)]
following adjuvant treatment. They detected that nuclear Nrf2
expression in 26% of the NSCLC samples was significantly more
common in SqCC (38%) than in ADC (18%; P<0.0001) and established
an association of a nuclear Nrf2 abundance with worse
progression-free survival. Li et al (45) reported that the frequency of Keap1
alterations was significantly higher in papillary ADC tumors (60%)
than that reported previously for NSCLC (3–19%) (46). In a recent study, Frank et al
(47) analyzed the tumour tissues of
1,391 patients with NSCLC and identified that the frequency for
Nrf2 mutations was 3.5% (n=49), while for Keap1 mutations was 11.3%
(n=157). Nrf2 mutations were most frequent in SqCC (59.2%) whereas
Keap1 mutations were predominantly detected in ADC (72.2%).
Furthermore, patients with tumors containing KEAP1 mutation
presented a worse Eastern Cooperative Oncology Group performance
status and the response on application to different chemotherapy
regimens was notably poor (47). A
large-scale genomic study involving The Cancer Genome Atlas
Research Network examined the exome sequences and copy number
profiles of 660 ADC and 484 SqCC tumour/normal tissue pairs
(48). This report estimated a
KEAP1-NRF2 mutation frequency of 34% in 178 tumors from affected
individuals and revealed that the KEAP1 gene was significantly
mutated exclusively in ADC, whereas NRF2 was significantly mutated
in SqCC. CUL3, the protein product of which is a known interaction
partner of Keap1, also reached statistical significance as a
mutated gene in the lung SqCC cohort (48).
Studies have also investigated whether the frequency
of KEAP1-NRF2 mutations differs by race and ethnicity. Ohta et
al (11) reported loss of
heterozygosity (LOH) in KEAP1 in 5 of 65 (8%) Japanese patients
with lung cancer, and Singh et al (10) performed a systematic analysis of the
KEAP1 genomic locus in a Caucasian population of 54 cases of NSCLC
samples, and in 12 lung cancer cell lines, in which deletions,
insertions, missense mutations of KEAP1 and LOH at 19p13.2 were
commonly found. The KEAP1 somatic mutations were detected in 19%
(10 of 54) of all lung cancer cases and in 26% (9 of 35) of ADC
cases (10).
Genomic profiling analysis has suggested the
prevalence of NRF2 exon deletions to be 1–2% in a panel of 113
NSCLC cell lines (38), and the
existence of two NRF2 mutation ‘hot-spots’ in ~10% of patients with
SqCC, which may enable the transcription factor to evade
Keap1-mediated repression (38,49).
Besides, the mutation burden in Keap1 is spread across the protein
domains, while those in Nrf2 only occur in codons for amino acids
around the DLG and ETGE motifs (the domains of Nrf2 that interact
with Keap1) (10,11,39,50)
(Fig. 2).
 | Figure 2.Schematic representation of domain
architecture of the Keap1 and Nrf2 proteins and mutations found in
NSCLC. (A) Human NRF2 is a polypeptide of 605 amino acids and
contains 7 Neh domains. Neh1 contains the signature CNC motif,
which is a highly conserved bZIP domain for DNA binding. The Neh1
CNC-bZIP domain is responsible for dimerization with sMaf protein
and is required for binding to ARE sequences in DNA; the Neh2
domain controls the interaction with Keap1 through the DLG and ETGE
motifs; the Neh6 domain contains two binding sites (DSGIS and
DSAPGS motifs), and the phosphorylation of the DSGIS motif by
GSK-3β increases binding ability to the β-TrCP1 adaptor protein.
(B) Human Keap1 is a polypeptide of 644 amino acids. The BTB domain
is required for the formation of Keap1 homodimers as well as the
recruitment of Cul3-based E3-ligase. The Kelch-repeat domain
controls Nrf2 interaction. The region between the BTB and Kelch
repeat domains constitutes the IVR. Amino acid positions of the
identified mutations of Nrf2 and Keap1 are shown for ADC, ADC/SqCC
and SqCC. All the indicated mutations are listed in the Catalogue
of Somatic Mutations in Cancer database (http://cancer.sanger.ac.uk/cosmic). ADC,
adenocarcinoma; ADC/SqCC, adenocarcinoma/squamous cell carcinoma;
ARE, antioxidant response element; bZIP, basic leucine zipper; BTB,
broad complex, tram-track and bric-a-brac; CNC, cap'n'collar; Cul3,
Cullin3; GSK-3β, glycogen synthase kinase 3; IVR, intervening
linker; KEAP1, kelch-like ECH-associated protein 1; sMaf,
musculoaponeurotic fibrosarcoma oncogene; Neh, Nrf2-ECH homologous
structure; NRF2, nuclear factor erythroid-2-related factor-2; SqCC,
squamous cell carcinoma; β-TrCP1, β-T-complex protein 1. |
Despite these data, the frequency of KEAP1 or NRF2
mutation in NSCLC remains uncertain, which thus warrants further
studies to provide discriminating differences between the NSCLC
subtypes as well as a comparison between racial/ethnic groups.
Co-occurring aberrations
Comparative genomic analyses have determined that
co-occurring genomic aberrations are present in 83.7% of patients
with tumors containing a mutation in Nrf2 and 87.3% of those with a
mutation in Keap1 (47). Mutations in
the gene for p53 (TP53) are the most common concurrent aberration,
with a frequency of 40.8% in tumors harboring mutations in Nrf2 and
44.9% in tumors with mutations in Keap1. Similar distribution
patterns have been observed for epidermal growth factor receptor
(EGFR) mutations, with a frequency of 6.1% reported in tumors with
mutation in Nrf2 and 6.3% in those with mutation in Keap1. For
c-MET amplification, a mutation frequency of 26.3% has been
observed in tumors with Nrf2 mutation, while for Keap1 mutation the
frequency was 18.3% (47). NSCLC with
Keap1 mutation which also harbors an activating KRAS mutation is
associated with worse prognosis compared with KRAS-mutated patients
without Keap1 mutation (51).
Interestingly, patients with ADC that have high mutational load and
which could benefit from anti-programmed cell death-1 treatment
have shown a high prevalence of Keap1 mutations (52). By contrast, SqCC and mutations in Nrf2
have been associated with high programmed death-ligand 1 expression
(53).
Altogether these studies further suggest that in
NSCLC, the mutations in KEAP1 or NRF2 may each give rise to a
different subset of cancer, opening the opportunity to select a
novel series of druggable molecules distinct between the two main
lung cancer histological types, for overall improved treatment.
Therefore, it appears necessary to implement next-generation
sequencing (NGS)-based multiplex diagnostics for cancer, which is
capable of capturing and amplifying ~10,000 human exons in a single
multiplex reaction. Through use of such novel technology, an
improved understanding may be gained of the molecular interactions
of mutations in KEAP1 and NRF2 with co-occurring, targetable
genetic aberrations.
Crosstalk between the Keap1-Nrf2 and EGFR
pathways
In lung ADC the tyrosine kinase activity of EGFR is
frequently overexpressed or highly activated and has been
associated with growth, survival and therapeutic resistance
(54). EGFR aberrations can
overactivate downstream pro-oncogenic signaling pathways, including
the PI3K/Akt and mitogen-activated protein kinase/extracellular
signal-regulated kinase pathways, leading to cell growth and
proliferation (55). As a result,
EGFR has become viewed as an important specific molecule for
targeted therapy with specific tyrosine kinase inhibitors (TKIs)
(56). In cancer cells, EGFR-mediated
stimulation of the PI3K/Akt pathway may inhibit the constitutive
activity of GSK-3β, decreasing the GSK-3β-dependent degradation of
Nrf2, enabling it to translocate to the nucleus, where
Nrf2-mediated transcriptional activity of cytoprotective genes is
initiated (57). Conversely, the
activation of GSK-3β by inhibition of PI3K/Akt was reported to
decrease Nrf2 protein levels in human A549 lung cells that lack
functional Keap1 (48). Activation of
the PI3/Akt pathway provides a rationalized explanation of how
ARE-driven genes may be induced by aberrant activity of growth
factor receptors, and of why this pathway may be implicated as an
important mechanism in resistance to EGFR inhibitors (58).
Furthermore, EGFR itself may directly regulate Nrf2
activity in cancer cells. Huo et al (59) showed that nuclear EGFR induced
phosphorylation and ubiquitination of Keap1, leading to
stabilization of Nrf2 and stimulation of transcriptional activity,
which contributed to cancer cell resistance to chemotherapy. Thus,
the function of the EGFR-PI3K-AKT pathway in regulating the
Keap1-Nrf2 axis may have an important role not only in cancer cell
growth but also in the expression of genes that confer drug
resistance in human cancers (60).
Notably, in lung tumors, mutations of EGFR or
overactivity of Nrf2 appear to be mutually exclusive (61); it has been observed that the frequency
of nuclear Nrf2 expression is significantly higher in EGFR
wild-type ADC (21%) than in tumors with EGFR mutation (0%)
(44,62). In this regard, it has been speculated
that the lack of nuclear Nrf2 expression in ADC containing EGFR
mutations may be an important factor that contributes to the
chemosensitivity observed with platinum-based chemotherapy
(44). This crosstalk between the
EGFR-PI3K-AKT and Keap1-Nrf2 pathways may explain why for certain
cases of NSCLC, mortality rate remains among the highest of all
cancers, despite the availability of improved therapeutics
including EGFR-TKIs (56).
The interaction of mutations in KEAP1 and NRF2 with
co-occurring targetable genetic aberrations requires an improved
understanding, and highlights the need for NGS-based molecular
cancer diagnostics to cover co-occurring mutations at least in the
setting of clinical research.
Drug resistance and clinical
implications
There is data to suggest that the activation of the
Keap1-Nrf2 pathway is associated with the emergence of resistance
to chemotherapy drugs via transcriptional activation of genes
conferring resistance to such drugs, including: Multidrug
resistance-associated protein 1 [also known as ATP binding cassette
(ABC) subfamily C member 1] and γ-glutamylcysteine synthetase
genes, which may be involved in resistance against cisplatin and
alkylating agents (63,64); breast cancer resistance protein (also
known as ABC subfamily G member 2), which is considered to mediate
the efflux of gefitinib (an EGFR inhibitor), conferring resistance
to TKI regardless of EGFR mutation (65–67); and
several other cytoprotective genes (heme oxygenase 1, NAD(P)H
quinone dehydrogenase 1 and glutathione S-transferases) (68).
In support of these observations, it has been
reported that the inhibition of Nrf2 in lung cancer cells resulted
in enhanced intracellular accumulation of carboplatin and
etoposide, and consequently in enhanced chemotherapy-induced cell
death (69,70). Furthermore, Tian et al
(71) reported that small interfering
RNA knockdown of Nrf2 significantly disrupted Nrf2 signaling in
vitro and led to sensitization of the H292 cell line to
platinum-based drugs, and Singh et al (70) demonstrated that xenografts derived
from Nrf2-silenced lung cancer cells had an enhanced response to
carboplatin in vivo compared with control knockdown
cells.
In clinical practice, the repercussion of
deregulation in the Keap1-Nrf2 pathway is a negative effect for
patients with NSCLC, generally manifesting as reduction in overall
survival and poorer prognosis. Several reports support this; for
example, Solis et al (44)
reported that Nrf2 overexpression [P=0.0139; hazard ratio (HR),
1.75] or low or absent Keap1 expression (P=0.0181; HR, 2.09) was
associated with worse overall survival in SqCC. Takahashi et
al (62) identified that KEAP1
gene mutations were likely associated with a worse prognosis and
lower postoperative disease-free survival rate in pathological
stage I–II NSCLC. These observations were also confirmed in other
reports, where the nuclear expression of Nrf2 was indicated to
serve a role in resistance to platinum-based treatment in SqCC
(61,72,73). A
previous meta-analysis of microarray data on the expression
signatures of 240 NRF2-mediated genes identified a group of 50
genes that predicted a worse clinical outcome in 60% of NSCLC
cohorts analyzed (72). In a recent
study of patients with NSCLC, the median overall survival of
patients with Keap1 mutation was 19.1 months (95% CI, 1.8–36.3
months), and 14.0 months (95% CI, 5.6–22.3 months) for those
patients with Nrf2 mutation (47).
Overall, the available data indicates that
Keap1-Nrf2 pathway activation serves an important role in the
acquisition of resistance to chemotherapy in NSCLC, and provides an
explanation for the poor outcomes observed clinically. It has
therefore been suggested that the dysregulation of Keap1-Nrf2
pathway may be a clinically useful biomarker of prognosis in lung
cancer patients (74,75).
Future directions and conclusions
In recent years, pharmaceutical companies have
focused on Keap1-Nrf2 pathway targets in order to identify novel
and effective molecules capable of inhibiting the Keap1-Nrf2
interaction (76). An obvious
advantage of targeting Keap1 or Nrf2 molecules would be the
expected increase in effectiveness of standard antitumor
chemotherapies; targeting of the detoxification pathways involved
in detoxification of various chemotherapy drugs would also be
required to reduce the possibility of alternative or redundant
detoxification pathway activation. Two reports in the last 5 years
have provided a comprehensive summary of current literature
relevant to small-molecule modulators of the Keap1-Nrf2 pathway and
their usefulness as potential preventive and therapeutic agents
(77,78).
Recently, the inhibition of NRF2 has become a
promising therapeutic approach for cancers displaying Nrf2
overactivation, and consequently, the clinical implementation of
Nrf2 inhibitors in NSCLC patients with Keap1-Nrf2 pathway
deregulation may be a useful therapeutic strategy (76). There have been few Nrf2 inhibitors
identified to date. Brusatol, a quassinoid isolated from the
Brucea javanica shrub, has been identified as a unique
inhibitor of the Nrf2 through enhancing ubiquitination and
degradation of Nrf2 (79). Notably,
treatment with brusatol sensitized a broad spectrum of cancer cells
to antitumor drugs, reduced tumour burden and improved survival in
murine A549 ×enograft models (80,81).
Nevertheless, while the use of targeted inhibitors may be a novel
therapeutic strategy to treat several types of cancer, it is
noteworthy that administration of systemic NRF2 inhibitors may have
undesirable effects on cancer patients, considering the central
roles of NRF2 in cytoprotection (82).
Novel potential therapeutic targets in cancers
exhibiting Keap1-Nrf2 pathway deregulation are being identified in
addition to Nrf2 inhibitors. Some of these targets, including the
glutathione synthesis, serine synthesis and pentose phosphate
pathways, and IL-11, are direct or indirect downstream effectors of
Nrf2 in mediating malignant phenotypes. However, efforts are still
required to search for novel compounds and engineered small
molecules that target Nrf2 more specifically, and to establish Nrf2
inhibitors suitable for safe clinical use and temporary use as a
sensitizer in chemo- and radiotherapy regimens.
To date, considerable progress has been made to
understand the mechanisms involved in the fine regulation of the
Keap1-Nrf2 pathway and its downstream genes. However, due to the
complexity of the cross-talk between Nrf2 and its numerous
signaling-network partners, further studies are still required to
determine the optimum markers for predicting the clinical outcome
of patients with NSCLC. An improved understanding of the molecular
mechanisms activating the Keap1-Nrf2 pathway may provide a basis
for improvement in the selection of patients with poor prognosis,
to be treated only with supportive care and thus avoiding
unnecessary adverse effects and complications of systemic
chemotherapy.
In conclusion, the findings reviewed herein suggest
that evaluation of Keap1-Nrf2 pathway dysregulation in patients
with NSCLC may provide predictive biomarkers of chemotherapy
resistance and improve the accuracy of diagnosis.
Acknowledgements
The author would like to thank Dr Francisco
Hernandez Gómez-Crespo at the Department of Biochemistry and
Environmental Medicine, National Institute of Respiratory Diseases
(Mexico City, Mexico), for his critique of this manuscript. Thanks
are also afforded to Mrs Cristina Wilson at ‘Gastro One’ medical
clinic (Memphis, TN, USA), for her editing of the English
language.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Authors' contributions
RBR was responsible for all aspects involved in the
design, performance and writing of the literature review.
Ethics approval and consent to
participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
References
|
1
|
Siegel RL, Miller KD and Jemal A: Cancer
Statistics, 2017. CA Cancer J Clin. 67:7–30. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cheng TY, Cramb SM, Baade PD, Youlden DR,
Nwogu C and Reid ME: The international epidemiology of lung cancer:
Latest trends, disparities, and tumor characteristics. J Thorac
Oncol. 11:1653–1671. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forde PM and Ettinger DS: Targeted therapy
for non-small-cell lung cancer: Past, present and future. Expert
Rev Anticancer Ther. 13:745–758. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Novello S, Barlesi F, Califano R, Cufer T,
Ekman S, Levra MG, Kerr K, Popat S, Reck M, Senan S, et al: ESMO
Guidelines Committee: Metastatic non-small-cell lung cancer: ESMO
Clinical Practice Guidelines for diagnosis, treatment and
follow-up. Ann Oncol. 27 Suppl 5:v1–v27. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim ES: Chemotherapy Resistance in Lung
Cancer. Adv Exp Med Biol. 893:189–209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rahman I, Biswas SK and Kode A: Oxidant
and antioxidant balance in the airways and airway diseases. Eur J
Pharmacol. 533:222–239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhang JY, Wang Y and Prakash C:
Xenobiotic-metabolizing enzymes in human lung. Curr Drug Metab.
7:939–948. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jaramillo MC and Zhang DD: The emerging
role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev.
27:2179–2191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ma Q: Role of nrf2 in oxidative stress and
toxicity. Annu Rev Pharmacol Toxicol. 53:401–426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Singh A, Misra V, Thimmulappa RK, Lee H,
Ames S, Hoque MO, Herman JG, Baylin SB, Sidransky D, Gabrielson E,
et al: Dysfunctional KEAP1-NRF2 interaction in non-small-cell lung
cancer. PLoS Med. 3:e4202006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ohta T, Iijima K, Miyamoto M, Nakahara I,
Tanaka H, Ohtsuji M, Suzuki T, Kobayashi A, Yokota J, Sakiyama T,
et al: Loss of Keap1 function activates Nrf2 and provides
advantages for lung cancer cell growth. Cancer Res. 68:1303–1309.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang R, An J, Ji F, Jiao H, Sun H and Zhou
D: Hypermethylation of the Keap1 gene in human lung cancer cell
lines and lung cancer tissues. Biochem Biophys Res Commun.
373:151–154. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vollrath V, Wielandt AM, Iruretagoyena M
and Chianale J: Role of Nrf2 in the regulation of the Mrp2 (ABCC2)
gene. Biochem J. 395:599–609. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lu MC, Ji JA, Jiang ZY and You QD: The
Keap1-Nrf2-ARE pathway as a potential preventive and therapeutic
target: An update. Med Res Rev. 36:924–963. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sporn MB and Liby KT: NRF2 and cancer: The
good, the bad and the importance of context. Nat Rev Cancer.
12:564–571. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Villeneuve NF, Lau A and Zhang DD:
Regulation of the Nrf2-Keap1 antioxidant response by the ubiquitin
proteasome system: An insight into cullin-ring ubiquitin ligases.
Antioxid Redox Signal. 13:1699–1712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Menegon S, Columbano A and Giordano S: The
dual roles of NRF2 in cancer. Trends Mol Med. 22:578–593. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Theodore M, Kawai Y, Yang J, Kleshchenko
Y, Reddy SP, Villalta F and Arinze IJ: Multiple nuclear
localization signals function in the nuclear import of the
transcription factor Nrf2. J Biol Chem. 283:8984–8994. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Padmanabhan B, Tong KI, Ohta T, Nakamura
Y, Scharlock M, Ohtsuji M, Kang MI, Kobayashi A, Yokoyama S and
Yamamoto M: Structural basis for defects of Keap1 activity provoked
by its point mutations in lung cancer. Mol Cell. 21:689–700. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim YR, Oh JE, Kim MS, Kang MR, Park SW,
Han JY, Eom HS, Yoo NJ and Lee SH: Oncogenic NRF2 mutations in
squamous cell carcinomas of oesophagus and skin. J Pathol.
220:446–451. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Nioi P and Nguyen T: A mutation of Keap1
found in breast cancer impairs its ability to repress Nrf2
activity. Biochem Biophys Res Commun. 362:816–821. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taguchi K and Yamamoto M: The KEAP1-NRF2
system in cancer. Front Oncol. 7:852017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Muscarella LA, Parrella P, D'Alessandro V,
la Torre A, Barbano R, Fontana A, Tancredi A, Guarnieri V, Balsamo
T, Coco M, et al: Frequent epigenetics inactivation of KEAP1 gene
in non-small cell lung cancer. Epigenetics. 6:710–719. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Khor TO, Fuentes F, Shu L,
Paredes-Gonzalez X, Yang AY, Liu Y, Smiraglia DJ, Yegnasubramanian
S, Nelson WG and Kong AN: Epigenetic DNA methylation of
antioxidative stress regulator NRF2 in human prostate cancer.
Cancer Prev Res (Phila). 7:1186–1197. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo Y, Yu S, Zhang C and Kong AN:
Epigenetic regulation of Keap1-Nrf2 signaling. Free Radic Biol Med.
88:337–349. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chen W, Sun Z, Wang XJ, Jiang T, Huang Z,
Fang D and Zhang DD: Direct interaction between Nrf2 and
p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response.
Mol Cell. 34:663–673. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Williams AB and Schumacher B: p53 in the
DNA-damage-repair process. Cold Spring Harb Perspect Med.
6:a0260702016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Faraonio R, Vergara P, Di Marzo D,
Pierantoni MG, Napolitano M, Russo T and Cimino F: p53 suppresses
the Nrf2-dependent transcription of antioxidant response genes. J
Biol Chem. 281:39776–39784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ichimura Y, Waguri S, Sou YS, Kageyama S,
Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et
al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during
selective autophagy. Mol Cell. 51:618–631. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Taguchi K, Fujikawa N, Komatsu M, Ishii T,
Unno M, Akaike T, Motohashi H and Yamamoto M: Keap1 degradation by
autophagy for the maintenance of redox homeostasis. Proc Natl Acad
Sci USA. 109:13561–13566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
DeNicola GM, Karreth FA, Humpton TJ,
Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES,
et al: Oncogene-induced Nrf2 transcription promotes ROS
detoxification and tumorigenesis. Nature. 475:106–109. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kinch L, Grishin NV and Brugarolas J:
Succination of Keap1 and activation of Nrf2-dependent antioxidant
pathways in FH-deficient papillary renal cell carcinoma type 2.
Cancer Cell. 20:418–420. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Adam J, Hatipoglu E, O'Flaherty L,
Ternette N, Sahgal N, Lockstone H, Baban D, Nye E, Stamp GW,
Wolhuter K, et al: Renal cyst formation in Fh1-deficient mice is
independent of the Hif/Phd pathway: Roles for fumarate in KEAP1
succination and Nrf2 signaling. Cancer Cell. 20:524–537. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
DeNicola GM, Chen PH, Mullarky E, Sudderth
JA, Hu Z, Wu D, Tang H, Xie Y, Asara JM, Huffman KE, et al: NRF2
regulates serine biosynthesis in non-small cell lung cancer. Nat
Genet. 47:1475–1481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mitsuishi Y, Taguchi K, Kawatani Y,
Shibata T, Nukiwa T, Aburatani H, Yamamoto M and Motohashi H: Nrf2
redirects glucose and glutamine into anabolic pathways in metabolic
reprogramming. Cancer Cell. 22:66–79. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Goldstein LD, Lee J, Gnad F, Klijn C,
Schaub A, Reeder J, Daemen A, Bakalarski CE, Holcomb T, Shames DS,
et al: Recurrent loss of NFE2L2 Exon 2 is a mechanism for Nrf2
pathway activation in human cancers. Cell Rep. 16:2605–2617. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shibata T, Ohta T, Tong KI, Kokubu A,
Odogawa R, Tsuta K, Asamura H, Yamamoto M and Hirohashi S: Cancer
related mutations in NRF2 impair its recognition by Keap1-Cul3 E3
ligase and promote malignancy. Proc Natl Acad Sci USA.
105:13568–13573. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chowdhry S, Zhang Y, McMahon M, Sutherland
C, Cuadrado A and Hayes JD: Nrf2 is controlled by two distinct
β-TrCP recognition motifs in its Neh6 domain, one of which can be
modulated by GSK-3 activity. Oncogene. 32:3765–3781. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koundouros N and Poulogiannis G:
Phosphoinositide 3-kinase/Akt signaling and redox metabolism in
cancer. Front Oncol. 8:1602018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Inoue D, Suzuki T, Mitsuishi Y, Miki Y,
Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, et
al: Accumulation of p62/SQSTM1 is associated with poor prognosis in
patients with lung adenocarcinoma. Cancer Sci. 103:760–766. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yang H, Wang W, Zhang Y, Zhao J, Lin E,
Gao J and He J: The role of NF-E2-related factor 2 in predicting
chemoresistance and prognosis in advanced non-small-cell lung
cancer. Clin Lung Cancer. 12:166–171. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Solis LM, Behrens C, Dong W, Suraokar M,
Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN,
et al: Nrf2 and Keap1 abnormalities in non-small cell lung
carcinoma and association with clinicopathologic features. Clin
Cancer Res. 16:3743–3753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Li QK, Singh A, Biswal S, Askin F and
Gabrielson E: KEAP1 gene mutations and NRF2 activation are common
in pulmonary papillary adenocarcinoma. J Hum Genet. 56:230–234.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Kim Y, Hammerman PS, Kim J, Yoon JA, Lee
Y, Sun JM, Wilkerson MD, Pedamallu CS, Cibulskis K, Yoo YK, et al:
Integrative and comparative genomic analysis of lung squamous cell
carcinomas in East Asian patients. J Clin Oncol. 32:121–128. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Frank R, Scheffler M, Merkelbach-Bruse S,
Ihle MA, Kron A, Rauer M, Ueckeroth F, König K, Michels S, Fischer
R, et al: Clinical and pathological characteristics of KEAP1- and
NFE2L2-mutated non-small cell lung carcinoma (NSCLC). Clin Cancer
Res. 24:3087–3096. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Campbell JD, Alexandrov A, Kim J, Wala J,
Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et
al: Cancer Genome Atlas Research Network: Distinct patterns of
somatic genome alterations in lung adenocarcinomas and squamous
cell carcinomas. Nat Genet. 48:607–616. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hayes JD and McMahon M: NRF2 and KEAP1
mutations: Permanent activation of an adaptive response in cancer.
Trends Biochem Sci. 34:176–188. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cancer Genome Atlas Research Network:
Comprehensive genomic characterization of squamous cell lung
cancers. Nature. 489:519–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Riely GJ, Jordan E, Kim HR, Yu HA, Berger
MF and Solit DB: Association of outcomes and co-occurring genomic
alterations in patients with KRAS-mutant non-small cell lung
cancer. J Clin Oncol. 34:90192016. View Article : Google Scholar
|
|
52
|
Rizvi NA, Hellmann MD, Snyder A, Kvistborg
P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al: Cancer
immunology. Mutational landscape determines sensitivity to PD-1
blockade in non-small cell lung cancer. Science. 348:124–128. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Scheel AH, Ansén S, Schultheis AM,
Scheffler M, Fischer RN, Michels S, Hellmich M, George J, Zander T,
Brockmann M, et al: PD-L1 expression in non-small cell lung cancer:
Correlations with genetic alterations. Oncoimmunology.
5:e11313792016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Bethune G, Bethune D, Ridgway N and Xu Z:
Epidermal growth factor receptor (EGFR) in lung cancer: An overview
and update. J Thorac Dis. 2:48–51. 2010.PubMed/NCBI
|
|
55
|
Seshacharyulu P, Ponnusamy MP, Haridas D,
Jain M, Ganti AK and Batra SK: Targeting the EGFR signaling pathway
in cancer therapy. Expert Opin Ther Targets. 16:15–31. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Papaiahgari S, Yerrapureddy A, Hassoun PM,
Garcia JG, Birukov KG and Reddy SP: EGFR-activated signaling and
actin remodeling regulate cyclic stretch-induced NRF2-ARE
activation. Am J Respir Cell Mol Biol. 36:304–312. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Jacobsen K, Bertran-Alamillo J, Molina MA,
Teixidó C, Karachaliou N, Pedersen MH, Castellví J, Garzón M,
Codony-Servat C, Codony-Servat J, et al: Convergent Akt activation
drives acquired EGFR inhibitor resistance in lung cancer. Nat
Commun. 8:4102017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Huo L, Li CW, Huang TH, Lam YC, Xia W, Tu
C, Chang WC, Hsu JL, Lee DF, Nie L, et al: Activation of Keap1/Nrf2
signaling pathway by nuclear epidermal growth factor receptor in
cancer cells. Am J Transl Res. 6:649–663. 2014.PubMed/NCBI
|
|
60
|
Denduluri SK, Idowu O, Wang Z, Liao Z, Yan
Z, Mohammed MK, Ye J, Wei Q, Wang J, Zhao L, et al: Insulin-like
growth factor (IGF) signaling in tumorigenesis and the development
of cancer drug resistance. Genes Dis. 2:13–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sasaki H, Hikosaka Y, Okuda K, Kawano O,
Moriyama S, Yano M and Fujii Y: NFE2L2 gene mutation in male
Japanese squamous cell carcinoma of the lung. J Thorac Oncol.
5:786–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Takahashi T, Sonobe M, Menju T, Nakayama
E, Mino N, Iwakiri S, Nagai S, Sato K, Miyahara R, Okubo K, et al:
Mutations in Keap1 are a potential prognostic factor in resected
non-small cell lung cancer. J Surg Oncol. 101:500–506.
2010.PubMed/NCBI
|
|
63
|
Ishikawa T, Bao JJ, Yamane Y, Akimaru K,
Frindrich K, Wright CD and Kuo MT: Coordinated induction of
MRP/GS-X pump and gamma-glutamylcysteine synthetase by heavy metals
in human leukemia cells. J Biol Chem. 271:14981–14988. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Adachi T, Nakagawa H, Chung I, Hagiya Y,
Hoshijima K, Noguchi N, Kuo MT and Ishikawa T: Nrf2-dependent and
-independent induction of ABC transporters ABCC1, ABCC2, and ABCG2
in HepG2 cells under oxidative stress. J Exp Ther Oncol. 6:335–348.
2007.PubMed/NCBI
|
|
65
|
Singh A, Wu H, Zhang P, Happel C, Ma J and
Biswal S: Expression of ABCG2 (BCRP) is regulated by Nrf2 in cancer
cells that confers side population and chemoresistance phenotype.
Mol Cancer Ther. 9:2365–2376. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Saito H, Hirano H, Nakagawa H, Fukami T,
Oosumi K, Murakami K, Kimura H, Kouchi T, Konomi M, Tao E, et al: A
new strategy of high-speed screening and quantitative
structure-activity relationship analysis to evaluate human
ATP-binding cassette transporter ABCG2-drug interactions. J
Pharmacol Exp Ther. 317:1114–1124. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
zu Schwabedissen Meyer HE, Grube M,
Dreisbach A, Jedlitschky G, Meissner K, Linnemann K, Fusch C,
Ritter CA, Völker U and Kroemer HK: Epidermal growth
factor-mediated activation of the map kinase cascade results in
altered expression and function of ABCG2 (BCRP). Drug Metab Dispos.
34:524–533. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Hayes JD, McMahon M, Chowdhry S and
Dinkova-Kostova AT: Cancer chemoprevention mechanisms mediated
through the Keap1-Nrf2 pathway. Antioxid Redox Signal.
13:1713–1748. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Zhang P, Singh A, Yegnasubramanian S,
Esopi D, Kombairaju P, Bodas M, Wu H, Bova SG and Biswal S: Loss of
Kelch-like ECH-associated protein 1 function in prostate cancer
cells causes chemoresistance and radioresistance and promotes tumor
growth. Mol Cancer Ther. 9:336–346. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Singh A, Boldin-Adamsky S, Thimmulappa RK,
Rath SK, Ashush H, Coulter J, Blackford A, Goodman SN, Bunz F,
Watson WH, et al: RNAi-mediated silencing of nuclear factor
erythroid-2-related factor 2 gene expression in non-small cell lung
cancer inhibits tumor growth and increases efficacy of
chemotherapy. Cancer Res. 68:7975–7984. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Tian Y, Liu Q, He X, Yuan X, Chen Y, Chu Q
and Wu K: Emerging roles of Nrf2 signal in non-small cell lung
cancer. J Hematol Oncol. 9:142016. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Qian Z, Zhou T, Gurguis CI, Xu X, Wen Q,
Lv J, Fang F, Hecker L, Cress AE, Natarajan V, et al: Nuclear
factor, erythroid 2-like 2-associated molecular signature predicts
lung cancer survival. Sci Rep. 5:168892015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Mahaffey CM, Zhang H, Rinna A, Holland W,
Mack PC and Forman HJ: Multidrug-resistant protein-3 gene
regulation by the transcription factor Nrf2 in human bronchial
epithelial and non-small-cell lung carcinoma. Free Radic Biol Med.
46:1650–1657. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Liu X, Sun C, Liu B, Jin X, Li P, Zheng X,
Zhao T, Li F and Li Q: Genistein mediates the selective
radiosensitizing effect in NSCLC A549 cells via inhibiting
methylation of the keap1 gene promoter region. Oncotarget.
7:27267–27279. 2016.PubMed/NCBI
|
|
75
|
Cho JM, Manandhar S, Lee HR, Park HM and
Kwak MK: Role of the Nrf2-antioxidant system in cytotoxicity
mediated by anticancer cisplatin: Implication to cancer cell
resistance. Cancer Lett. 260:96–108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Copple IM, Dinkova-Kostova AT, Kensler TW,
Liby KT and Wigley WC: NRF2 as an emerging therapeutic target. Oxid
Med Cell Longev. 2017:81654582017. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Magesh S, Chen Y and Hu L: Small molecule
modulators of Keap1-Nrf2-ARE pathway as potential preventive and
therapeutic agents. Med Res Rev. 32:687–726. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Abed DA, Goldstein M, Albanyan H, Jin H
and Hu L: Discovery of direct inhibitors of Keap1-Nrf2
protein-protein interaction as potential therapeutic and preventive
agents. Acta Pharm Sin B. 5:285–299. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Harder B, Tian W, La Clair JJ, Tan AC, Ooi
A, Chapman E and Zhang DD: Brusatol overcomes chemoresistance
through inhibition of protein translation. Mol Carcinog.
56:1493–1500. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Ren D, Villeneuve NF, Jiang T, Wu T, Lau
A, Toppin HA and Zhang DD: Brusatol enhances the efficacy of
chemotherapy by inhibiting the Nrf2-mediated defense mechanism.
Proc Natl Acad Sci USA. 108:1433–1438. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Olayanju A, Copple IM, Bryan HK, Edge GT,
Sison RL, Wong MW, Lai ZQ, Lin ZX, Dunn K, Sanderson CM, et al:
Brusatol provokes a rapid and transient inhibition of Nrf2
signaling and sensitizes mammalian cells to chemical
toxicity-implications for therapeutic targeting of Nrf2. Free Radic
Biol Med. 78:202–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kitamura H and Motohashi H: NRF2 addiction
in cancer cells. Cancer Sci. 109:900–911. 2018. View Article : Google Scholar : PubMed/NCBI
|














