Cox-2 inhibitors and PPARγ ligands can act
synergistically to suppress Cox-2 and activate PPARγ
Despite extensive research during the last decade,
the role of cyclooxygenase-2 (Cox-2) and peroxisome
proliferator-activated receptor γ (PPARγ) in cancerogenesis remains
controversial. Therefore, potential clinical outcomes of their
respective inhibitors and activators are still elusive.
Nevertheless, the effects of these agents are promising enough to
prompt further research of the involved cell signaling pathways.
Recently, this research has revealed multiple interactions between
Cox-2 and PPARγ pathways that may be important for anti-cancer
therapies.
Cyclooxygenase is the rate-limiting enzyme involved
in the synthesis of prostaglandins (PGs). There are two isoforms of
this enzyme, the constitutive Cox-1 and the inducible one, Cox-2.
cox-2 gene expression is induced by a wide variety of
stimuli in cells of organisms fighting inflammatory disorders and
cancer. Therefore, the level of the Cox-2 protein is elevated in
various types of cancer cells in comparison with non-malignant
tissues (1). A growing body of
evidence suggests an association of Cox-2 with tumor development,
aggressivity, resistance to standard therapy and unfavorable
patient outcome. Cox-2 may participate in cancer development
through multiple mechanisms, including stimulation of growth,
migration, invasiveness, resistance to apoptosis and enhancement of
angiogenesis (2).
In addition to a number of pre-clinical studies
revealing the anti-proliferative and pro-apoptotic effects of
nonsteroidal anti-inflammatory drugs (NSAIDs) and specific Cox-2
inhibitors, multiple population studies have documented that
chronic intake of NSAIDs is associated with a decreased incidence
of colorectal, prostate, bladder, breast and lung cancers (3–8).
There is also clinical evidence demonstrating the reduction of
colorectal polyps by the Cox-2 inhibitor celecoxib (9). Several pre-clinical and clinical
studies have repeatedly demonstrated that specific Cox-2 inhibitors
are promising enhancers of chemotherapy (10–13).
Nevertheless, the safety of Cox-2 inhibitors in
anti-cancer therapies is still a matter of debate. Although the
tumor-suppressive effects of NSAIDs were attributed to their
ability to act as Cox-2 inhibitors, some effects of these agents
cannot be explained by inhibition of Cox-2, as these drugs can also
provoke responses in Cox-2-negative cells. This suggests that there
are some Cox-2-independent pathways involved in the anti-cancer
effects of these agents. Therefore, inhibition of Cox-2 activity
and PG synthesis is not necessarily beneficial in general;
moreover, it can induce even adverse effects (14,15).
Considering both the benefits and risks of Cox-2 inhibition, there
is still great concern regarding the potential use of
Cox-2-specific inhibitors in combination with other anti-cancer
therapeutics, including the PPAR ligands.
PPARγ is a member of the nuclear hormone receptor
superfamily functioning as a ligand-dependent transcription factor
(16). PPAR affects gene
expression either directly through binding to peroxisome
proliferator response elements (PPREs) located upstream of
controlled genes or indirectly by interfering with other pathways
driven by transcription factors resulting in the silencing of gene
transcription.
Natural ligands of PPARγ are mostly metabolites of
arachidonic acid; they include polyunsaturated fatty acids,
cyclopentenone prostaglandin 15-deoxy-D12,14 prostaglandin J2
(15d-PGJ2) and oxidized lipids (17,18).
Synthetic ligands include the thiazolidinediones (such as
troglitazone, pioglitazone and rosiglitazone) that have been
clinically used in the treatment of type II diabetes (19–21).
Recently, the role of PPARγ in various human cancers
has been intensively studied. PPARγ expression has been reported in
a variety of tumors, including colon (22), breast (23), prostate (24–26),
stomach (27), lung (28), pancreas (29), ovarian (30) and cervical tumors (31). Both natural and synthetic PPARγ
ligands inhibit cancer cell growth in vitro and in
vivo (32,33). These studies, coupled with clinical
trials (34,35), suggest that PPARγ is a novel target
for the development of novel and effective anti-cancer
therapies.
However, there is considerable concern regarding the
significance and safety of PPARγ ligands used as anti-cancer drugs
(36). The mechanism of their
action is still elusive, since both PPARγ-dependent and
PPARγ-independent pathways mediate their anti-proliferative and
pro-apoptotic effects. Furthermore, the biological significance of
PPARγ is still a controversial issue. There are studies
illustrating even tumor-promoting effects of PPARγ, in particular
in colon and breast cancer models (37–39).
Therefore, both Cox-2 and PPARγ are considered as
possible targets for anti-cancer therapy and prevention, but
applications of Cox-2 inhibitors as well as PPARγ ligands in
therapy remain controversial. Detailed understanding of the
molecular mechanisms and signaling pathways may elucidate the pros
and cons of their action and provide more effective therapeutical
approaches. Recent findings involving the cross-talk between Cox-2
and PPAR signaling may have such therapeutically relevant
implications. This review summarizes the current knowledge on the
interplay between Cox-2 and PPARγ signaling pathways and focuses on
the benefits and risks of the combined application of Cox-2
inhibitors and PPARγ ligands in anti-cancer therapy.
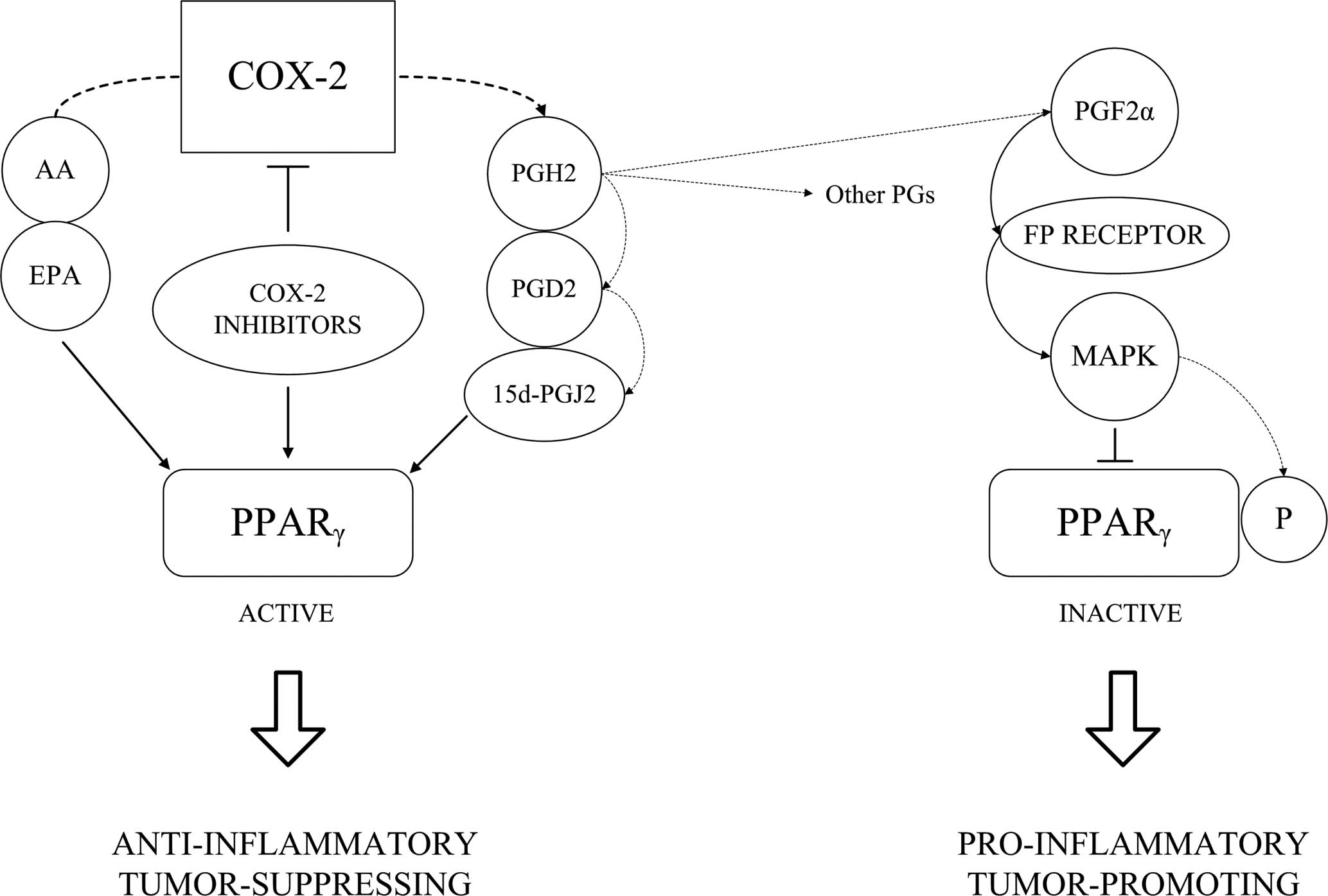
Various Cox-2 products can also bind and activate
PPARγ. Cox-2 catalyzes formation of a chemically unstable
prostaglandin H2 (PGH2) which can be further converted to various
prostanoids (e.g., PGE2, PGD2 and PGF2α) by tissue-specific
isomerases. Dehydration of these PGs leads to the formation of
cyclopentenone prostaglandins PGA2, PGA1 and PGJ2 (43). 15d-PGJ2 is formed from PGJ2 by
further nonenzymatic rearrangements and dehydration. While
prostaglandins PGE2, PGF2α and PGD2 transduce their signals through
binding to the G-protein-coupled cell surface receptors (44), cyclopentenone prostaglandins (e.g.,
15d-PGJ2) are known ligands of PPARγ.
While PGE2, which is considered to be the major
Cox-2 product, possesses pro-inflammatory and tumor-promoting
effects (45,46), accumulating data suggest that
15d-PGJ2 acts as an anti-inflammator (47). Therefore, both pro- and
anti-inflammatory effects can be controlled by Cox-2. During the
early phase of inflammation, Cox-2 expression and activity is
induced and associated with increased synthesis of PGE2. During the
later phase, Cox-2 may be involved in the resolution of acute
inflammation by generating an alternate set of PGs, such as those
of the cyclopentenone family (15). Anti-inflammatory effects of
cyclopentanone PGs are mediated either by binding/activating PPARγ
or by interaction with other target molecules, such as NF-κB or IκB
kinase (43).
Although the anti-inflammatory effect of 15d-PGJ2 is
well known and accepted, the results concerning the effects of
cyclopentanone PGs on tumor growth are still conflicting. 15d-PGJ2
was found to possess anti-neoplastic properties; it inhibits cell
growth, induces terminal differentiation and apoptotic cell death
in a variety of tumor cells, thereby promoting phenotypic changes
associated with a less malignant status (23,35,48).
In contrast, there are reports demonstrating the tumor-promoting
action of 15d-PGJ2 as well (49,50).
On the other hand, Cox-2 can produce metabolites
inhibiting PPARγ. PGF2α, acting through its cell surface
G-protein-coupled receptor, inhibits PPARγ through MAP
kinase-dependent phosphorylation. The antagonistic effects of PGJ2
and PGF2α on the activity of PPARγ result in opposing effects of
these compounds on adipocyte differentiation. PGJ2 stimulates,
while PGF2α blocks, adipogenesis (51). Similarly, antagonistic effects of
15d-PGJ2 and PGF2α were observed in B lymphoma cells; 15d-PGJ2
induced apoptosis via PPARγ activation, while PGF2α pretreatment
attenuated its cytotoxic effect (52).
Moreover, not only the Cox-2 substrates and products
can be PPAR ligands, PPARγ activity can also be stimulated by Cox-2
inhibitors. Ibuprofen, indomethacin and some other NSAIDs can both
inhibit Cox-1/Cox-2 and function as PPARγ ligands in various cell
systems as well (53,54). Celecoxib, a selective Cox-2
inhibitor, binds and activates PPARγ in rat mesangial cells
(55). NS-398, another selective
inhibitor of Cox-2, has been found to increase expression of PPARγ,
PPARα and PPARβ in human fibroblasts (56). PPARγ expression was up-regulated in
lung tumors in mice treated with nimesulfide, another
Cox-2-specific inhibitor, when compared to tumor tissue of
untreated mice (57). Indomethacin
and other NSAIDs as well as NS-398 induced growth suppression and
apoptosis associated with activation of PPARγ in rheumatoid
synovial cells. 15d-PGJ2 and troglitazone, other PPARγ ligands have
a similar inhibitory effect on the growth of synovial cells
(58). Mechanisms of
celecoxib-induced inhibition of hepatocellular carcinoma cell
growth involve up-regulation of PPARγ (59). Therefore, activation of PPARγ is
considered as one of the Cox-2-independent mechanisms responsible
for the anti-inflammatory and anti-neoplastic effects of NSAIDs.
Induction of PPARγ can account for the puzzling fact that selective
Cox-2 inhibitors display anti-proliferative properties in cells
lacking Cox-2 expression. It has been demonstrated that JTE-522, a
Cox-2-specific inhibitor, interferes with the growth of
Cox-2-negative HCC cells. This growth arrest is, in part, mediated
by up-regulation of PPARγ protein expression (60). We conclude that PPARγ activity can
be induced by several Cox-2 inhibitors and possibly participates in
mediating the effects that cannot be attributed to the Cox-2
inhibition itself.
There are numerous studies documenting PPARγ
ligand-induced Cox-2 up-regulation. Endogenous PPARγ ligand
15d-PGJ2, as well as synthetic PPARγ agonists, stimulate
cox-2 expression and activity in several cell types
(49,61–66).
However, the mechanism of this up-regulation varies significantly
in different cell types and according to the specificity of the
activating stimulus. cox-2 transcription can be directly
activated by PPARγ itself, and the peroxisome proliferator
responsive element (PPRE) was indentified in the cox-2
promoter sequence (61). The
artificial construct containing the cox-2 promoter including
PPRE was activated in cells cotransfected with vectors encoding
PPARα, δ and γ. Similarly, PPRE in the cox-2 promoter was
required for the PPARγ ligand rosiglitazone-induced activation of
the reporter (62,67). PPARγ-dependent activation of Cox-2
by rosiglitazone was observed in smooth muscle cells, and it was
sensitive to the PPARγ antagonist (63).
Notably, several Cox-2 inhibitors (such as
ibuprofen, sulindac sulfide, NS-398 and mefenamic acid) while
inhibiting Cox-2 activity, also enhance its expression, possibly by
binding and activating PPARγ (61). It was demonstrated that
indomethacin and naproxen stimulate cox-2 expression at
concentrations that were shown to activate PPARγ (64). Detailed study of the mechanism of
indomethacin-, flurbiprofen- and NS-398-induced Cox-2 expression
was performed by Pang et al (68). They found that NSAIDs as well as
15d-PGJ2 induced the transcriptional activity of the Cox-2-reporter
construct containing the PPRE, but had no effect on the
Cox-2-reporter construct lacking the PPRE. These results revealed
that stimulation of cox-2 expression by NSAIDs involves
PPARγ activation and provide the first direct evidence that the
PPRE in the promoter is required for NSAID-induced Cox-2
expression.
On the other hand, there are multiple studies
suggesting that Cox-2 activation induced by some PPARγ ligands is
PPARγ-independent. In human synovial fibroblasts treated with both
natural and synthetic PPAR ligands, Cox-2 mRNA and protein
synthesis were up-regulated in a dose-dependent manner. It is
interesting to note that synthetic ligands WY-14,643 and
ciglitazone induce Cox-2 expression via PPAR/PPRE-dependent,
promoter-based transcriptional activation, but 15d-PGJ2 probably
does so by a PPAR-independent mechanism (64). Results obtained by Lee et al
(65) in articular chondrocytes
are in agreement with this observation; PPARγ antagonists do not
block 15d-PGJ2-induced Cox-2 expression. However, not only
15d-PGJ2, but even synthetic PPARγ ligands perform PPAR-independent
cox-2 induction. Troglitazone-induced Cox-2 expression in
human lung epithelial A549 cells was not mediated via PPARγ but via
activation of the ERK and PI3K pathways instead (66). Another signaling transducer
involved in cox-2 up-regulation by PPARγ ligands is MAPK
p38. Both 15d-PGJ2 and synthetic PPARγ ligand GW7845 induced Cox-2
synthesis in the MC615 cartilage cell line. Pretreatment of the
cells with the p38-specific inhibitor repressed expression of Cox-2
induced by both 15d-PGJ2 and GW7845 (69). In neuronal cells, p38 was also
involved in Cox-2 induction by 15d-PGJ2, and again an involvement
of PPARγ was excluded (70). These
findings correspond with the fact, that p38 is an activator of
NF-κB during inflammation and cox-2 belongs among
theNF-κB-regulated genes (71,72).
This suggests a possible signaling pathway leading to Cox-2
up-regulation by 15d-PGJ2 without PPARγ participation.
There are also studies reporting that PPARγ ligands
have two opposing effects on cox-2 expression. Although
NSAIDs can increase the basal Cox-2 level, they inhibit
cytokine-induced cox-2 expression. For example, flufenamic
acid inhibits lipopolysaccharide (LPS)- and tumor necrosis factor α
(TNFα)-induced cox-2 expression in RAW 264.7 and HT-29
cells, whereas it induces cox-2 expression in the absence of
LPS or TNFα. However, the inhibitory effect of NSAIDs on
cytokine-induced cox-2 expression is mediated rather via
NF-κB inhibition than PPARγ activation, while NSAID-induced
cox-2 expression is mediated through signaling pathways that
do not require the activation of MAPKs and NF-κB, but might involve
activation of PPARγ (73). Not
only NSAIDs but also endogenous PPARγ ligand 15d-PGJ2 inhibits
IL-β-induced Cox-2 up-regulation. Also in this case, Cox-2
down-regulation is mediated by NF-κB inhibiton but not by PPARγ
activation (74).
However, in cells with overexpressed and
constitutively active Cox-2, some PPARγ activators can inhibit
cox-2 expression as well (75,76).
It is notable that some studies proved PPARγ involvement in Cox-2
down-regulation (77), while
others described Cox-2 down-regulation as a PPARγ-independent
phenomenon (76). Hazra and
Dubinett (76) used dominant
negative PPARγ to show that ciglitazone decreases cox-2
promoter activity in a PPARγ-independent manner. On the other hand,
Bren-Mattison et al (77)
showed that PPARγ overexpression suppresses cox-2
transcription. This discrepancy is explained by the fact that Cox-2
is not down-regulated due to PPARγ trans-repressing effect but due
to the inhibition of some other transcription factors such as NF-κB
or C/EBP. The cox-2 gene is under the control of NF-κB and
is negatively regulated by various PPARγ ligands via either
PPARγ-dependent or -independent repression of NF-κB (17). PPARγ can inhibit NF-κB by
stimulation of IκB transcription (78). PPARγ-induced IκB synthesis accounts
for at least some of the anti-inflammatory effects of PPARγ ligands
(79–81). 15d-PGJ2 can inhibit NF-κB
independently of PPARγ as well, either by inhibiting the IκB
kinase, therefore preventing IκB phosphorylation and degradation
(82,83), or directly by interacting with
NF-κB (84).
In conclusion, 15d-PGJ and some synthetic PPARγ
ligands can down-regulate the cytokine-stimulated and in some cases
unstimulated cox-2 expression through inhibition of NF-κB or
other transcription factors which can occur either via
PPARγ-dependent or PPARγ-indepedent mechanisms (Fig. 2B).
Despite the facts disclosed in the previous sections
documenting the complex and somewhat ambivalent interplay between
Cox-2 and PPARγ pathways, several studies indicate a possible
coordinated effects of Cox-2 inhibitors and PPARγ activators and
suggest the combined treatment as a promising therapeutic
strategy.
Simultaneous targeting of Cox-2 and PPARγ was found
to result in the synergistic inhibition of mammary cancer
development (85). Treatment of
MDA-MB-231 breast cancer cells with NS-398 (a Cox-2 inhibitor) or
ciglitazone (a PPARγ ligand) inhibited cell proliferation and
markedly increased rates of apoptosis. Compared to using both
agents separately, combined treatment resulted in the synergistic
inhibition of cell proliferation and induction of apoptosis. Thus,
the combinatorial targeting of Cox-2 and PPARγ possesses a stronger
anti-neoplastic effect in vitro than targeting each molecule
separately (86). This result was
confirmed with a different combination of the Cox-2 inhibitor
(celecoxib) and PPARγ agonist (F-L-Leu) in animal breast cancer
models (87,88). Celecoxib and F-L-Leu cooperated in
the growth inhibition of a mouse mammary adenocarcinoma cell
(MMAC-1) line in vitro. In mice the combined diet of
celecoxib and F-L-Leu delayed the median age of death due to
mammary tumors more effectively than celecoxib alone (88).
Breast cancer is not the only possible candidate for
combinatorial therapy with Cox-2 inhibitors and PPARγ ligands, as
the combination of NS-398 and rosiglitazone exerted synergistic
effects in the inhibition of proliferation and induction of
apoptosis of human pancreatic carcinoma cells as well (89). Narayanan et al (90) showed that low doses of celecoxib in
combination with DHA which functions as a PPAR ligand in prostate
cancer cells could be a highly promising strategy for prostate
cancer chemoprevention while minimizing undesired side effects.
Combined treatment with DHA and celecoxib increased PPARγ
expression and activity, decreased the Cox-2 level, inhibited cell
growth and induced apoptosis more efficiently than each agent
alone.
There is cross-talk between the Cox-2- and
PPARγ-driven pathways. An inverse correlation between Cox-2 and
PPARγ expression/activity was demostrated to occur in various types
of human cancers, and it significantly affects carcinogenesis
(22,23,91,92);
the weaker the expression of PPARγ, the higher the level of
Cox-2/PGE2 and the more tumor development progresses (23,93).
Inhibition of Cox-2 and activation of PPARγ prevent cancer growth
in vitro and in vivo. There is now strong evidence
documenting that both Cox-2 inhibitors and PPARγ agonists exert
their anti-tumor effects not only via their respective targets,
Cox-2 and PPARγ. Various Cox-2-independent anti-inflammatory and
anti-neoplastic effects of NSAIDs can be mediated via PPARγ
activation (60), and Cox-2
suppression might be responsible for the anti-cancer effects of
PPARγ ligands (77). Combined
treatment with both classes of agents can exert an additive, if not
synergistic, inhibition in human cancer (87). However, the interplay between these
systems is very complex. Several components of the Cox-2 metabolic
pathway regulate PPARγ activity, and PPARγ ligands modulate
cox-2 expression, both positively and negatively, both in
PPARγ-dependent and PPARγ-independent manners. Although several
studies have demonstrated the synergistic anti-cancer effects of
PPARγ ligands in combination with Cox-2 inhibitors, particularly in
breast cancer models, further pre-clinical and clinical trials are
required to clarify the role that simultaneous Cox-2 inhibition and
PPARγ activation may play in the treatment of human cancer.
We thank Filip Trčka for drawing the
schemes. This work was supported by grants no. 301/09/1115 and
204/08/H054 of the Czech Science Foundation, MSM0021622415 of the
Ministry of Education, Youth and Sports of the Czech Republic and
MUNI/0099/2009 of Masaryk University.
|
1.
|
Zha S, Yegnasubramanian V, Nelson WG,
Isaacs WB and De Marzo AM: Cyclooxygenases in cancer: progress and
perspective. Cancer Lett. 215:1–20. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Liao Z, Mason KA and Milas L:
Cyclo-oxygenase-2 and its inhibition in cancer: is there a role?
Drugs. 67:821–845. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Koehne CH and Dubois RN: COX-2 inhibition
and colorectal cancer. Semin Oncol. 31:12–21. 2004. View Article : Google Scholar
|
|
4.
|
Khuder SA and Mutgi AB: Breast cancer and
NSAID use: a meta-analysis. Br J Cancer. 84:1188–1192. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Castelao JE, Yuan JM, Gago-Dominguez M, Yu
MC and Ross RK: Non-steroidal anti-inflammatory drugs and bladder
cancer prevention. Br J Cancer. 82:1364–1369. 2000.PubMed/NCBI
|
|
6.
|
Sooriakumaran P, Langley SE, Laing RW and
Coley HM: COX-2 inhibition: a possible role in the management of
prostate cancer? J Chemother. 19:21–32. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Muscat JE, Chen SQ, Richie JP Jr, Altorki
NK, Citron M, Olson S, Neugut AI and Stellman SD: Risk of lung
carcinoma among users of nonsteroidal antiinflammatory drugs.
Cancer. 97:1732–1736. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Sandler AB and Dubinett SM: COX-2
inhibition and lung cancer. Semin Oncol. 31:45–52. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Steinbach G, Lynch PM, Phillips RK,
Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y,
Fujimura T, Su LK and Levin B: The effect of celecoxib, a
cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N
Engl J Med. 342:1946–1952. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Suzuki R, Yamamoto M, Saka H, Taniguchi H,
Shindoh J, Tanikawa Y, Nomura F, Gonda H, Imaizumi K, Hasegawa Y
and Shimokata K: A phase II study of carboplatin and paclitacel
with meloxicam. Lung Cancer. 63:72–76. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Soriano F, Helfrich B, Chan DC, Heasley
LE, Bunn PA Jr and Chou TC: Synergistic effects of new
chemopreventive agents and conventional cytotoxic agents against
human lung cancer cell lines. Cancer Res. 59:6178–6184.
1999.PubMed/NCBI
|
|
12.
|
Tuettenberg J, Grobholz R, Korn T, Wenz F,
Erber R and Vajkoczy P: Continuous low-dose chemotherapy plus
inhibition of cyclooxygenase-2 as an antiangiogenic therapy of
glioblastoma multiforme. J Cancer Res Clin Oncol. 131:31–40. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Tachimori A, Yamada N, Amano R, Ohira M
and Hirakawa K: Combination therapy of S-1 with selective
cyclooxygenase-2 inhibitor for liver metastasis of colorectal
carcinoma. Anticancer Res. 28:629–638. 2008.PubMed/NCBI
|
|
14.
|
Eichele K, Ramer R and Hinz B: Decisive
role of cyclooxygenase-2 and lipocalin-type prostaglandin D
synthase in chemotherapeutic-induced apoptosis of human cervical
carcinoma cells. Oncogene. 27:3032–3044. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Gilroy DW, Colville-Nash PR, Willis D,
Chivers J, Paul-Clark MJ and Willoughby DA: Inducible
cyclooxygenase may have anti-inflammatory properties. Nat Med.
5:698–701. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Issemann I and Green S: Activation of a
member of the steroid hormone receptor superfamily by peroxisome
proliferators. Nature. 347:645–650. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Nosjean O and Boutin JA: Natural ligands
of PPARγ: Are prostaglandin J2 derivatives really playing the part?
Cell Signal. 14:573–583. 2002.
|
|
18.
|
Bull AW, Steffensen KR, Leers J and Rafter
JJ: Activation of PPAR gamma in colon tumor cell lines by oxidized
metabolites of linoleic acid, endogenous ligands for PPAR gamma.
Carcinogenesis. 24:1717–1722. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Kepez A, Oto A and Dagdelen S: Peroxisome
proliferator-activated receptor-gamma: novel therapeutic target
linking adiposity, insulin resistance and atherosclerosis.
BioDrugs. 20:121–135. 2006.
|
|
20.
|
Chiarelli F and Di Marzio D: Peroxisome
proliferator-activated receptor-gamma agonists and diabetes:
current evidence and future perspectives. Vasc Health Risk Manag.
4:297–304. 2008.PubMed/NCBI
|
|
21.
|
Quinn CE, Hamilton PK, Lockhart CJ and
McVeigh GE: Thiazolidinediones: effects on insulin resistance and
the cardiovascular system. Br J Pharmacol. 153:636–645. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Konstantinopoulos PA, Vandoros GP,
Sotiropoulou-Bonikou G, Kominea A and Papavassiliou AG: NF-κB/PPARγ
and/or AP-1/PPARγ ‘on/off’ switches and induction of CBP in colon
adenocarcinomas: correlation with COX-2 expression. Int J
Colorectal Dis. 22:57–68. 2007.
|
|
23.
|
Badawi AF and Badr MZ: Expression of
cyclooxygenase-2 and peroxisome proliferator-activated
receptor-gamma and levels of prostaglandin E2 and
15-deoxy-delta12,14-prostaglandin J2 in human breast cancer and
metastasis. Int J Cancer. 103:84–90. 2003. View Article : Google Scholar
|
|
24.
|
Nagata D, Yoshihiro H, Nakanishi M,
Naruyama H, Okada S, Ando R, Tozawa K and Kohri K: Peroxisome
proliferator-activated receptor-gamma and growth inhibition by its
ligands in prostate cancer. Cancer Detect Prev. 32:259–266. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Matsuyama M and Yoshimura R: Peroxisome
proliferator-activated receptor-gamma is a potent target for
prevention and treatment in human prostate and testicular cancer.
PPAR Res. 2008:2498492008. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Segawa Y, Yoshimura R, Hase T, Nakatani T,
Wada S, Kawahito Y, Kishimoto T and Sano H: Expression of
peroxisome proliferator-activated receptor (PPAR) in human prostate
cancer. Prostate. 51:108–116. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Sato H, Ishihara S, Kawashima K, Moriyama
N, Suetsugu H, Kazumori H, Okuyama T, Rumi MA, Fukuda R, Nagasue N
and Kinoshita Y: Expression of peroxisome proliferator-activated
receptor (PPAR)gamma in gastric cancer and inhibitory effects of
PPARgamma agonists. Br J Cancer. 83:1394–1400. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Inoue K, Kawahito Y, Tsubouchi Y, Yamada
R, Kohno M, Hosokawa Y, Katoh D, Bishop-Bailey D, Hla T and Sano H:
Expression of peroxisome proliferator-activated receptor
(PPAR)-gamma in human lung cancer. Anticancer Res. 21:2471–2476.
2001.PubMed/NCBI
|
|
29.
|
Kristiansen G, Jacob J, Buckendahl AC,
Grützmann R, Alldinger I, Sipos B, Klöppel G, Bahra M, Langrehr JM,
Neuhaus P, Dietel M and Pilarsky C: Peroxisome
proliferator-activated receptor gamma is highly expressed in
pancreatic cancer and is associated with shorter overall survival
times. Clin Cancer Res. 12:6444–6451. 2006. View Article : Google Scholar
|
|
30.
|
Vignati S, Albertini V, Rinaldi A, Kwee I,
Riva C, Oldrini R, Capella C, Bertoni F, Carbone GM and Catapano
CV: Cellular and molecular consequences of peroxisome
proliferator-activated receptor-gamma activation in ovarian cancer
cells. Neoplasia. 8:851–861. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Jung TI, Baek WK, Suh SI, Jang BC, Song
DK, Bae JH, Kwon KY, Bae JH, Cha SD, Bae I and Cho CH:
Down-regulation of peroxisome proliferator-activated receptor gamma
in human cervical carcinoma. Gynecol Oncol. 97:365–373. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Grommes C, Landreth GE and Heneka MT:
Antineoplastic effects of peroxisome proliferator-activated
receptor gamma agonists. Lancet Oncol. 5:419–429. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Keshamouni VG, Reddy RC, Arenberg DA, Joel
B, Thannickal VJ, Kalemkerian GP and Standiford TJ: Peroxisome
proliferator-activated receptor-gamma activation inhibits tumor
progression in non-small-cell lung cancer. Oncogene. 23:100–108.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Yasui Y, Kim M and Tanaka T: PPAR ligands
for cancer chemoprevention. PPAR Res. 2008:5489192008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Mueller E, Smith M, Sarraf P, Kroll T,
Aiyer A, Kaufman DS, Oh W, Demetri G, Figg WD, Zhou XP, Eng C,
Spiegelman BM and Kantoff PW: Effects of ligand activation of
peroxisome proliferator-activated receptor gamma in human prostate
cancer. Proc Natl Acad Sci USA. 97:10990–10995. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Rumi MA, Ishihara S, Kazumori H, Kadowaki
Y and Kinoshita Y: Can PPAR gamma ligands be used in cancer
therapy? Curr Med Chem Anticancer Agents. 4:465–477. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Lefebvre AM, Chen I, Desreumaux P, Najib
J, Fruchart JC, Geboes K, Briggs M, Heyman R and Auwerx J:
Activation of the peroxisome proliferator-activated receptor gamma
promotes the development of colon tumors in C57BL/6J-APCMin/+ mice.
Nat Med. 4:1053–1057. 1998.PubMed/NCBI
|
|
38.
|
Saez E, Tontonoz P, Nelson MC, Alvarez JG,
Ming UT, Baird SM, Thomazy VA and Evans RM: Activators of the
nuclear receptor PPARgamma enhance colon polyp formation. Nat Med.
4:1058–1061. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Saez E, Rosenfeld J, Livolsi A, Olson P,
Lombardo E, Nelson M, Banayo E, Cardiff RD, Izpisua-Belmonte JC and
Evans RM: PPAR gamma signaling exacerbates mammary gland tumor
development. Genes Dev. 18:528–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Eibl G: The role of PPAR-gamma and its
interaction with COX-2 in pancreatic cancer. PPAR Res.
2008:3269152008. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Kawashima A, Harada T, Imada K, Yano T and
Mizuguchi K: Eicosapentaenoic acid inhibits interleukin-6
production in interleukin-1beta-stimulated C6 glioma cells through
peroxisome proliferator-activated receptor-gamma. Prostaglandins
Leukot Essent Fatty Acids. 79:59–65. 2008. View Article : Google Scholar
|
|
42.
|
Allred CD, Talbert DR, Southard RC, Wang X
and Kilgore MW: PPARgamma1 as a molecular target of
eicosapentaenoic acid in human colon cancer (HT-29) cells. J Nutr.
138:250–256. 2008.PubMed/NCBI
|
|
43.
|
Straus DS and Glass CK: Cyclopentenone
prostaglandins: new insights on biological activities and cellular
targets. Med Res Rev. 21:185–210. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Matsuoka T and Narumiya S: Prostaglandin
receptor signaling in disease. ScientificWorldJournal. 7:1329–1347.
2007. View Article : Google Scholar
|
|
45.
|
Castellone MD, Teramoto H, Williams BO,
Druey KM and Gutkind JS: Prostaglandin E2 promotes colon cancer
cell growth through a Gs-axin-beta-catenin signaling axis. Science.
310:1504–1510. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Chan TA: Prostaglandins and the colon
cancer connection. Trends Mol Med. 12:240–244. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Scher JU and Pillinger MH: 15d-PGJ2: the
anti-inflammatory prostaglandin? Clin Immunol. 114:100–109. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Shimada T, Kojima K, Yoshiura K, Hiraishi
H and Terano A: Characteristics of the peroxisome proliferator
activated receptor gamma (PPARgamma) ligand-induced apoptosis in
colon cancer cells. Gut. 50:658–664. 2002. View Article : Google Scholar
|
|
49.
|
Millan O, Rico D, Peinado H, Zarich N,
Stamatakis K, Pérez-Sala D, Rojas JM, Cano A and Boscá L:
Potentiation of tumor formation by topical administration of
15-deoxy-delta12,14-prostaglandin J2 in a model of skin
carcinogenesis. Carcinogenesis. 27:328–336. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50.
|
Chinery R, Coffey RJ, Graves-Deal R,
Kirkland SC, Sanchez SC, Zackert WE, Oates JA and Morrow JD:
Prostaglandin J2 and 15-deoxy-delta12,14-prostaglandin J2 induce
proliferation of cyclooxygenase-depleted colorectal cancer cells.
Cancer Res. 59:2739–2746. 1999.PubMed/NCBI
|
|
51.
|
Reginato MJ, Krakow SL, Bailey ST and
Lazar MA: Prostaglandins promote and block adipogenesis through
opposing effects on peroxisome proliferator-activated receptor
gamma. J Biol Chem. 273:1855–1858. 1998. View Article : Google Scholar
|
|
52.
|
Padilla J, Kaur K, Cao HJ, Smith TJ and
Phipps RP: Peroxisome proliferator activator receptor-gamma
agonists and 15-deoxy-delta(12,14)(12,14)-PGJ(2) induce apoptosis
in normal and malignant B-lineage cells. J Immunol. 165:6941–6948.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
53.
|
Jaradat MS, Wongsud B, Phornchirasilp S,
Rangwala SM, Shams G, Sutton M, Romstedt KJ, Noonan DJ and Feller
DR: Activation of peroxisome proliferator-activated receptor
isoforms and inhibition of prostaglandin H(2) synthases by
ibuprofen, naproxen and indomethacin. Biochem Pharmacol.
62:1587–1595. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54.
|
Lehmann JM, Lenhard JM, Oliver BB, Ringold
GM and Kliewer SA: Peroxisome proliferator-activated receptors
alpha and gamma are activated by indomethacin and other
non-steroidal anti-inflammatory drugs. J Biol Chem. 272:3406–3410.
1997. View Article : Google Scholar
|
|
55.
|
López-Parra M, Clària J, Titos E,
Planagumà A, Párrizas M, Masferrer JL, Jiménez W, Arroyo V, Rivera
F and Rodés J: The selective cyclooxygenase-2 inhibitor celecoxib
modulates the formation of vasoconstrictor eicosanoids and
activates PPARgamma. Influence of albumin. J Hepatol. 42:75–81.
2005.PubMed/NCBI
|
|
56.
|
Diamond MP and Saed G: Modulation of the
expression of peroxisome proliferator-activated receptors in human
fibroblasts. Fertil Steril. 87:706–709. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57.
|
Shaik MS, Chatterjee A and Singh M: Effect
of a selective cyclooxygenase-2 inhibitor, nimesulide, on the
growth of lung tumors and their expression of cyclooxygenase-2 and
peroxisome proliferator-activated receptor-gamma. Clin Cancer Res.
10:1521–1529. 2004. View Article : Google Scholar
|
|
58.
|
Yamazaki R, Kusunoki N, Matsuzaki T,
Hashimoto S and Kawai S: Nonsteroidal anti-inflammatory drugs
induce apoptosis in association with activation of peroxisome
proliferator-activated receptor gamma in rheumatoid synovial cells.
J Pharmacol Exp Ther. 302:18–25. 2002. View Article : Google Scholar
|
|
59.
|
Cui W, Yu CH and Hu KQ: In vitro and in
vivo effects and mechanisms of celecoxib-induced growth inhibition
of human hepatocellular carcinoma cells. Clin Cancer Res.
11:8213–8221. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
60.
|
Nagahara T, Okano J and Murawaki Y:
Mechanisms of anti-proliferative effect of JTE-522, a selective
cyclooxygenase-2 inhibitor, on human liver cancer cells. Oncol Rep.
18:1281–1290. 2007.PubMed/NCBI
|
|
61.
|
Meade EA, McIntyre TM, Zimmerman GA and
Prescott SM: Peroxisome proliferators enhance cyclooxygenase-2
expression in epithelial cells. J Biol Chem. 274:8328–8334. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
62.
|
Pontsler AV, St Hilaire A, Marathe GK,
Zimmerman GA and McIntyre TM: Cyclooxygenase-2 is induced in
monocytes by peroxisome proliferator activated receptor gamma and
oxidized alkyl phospholipids from oxidized low density lipoprotein.
J Biol Chem. 277:13029–13036. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63.
|
Bishop-Bailey D and Warner TD: PPARgamma
ligands induce prostaglandin production in vascular smooth muscle
cells: indomethacin acts as a peroxisome proliferator-activated
receptor-gamma antagonist. FASEB J. 17:1925–1927. 2003.
|
|
64.
|
Kalajdzic T, Faour WH, He QW, Fahmi H,
Martel-Pelletier J, Pelletier JP and Di Battista JA: Nimesulide, a
preferential cyclooxygenase 2 inhibitor, suppresses peroxisome
proliferator-activated receptor induction of cyclooxygenase 2 gene
expression in human synovial fibroblasts: evidence for receptor
antagonism. Arthritis Rheum. 46:494–506. 2002. View Article : Google Scholar
|
|
65.
|
Lee JH, Yu SM, Yoon EK, Lee WK, Jung JC
and Kim SJ: 15-Deoxy-delta 12,14-prostaglandin J2 regulates
dedifferentiation through peroxisome proliferator-activated
receptor-gamma-dependent pathway but not COX-2 expression in
articular chondrocytes. J Korean Med Sci. 22:891–897. 2007.
View Article : Google Scholar
|
|
66.
|
Patel KM, Wright KL, Whittaker P,
Chakravarty P, Watson ML and Ward SG: Differential modulation of
COX-2 expression in A549 airway epithelial cells by structurally
distinct PPAR(gamma) agonists: evidence for disparate functional
effects which are independent of NF-(kappa)B and PPAR(gamma). Cell
Signal. 17:1098–1110. 2005. View Article : Google Scholar
|
|
67.
|
Chêne G, Dubourdeau M, Balard P,
Escoubet-Lozach L, Orfila C, Berry A, Bernad J, Aries MF, Charveron
M and Pipy B: n-3 and n-6 polyunsaturated fatty acids induce the
expression of COX-2 via PPARgamma activation in human keratinocyte
HaCaT cells. Biochim Biophys Acta. 1771:576–589. 2007.PubMed/NCBI
|
|
68.
|
Pang L, Nie M, Corbett L and Knox AJ:
Cyclooxygenase-2 expression by nonsteroidal anti-inflammatory drugs
in human airway smooth muscle cells: role of peroxisome
proliferator-activated receptors. J Immunol. 170:1043–1051. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
69.
|
Ulivi V, Cancedda R and Cancedda FD:
15-Deoxy-delta 12,14-prostaglandin J(2) inhibits the synthesis of
the acute phase protein SIP24 in cartilage: involvement of COX-2 in
resolution of inflammation. J Cell Physiol. 217:433–441. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
70.
|
Li Z, Jansen M, Ogburn K, Salvatierra L,
Hunter L, Mathew S and Figueiredo-Pereira ME: Neurotoxic
prostaglandin J2 enhances cyclooxygenase-2 expression in neuronal
cells through the p38MAPK pathway: a death wish? J Neurosci Res.
78:824–836. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
71.
|
Ulivi V, Giannoni P, Gentili C, Cancedda R
and Descalzi F: p38/NF-κB-dependent expression of COX-2 during
differentiation and inflammatory response of chondrocytes. J Cell
Biochem. 104:1393–1406. 2008.PubMed/NCBI
|
|
72.
|
Tsatsanis C, Androulidaki A, Venihaki M
and Margioris AN: Signalling networks regulating cyclooxygenase-2.
Int J Biochem Cell Biol. 38:1654–1661. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73.
|
Paik JH, Ju JH, Lee JY, Boudreau MD and
Hwang DH: Two opposing effects of non-steroidal anti-inflammatory
drugs on the expression of the inducible cyclooxygenase. Mediation
through different signaling pathways. J Biol Chem. 275:28173–28179.
2000.
|
|
74.
|
Boyault S, Simonin MA, Bianchi A, Compe E,
Liagre B, Mainard D, Becuwe P, Dauca M, Netter P, Terlain B and
Bordji K: 15-Deoxy-Δ12;14-PGJ2, but not troglitazone, modulates
IL-1β effects in human chondrocytes by inhibiting NF-κB and AP-1
activation pathways. FEBS Lett. 501:24–30. 2001.
|
|
75.
|
Liu JJ, Liu PQ, Lin DJ, Xiao RZ, Huang M,
Li XD, He Y and Huang RW: Downregulation of cyclooxygenase-2
expression and activation of caspase-3 are involved in peroxisome
proliferator-activated receptor-gamma agonists induced apoptosis in
human monocyte leukemia cells in vitro. Ann Hematol. 86:173–183.
2007. View Article : Google Scholar
|
|
76.
|
Hazra S and Dubinett SM: Ciglitazone
mediates COX-2 dependent suppression of PGE2 in human non-small
cell lung cancer cells. Prostaglandins Leukot Essent Fatty Acids.
77:51–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
77.
|
Bren-Mattison Y, Meyer AM, van Putten V,
Li H, Kuhn K, Stearman R, Weiser-Evans M, Winn RA, Heasley LE and
Nemenoff RA: Antitumorigenic effects of peroxisome
proliferator-activated receptor-gamma in non-small cell lung cancer
cells are mediated by suppression of cyclooxygenase-2 via
inhibition of nuclear factor-kappaB. Mol Pharmacol. 73:709–717.
2008. View Article : Google Scholar
|
|
78.
|
Delerive P, Gervois P, Fruchart JC and
Staels B: Induction of IkappaBalpha expression as a mechanism
contributing to the anti-inflammatory activities of peroxisome
proliferator-activated receptor-alpha activators. J Biol Chem.
275:36703–36707. 2000. View Article : Google Scholar
|
|
79.
|
Moraes LA, Piqueras L and Bishop-Bailey D:
Peroxisome proliferator-activated receptors and inflammation.
Pharmacol Ther. 110:371–385. 2006. View Article : Google Scholar
|
|
80.
|
Wahli W: A gut feeling of the PXR, PPAR
and NF-κB connection. J Intern Med. 263:613–619. 2008.PubMed/NCBI
|
|
81.
|
Ricote M, Li AC, Willson TM, Kelly CJ and
Glass CK: The peroxisome proliferator-activated receptor-γ is a
negative regulator of macrophage activation. Nature. 391:79–82.
1998.
|
|
82.
|
Rossi A, Kapahi P, Natoli G, Takahashi T,
Chen Y, Karin M and Santoro MG: Anti-inflammatory cyclopentenone
prostaglandins are direct inhibitors of IkappaB kinase. Nature.
403:103–108. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
83.
|
Ackerman WE, Zhang XL, Rovin BH and Kniss
DA: Modulation of cytokine-induced cyclooxygenase 2 expression by
PPARG ligands through NFκB signal disruption in human WISH and
amnion cells. Biol Reprod. 73:527–535. 2005.PubMed/NCBI
|
|
84.
|
Straus DS, Pascual G, Li M, Welch JS,
Ricote M, Hsiang CH, Sengchanthalangsy LL, Ghosh G and Glass CK:
15-Deoxy-delta 12,14-prostaglandin J2 inhibits multiple steps in
the NF-κB signaling pathway. Proc Natl Acad Sci USA. 97:4844–4849.
2000.
|
|
85.
|
Badawi AF and Badr MZ: Chemoprevention of
breast cancer by targeting cyclooxygenase-2 and peroxisome
proliferator-activated receptor-γ. Int J Oncol. 20:1109–1122.
2002.
|
|
86.
|
Michael MS, Badr MZ and Badawi AF:
Inhibition of cyclooxygenase-2 and activation of peroxisome
proliferator-activated receptor-γ synergistically induces apoptosis
and inhibits growth of human breast cancer cells. Int J Mol Med.
11:733–736. 2003.
|
|
87.
|
Badawi AF, Eldeen MB, Liu Y, Ross EA and
Badr MZ: Inhibition of rat mammary gland carcinogenesis by
simultaneous targeting of cyclooxygenase-2 and peroxisome
proliferator-activated receptor gamma. Cancer Res. 64:1181–1189.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
88.
|
Mustafa A and Kruger WD: Suppression of
tumor formation by a cyclooxygenase-2 inhibitor and a peroxisome
proliferator-activated receptor gamma agonist in an in vivo mouse
model of spontaneous breast cancer. Clin Cancer Res. 14:4935–4942.
2008. View Article : Google Scholar
|
|
89.
|
Sun WH, Chen GS, Ou XL, Yang Y, Luo C,
Zhang Y, Shao Y, Xu HC, Xiao B, Xue YP, Zhou SM, Zhao QS and Ding
GX: Inhibition of COX-2 and activation of peroxisome
proliferator-activated receptor gamma synergistically inhibits
proliferation and induces apoptosis of human pancreatic carcinoma
cells. Cancer Lett. 275:247–255. 2009. View Article : Google Scholar
|
|
90.
|
Narayanan NK, Narayanan BA and Reddy BS: A
combination of docosahexaenoic acid and celecoxib prevents prostate
cancer cell growth in vitro and is associated with
modulation of nuclear factor-κB, and steroid hormone receptors. Int
J Oncol. 26:785–792. 2005.PubMed/NCBI
|
|
91.
|
Gustafsson A, Hansson E, Kressner U,
Nordgren S, Andersson M, Wang W, Lönnroth C and Lundholm K: EP1-4
subtype, COX and PPAR gamma receptor expression in colorectal
cancer in prediction of disease-specific mortality. Int J Cancer.
121:232–240. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92.
|
Hazra S, Peebles KA, Sharma S, Mao JT and
Dubinett SM: The role of PPARgamma in the cyclooxygenase pathway in
lung cancer. PPAR Res. 2008:7905682008. View Article : Google Scholar : PubMed/NCBI
|
|
93.
|
Sasaki H, Tanahashi M, Yukiue H, Moiriyama
S, Kobayashi Y, Nakashima Y, Kaji M, Kiriyama M, Fukai I, Yamakawa
Y and Fujii Y: Decreased peroxisome proliferator-activated receptor
gamma gene expression was correlated with poor prognosis in
patients with lung cancer. Lung Cancer. 36:71–76. 2002. View Article : Google Scholar
|