Introduction
Cigarette smoking is widely recognized as a primary
risk factor for cardiovascular diseases. For the year 2000, it was
estimated that >10% of cardiovascular mortality cases worldwide
were attributable to smoking (1).
The accumulated evidence of previous studies suggests that
cigarette smoking results in atherosclerosis, which is the main
pathological characteristic of smoking-associated peripheral
vascular diseases, coronary heart diseases, cerebral vascular
diseases and aortic aneurysm (2,3).
Abnormal proliferation of vascular smooth muscle cells (VSMCs) is
considered to be an important event in the development of
atherosclerosis (4); however, the
pathogenesis of cigarette smoking-induced VSMC proliferation
remains unclear.
Cell cycle progression is vital to cell
proliferation (5) and is a highly
regulated process involving a complex cascade of events, including
the activation of cyclin-dependent kinases (CDKs) (6). For example, extracellular signals
induce cyclin D1 expression, which activates CDK4 and CDK6
(7). These activated CDKs induce
phosphorylation of retinoblastoma protein (Rb), causing it to
dissociate from transcription factors such as members of the E2F
family, which then activate various genes associated with cell
cycle progression to S phase (8).
Therefore, CDK4 and CDK6 serve a crucial role in regulating cell
cycle progression from G1 phase to the S phase.
P16 is a CDK inhibitor, which inhibits CDK4 and CDK6
activation and their downstream Rb-E2F signaling, thereby
preventing cell cycle progression from G1 to S phase (9). Previous studies have demonstrated that
P16 serves an important role in the regulation of VSMC
proliferation; for example, P16 was found to participate in the
process of peroxisome proliferator-activated receptor (PPAR)
α-inhibited VSMC proliferation (10,11).
Additionally, it has been demonstrated that various genetic and
epigenetic abnormalities, including mutations and hypermethylation,
may be associated with the development and progression of
atherosclerosis (12). Epigenetic
silencing of some key genes, such as P16, appears to be a critical
event in the development of cardiovascular diseases (13). However, the roles of P16 in cigarette
smoking-induced proliferation of VSMCs remain to be fully
elucidated; therefore, in the present study, the expression of P16
and its downstream signal molecules, and its roles in the
proliferation of cigarette smoke extract (CSE)-treated VSMCs were
investigated.
Materials and methods
Cell Culture
Human aortic smooth muscle cell (HAOSMC) line was
obtained from the Shanghai Institute for Biological Sciences,
Chinese Academy of Sciences (Shanghai, China) and maintained in
Dulbecco's Modified Eagle Medium (DMEM; MP Biomedicals, LLC, Santa
Ana, CA, USA) 10% fetal bovine serum supplemented with 100 U/ml
penicillin and 100 µg/ml streptomycin at 37°C in an atmosphere
containing 5% CO2. A total of 2 ml HAOSMCs
(2×105 cells/ml) were seeded in 6-well plates. HASMCs
were incubated in DMEM containing 0.2% bovine serum albumin (Wuhan
Huamei Biotech Co., Ltd., Wuhan, China) at 37°C for 24 h, at which
point the cells had reached ~80% confluence.
CSE preparation
CSE was prepared according to the method previously
described with a few modifications (12). Briefly, cigarettes (Fu-rong; Tobacco
Hunan Industrial Corporation, Changsha, China) without filters were
combusted and the smoke was passed through a filter to remove
particles and bacteria. Smoke was subsequently collected in a tube
containing PBS (1 ml per cigarette) using a vacuum pump. The
CSE-PBS solution was freshly prepared for each set of
experiments.
Measurement of P16 promoter CpG island
methylation status by bisulfite genomic sequencing polymerase chain
reaction (PCR)
Genomic DNA was extracted from HAOSMCs with or
without CSE treatment by using a DNA Extraction kit according to
the manufacturer's protocols (Takara Biotechnology Co., Dalian,
China). After identification and quantification by UV
spectrophotometry, genomic DNA (1 µg per sample) was modified with
bisulfite using a CpGenome™ DNA Modification kit (Qiagen China Co.,
Ltd., Shanghai, China) according to the manufacturer's protocol,
and the modified DNA was amplified using the following primers:
P16, forward 5′-TTTTGTTTTTTAAATTTTTTGGAGG-3′ and reverse
5′-AAACCCAATCCTCCTTCCTTAC-3′. The PCR amplification conditions were
as follows: 94°C for 3 min, followed by 35 cycles of 94°C for 30
sec, 55°C for 30 sec and 72°C for 30 sec, and a final extension at
72°C for 7 min. The PCR products were cloned into a T-vector and
transformed into Escherichia coli cells (DH5α; MiaoLing
Biological Technology Co., Ltd., Wuhan, China). Subsequently, the
E. coli cells were inoculated in 100 µg/ml Ampicillin+
LB agar plates, incubated at 37°C for 12–16 h and then five
independent clones were sequenced for the amplified fragment by
Shenzhen Huada Gene Technology Co., Ltd. (Shenzhen, China). The
demethylation rate of the CpG pairs in the HAOSMC with or without
of 5 µmol/l 5-Zac was calculated from the sequencing results.
Luciferase reporter gene assays
The P16 promoter region was ligated into the
pGL3-enhancer vector (Promega Corporation, Madison, WI, USA)
generating pGL3-basic-P16 (pGL3-P16), performed by Shanghai
GenePharma Co., Ltd. (Shanghai, China). All vector sequences were
confirmed by DNA sequencing. Cells were transfected with the
pGL3-P16 reporter plasmid, stored at 4°C for 24 h and treated with
2.5% CSE or co-treated with 2.5% CSE and 15 nM 5′-Aza-deoxycytidine
(5-Aza; Sigma-Aldrich; Merck Millipore, Darmstadt, Germany) and
stored at 4°C for 24 h. Normal control (NC) cells were treated with
0.1 M PBS. Cells were subsequently collected and total proteins
were isolated using lysis buffer (Beyotime Institute of
Biotechnology, Haimen, China). Lysates were separated by
centrifugation at 12,000 × g for 10 min at 4°C. Then, 10 ml of the
supernatants were used to detect luciferase activities using
luciferase reporter gene assay kit (BioVision, Inc., Milpitas, CA,
USA) on a microplate luminometer.
Cell transfection and small
interfering (si)RNA
P16INK4a siRNA, P16INK4a overexpression plasmid and
transfected reagents were purchased from Shanghai GenePharma Co.,
Ltd. For the transfections, HAOSMCs were seeded onto 6-well plates
and allowed to reach 60% confluence on the day of transfections.
Transfections were performed with a Lipofectamine® 2000
kit (Shanghai GenePharma Co., Ltd.) according to the manufacturer's
protocols. A total of 50 nM empty plasmid, expression plasmid,
P16INK4a overexpression plasmid (GenePharma Biotechnology) or
P16INK4a siRNA were transfected into HAOSMCs. The siRNA sequence
used was as follows: Forward, 5′-CCCAACGCACCGAAUAGUUTT-3′ and
reverse, 5′-AACUAUUCGGUGCGUUGGGTT−3′. Cells were harvested 48 h
post-transfection.
MTT assay
Cell growth was determined using MTT kits (Beyotime
Institute of Biotechnology) according to published protocols
(14). HAOSMCs were re-seeded into
96-well plates (2×103 cells/well) following 24 h of
treatment with different concentrations (0, 1, 2.5, 5 and 10%) of
CSE and continuously cultured for one, two, three or four days.
Absorbance at 490 nm was measured using a plate reader (Thermo
Fisher Scientific, Inc., Waltham, MA, USA).
Ethynyl deoxyuridine (EdU)
incorporation analysis
Cell proliferation was measured using an EdU assay
kit (Wuhan Huamei Biotech Co., Ltd.), according to the
manufacturer's protocols. Briefly, HAOSMCs were seeded at
4×103-1×105 cells per well in 96-well plates
in triplicate and transfected with 50 nM P16 overexpression vector,
P16 siRNA expression vector or their corresponding control,
respectively. Cells were stored for 48 h and incubated with 50 µM
EdU for 4 h at 37°C, followed by 4% formaldehyde for 15 min at room
temperature. Cells were subsequently permeabilized with 0.5% Triton
X-100 for 20 min at room temperature, washed three times with PBS
and treated with 100 µl/well of 1x Apollop reaction cocktail (Wuhan
Huamei Biotech Co., Ltd.) at room temperature for 30 min. DNA was
stained with 100 µl of Hoechst 33342 (5 µg/ml) at 4°C for 30 min
and visualized using a fluorescence microscope (Olympus
Corporation, Tokyo, Japan).
Flow cytometry
Briefly, ~3×106 cells were seeded in
6-well plates, stored at 4°C for 14 h and treated with 2.5% CSE
with or without P16 expression plasmid or P16 siRNA plasmid. Cells
were harvested at 8 h post-transfection and fixed in 70% ice-cold
ethanol at 4°C for 24 h. Following the treatment, cultured cells
were trypsinized and washed with PBS twice, and subsequently
permeabilized with 0.05% Triton X-100 in PBS for 10 min. The
permeabilized cells were stained with 10 µg/ml propidium iodide
solution for 10 min in a dark box. Finally, the different phases of
the cell cycle were analyzed using FACS analysis (BD FACSCalibur;
BD Biosciences, Franklin Lakes, NJ, USA).
Western blot analysis
Cell lysates were prepared as previously described
(15). A total of 20 µg protein per
lane was separated on a 12% SDS-PAGE gel and transferred onto
polyvinylidene fluoride membranes. Membranes were blocked with
Tris-buffered saline (10 mM Tris, pH 7.5; 100 mM NaCl) plus 0.1%
Tween-20 containing 5% dry skimmed milk for 45 min and incubated
with primary antibodies of CDK4 (PA11659A0Rb, 1:200), CDK6
(PA564539, 1:100), P16INK (PA051713, 1:1,000), p-Rb (PA201739,
1:1,000) and GAPDH (PA00025C0Rb, 1:1,000) (all from Wuhan Huamei
Biotech Co., Ltd.) overnight at 4°C. Membranes were washed three
times with PBS and incubated with horseradish peroxidase-conjugated
goat anti-rabbit secondary antibody (MA000071M0m, 1:4,000; Wuhan
Huamei Biotech Co., Ltd.) for 2 h at room temperature. The
immunoreaction was visualized using the enhanced chemiluminescence
reagents. CDK4, CDK6, p-Rb and P16INK bands were quantified by
scanning densitometry using the Molecular Imager ChemiDoc X-Ray
Spectroscopy System (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
and normalized to GAPDH. The experiment was performed in three
replicates.
Statistical analysis
Data were analyzed using SPSS v.17.0 software
package (SPSS Inc., Chicago, IL, USA). Pearson's correlation was
used to evaluate the association between P16 expression and cell
proliferation. One-way analysis of variance was used to analyze
measurement data and a least significant differences t-test was
used for comparisons between groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
CSE treatment promotes the viability
and proliferation of HASMCs and decreases the P16 protein
expression in HAOSMCs
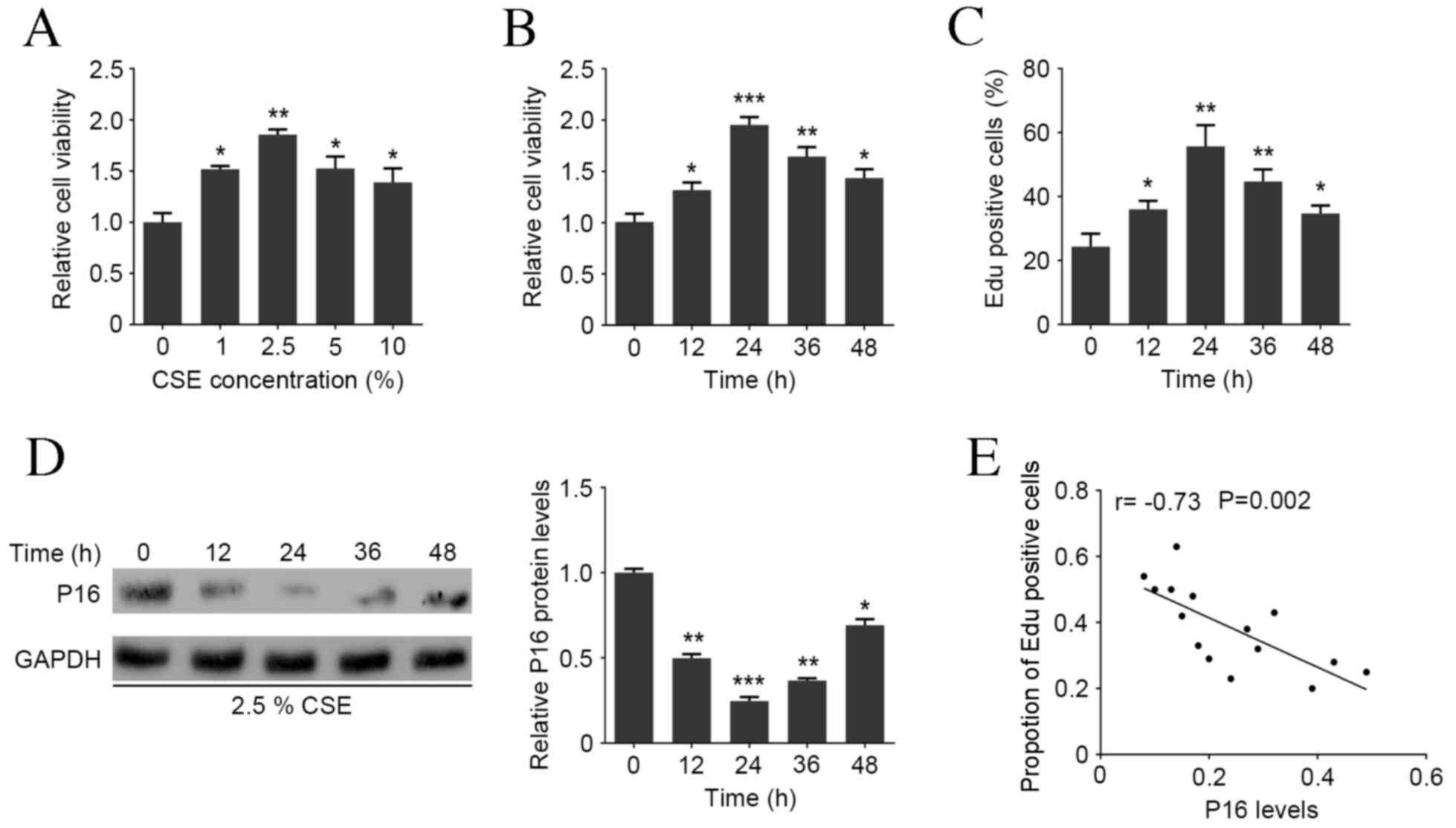
MTT assay revealed that the viability of HAOSMCs
significantly increased following treatment with 1–10% CSE compared
with the NC group, with a peak at 2.5% CSE (P<0.01; Fig. 1A). HAOSMCs were treated with 2.5% CSE
for 0, 12, 24, 36 or 48 h and MTT assay demonstrated that this
significantly induced cell viability with a peak at 24 h
(P<0.001; Fig. 1B). Additionally,
the Edu assay demonstrated that CSE treatment was able to
significantly increase the proportion of Edu positive cells
compared with the NC group, with a peak at 24 h (P<0.01;
Fig. 1C). Western blot analysis
demonstrated that P16 protein expression was markedly decreased
following 12 h 2.5% CSE treatment, with the largest decrease at 24
h (P<0.001; Fig. 1D). Pearson's
correlation analysis demonstrated that P16 expression was
negatively correlated with the ratio of Edu positive cells in
HAOSMCs at all time points (P=0.002; Fig. 1E), indicating that CSE induces HAOSMC
proliferation in a dose- and time-dependent manner and is
associated with P16 expression.
CSE treatment induces P16 promoter
hypermethylation in HAOSMCs
To investigate whether P16 silencing in CSE-treated
HAOSMCs was caused by promoter methylation, genomic DNA from was
extracted from HAOSMCs treated with or without CSE to perform
bisulfite genomic sequencing polymerase chain reaction. It was
demonstrated that the P16 promoter was hypermethylated in HAOSMCs
treated with CSE, particularly at the sites −786, −676, −31, +7 and
+32 (Fig. 2A). Additionally, it was
demonstrated that CSE was able to significantly reduce P16 promoter
activity, and this effect was significantly reversed by
demethylation agent 5-Aza (both P<0.01; Fig. 2B). Furthermore, CSE treatment was
able to markedly reduce P16 expression, whereas CDK4, CDK6 and p-Rb
expressions were markedly increased (Fig. 2C). Treatment with 5-Aza counteracted
the CSE mediated-downregulation of P16 protein, and reversed the
increased protein levels of CDK4, CDK6 and p-Rb (Fig. 2C). These results indicate that the
downregulation of P16 in HAOSMCs treated with CSE is associated
with its promoter hypermethylation.
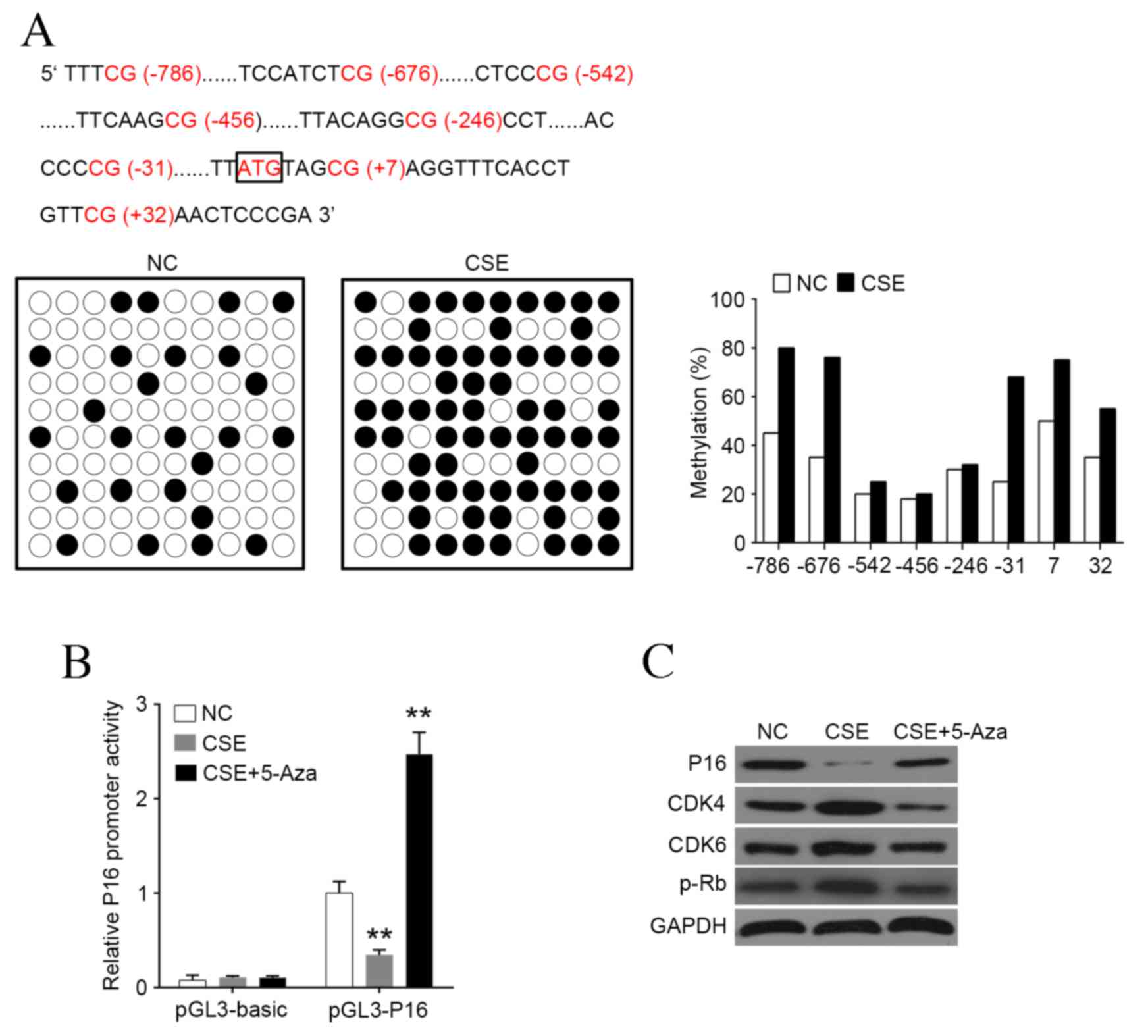 | Figure 2.CSE induces hypermethylation of P16
promoter. (A) P16 promoter was hypermethylated by CSE treatment,
most markedly at sites −786, −676, −31, +7 and +32. (B) Luciferase
reporter gene assay was used to detect the promoter activity of P16
following treatment with CSE or 5-Aza. (C) Western blot analysis of
P16, CDK4, CDK6 and p-Rb levels following treatment with CSE or
5-Aza. Data are expressed as the mean ± standard deviation. n=3,
*P<0.05, **P<0.01, ***P<0.001 vs. NC group. CSE, cigarette
smoke extract; Aza, azactadine; CDK, cyclin-dependent kinase; p-Rb,
phosphorylated retinoblastoma protein; NC, negative control. |
Downregulation of P16 enhances
CSE-mediated cell proliferation and cell cycle arrest in
HAOSMCs
To investigate the role of P16 in HAOSMC
proliferation, expression of P16 was significantly knocked down at
mRNA and protein levels (both P<0.001; Fig. 3A and B). The results of the MTT assay
demonstrated that P16 downregulation significantly enhanced
CSE-induced viability compared with scramble group (P<0.01;
Fig. 3C) and significantly increased
the ratio of Edu positive cells compared with scramble group
(P<0.01; Fig. 3D). The transition
from G1 phase to S phase is the earliest event in cell cycle
progression and serves an important role in cell proliferation.
Flow cytometry revealed that the ratio of cells in G0/G1 phase was
significantly decreased following P16 knockdown compared with the
scramble group, whereas the ratio of cells in S phase was
significantly increased compared with the scramble group
(P<0.001; Fig. 3E). To explore
the mechanism by which downregulation of P16 influences CSE-induced
proliferation, CDK4, CDK6, and p-Rb protein expression were
assessed. Protein levels of CDK4, CDK6 and p-Rb were significantly
increased in HAOSMCs co-treated with 2.5% CSE and siRNA P16
transfection (all P<0.001; Fig.
3F).
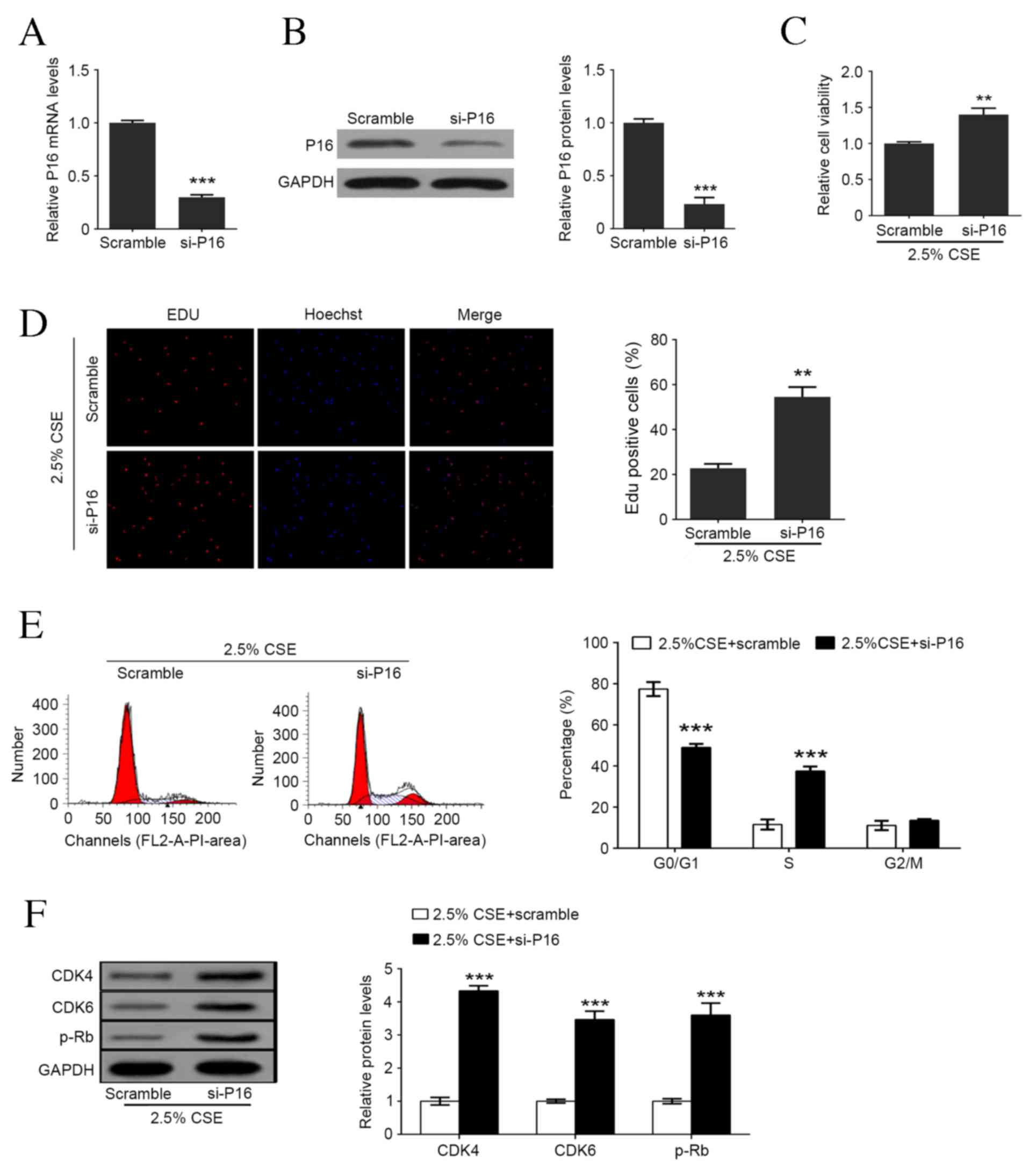 | Figure 3.Downregulation of P16 enhances
CSE-mediated cell proliferation and cell cycle arrest in human
aortic smooth muscle cells. (A) P16 mRNA levels following
transfection with scramble or siRNA P16. (B) P16 protein levels
following transfection with scramble or siRNA P16. (C) Cell
viability detection following P16 knockdown and 2.5% CSE treatment.
(D) Percentage of Edu positive cells following P16 knockdown and
2.5% CSE treatment. (E) Representative flow cytometry and the
percentage of cells in each cell cycle phase following P16
knockdown and 2.5% CSE treatment. (F) Western blot analysis of P16,
CDK4, CDK6 and p-Rb levels following P16 knockdown and 2.5% CSE
treatment. Data are expressed as the mean ± standard deviation.
n=3, *P<0.05, **P<0.01, ***P<0.001 vs. scramble group.
CSE, cigarette smoke extract; si, small interfering; Edu, ethynyl
deoxyuridine; CDK, cyclin-dependent kinase; p-Rb, retinoblastoma
protein. |
Overexpression of P16 reverses
CSE-stimulated cell proliferation and cell cycle progression in
HAOSMCs
To further illustrate the role of P16 on HAOSMC
proliferation, the overexpression of P16 at mRNA and protein levels
was induced (Fig. 4A and B). As
illustrated in Fig. 4C and D,
overexpression of P16 significantly inhibited CSE-induced viability
and reduced the ratio of Edu positive cells compared with the empty
vector (Ev) group (P<0.01). The result of flow cytometry
demonstrated that overexpression of P16 significantly increased the
ratio of cells in G0/G1 phase and also reduced the ratio of cells
in S phase compared with the Ev group (P<0.001; Fig. 4E). CDK4, CDK6, and p-Rb protein
levels were significantly decreased in HAOSMCs co-treated with 2.5%
CSE and P16 expressed vector compared with the Ev group (CDK4 and
CDK6, P<0.01; p-Rb, P<0.001; Fig.
4F).
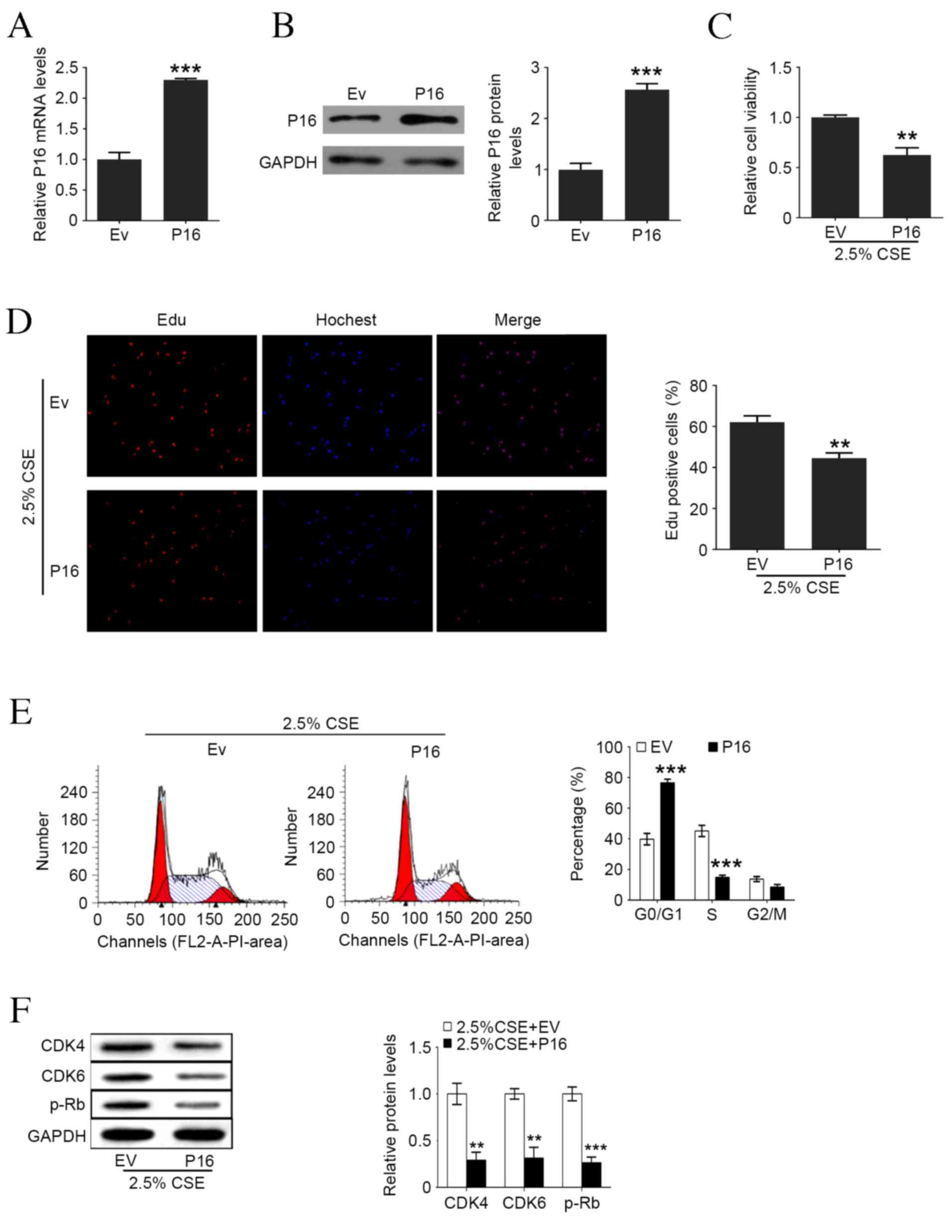 | Figure 4.Upregulation of P16 reverses
CSE-induced cell proliferation and cell cycle arrest in human
aortic smooth muscle cells. (A) P16 mRNA levels following Ev and
P16-expressed plasmid transfection. (B) P16 protein levels
following Ev and P16-expressed plasmid transfection. (C) Cell
viability following P16 overexpression and 2.5% CSE treatment. (D)
Percentage of Edu positive cells following P16 overexpression and
2.5% CSE treatment. (E) Representative flow cytometry and the
percentage of cells in each cell cycle phase following P16
overexpression and 2.5% CSE treatment. (F) Western blot analysis of
P16, CDK4, CDK6 and p-Rb levels following P16 overexpression and
2.5% CSE treatment measured by western blotting. Data are expressed
as the mean ± standard deviation. n=3, *P<0.05, **P<0.01,
***P<0.001 vs. Ev group. CSE, cigarette smoke extract; Ev, empty
vector; Edu, ethynyl deoxyuridine; CDK, cyclin-dependent kinase;
p-Rb, phosphorylated retinoblastoma protein. |
Discussion
It has been well documented that cigarette smoking
is able to cause atherosclerosis via the mechanisms of endothelial
dysfunction, inflammation, and VSMC proliferation (2). Although the molecular mechanisms
driving VSMC proliferation have been investigated (16), they remain unclear. The present study
demonstrated that CSE was able to stimulate VSMC proliferation via
downregulating the P16 expression by inducing P16 promoter
hypermethylation, upregulating the expression of P16 downstream
signal molecules and blocking cell cycle progression from G1 to S
phase.
P16 is an inhibitor of CDKs and an important
regulator of cell cycle progression in malignant tumor cells
(9). Downregulation of P16 leads to
a loss of cell cycle control, which promotes malignant cell
proliferation (17,18). In a previous study, P16 was found to
be overexpressed in VSMCs of aging mice, which may modify VSMC
response to injury and stress and therefore accelerate the
development of age-related cardiovascular diseases (10). P16 has also been demonstrated to
participate in the course of PPARα-inhibited VSMC proliferation
(11). It was reported that DNA
methylation levels at p15 (INK4b) was significantly increased in
patients with coronary artery disease (CAD), which was prevalent in
the lymphocytes, and there was a stepwise increase in p15 (INK4b)
and p16 (INK4a) methylation as levels of antisense non-coding RNA
in the INK4 locus (ANRIL) exon 1–5 expression were elevated
(19). Miki et al (20) demonstrated that the methylation
status of the P16 gene was important in the regulation of
angiogenesis associated with progression of lung cancer via
regulating vascular endothelial growth factor expression. The
results of the present study demonstrated that P16 expression in
HAOSMCs decreased following treatment with CSE in a concentration
and time-dependent manner, caused by hypermethylation of the
promoter and inhibition of promoter activity. Furthermore,
Pearson's correlation analysis revealed a negative correlation
between P16 protein expression and cell proliferation.
It has previously been suggested that CDKs are able
to positively regulate cell cycle progression via phosphorylating
Rb (6). Rb phosphorylation induces
the release of E2F in late G1 stage, which in turn enhances the
expression of genes that encode the regulatory proteins necessary
for cell cycle progression (8).
Restriction of cells at the G1-S and G2-M interphases ensure normal
cell cycle progression (21). In the
present study, it was demonstrated that CDK4, CDK6, and p-Rb
protein levels are elevated in CSE-treated HAOSMCs. Furthermore,
silencing P16 expression increased cell proliferation rate, whereas
overexpression of P16 induced a decrease in cell proliferation
rate, cell cycle progression, and the expression of CDK4, CDK6, and
p-Rb protein.
In conclusion, the results of the present study
suggest that CSE is able to downregulate P16 expression in HAOSMCs
by inducing hypermethylation of its promoter, which promotes VSMC
proliferation via activating the CDK/p-Rb pathway, preventing cell
cycle progression from G1 to S phase. These findings may provide a
new insight for cigarette smoking-induced VSMC proliferation.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (grant no. 81471896) and Hunan
Science Foundation (grant no. 10JJ5008).
References
|
1
|
Ezzati M, Henley SJ, Thun MJ and Lopez AD:
Role of smoking in global and regional cardiovascular mortality.
Circulation. 112:489–497. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ambrose JA and Barua RS: The
pathophysiology of cigarette smoking and cardiovascular disease: An
update. J Am Coll Cardiol. 43:1731–1737. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Burns DM: Epidemiology of smoking-induced
cardiovascular disease. Prog Cardiovasc Dis. 46:11–29. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hao H, Gabbiani G and Bochaton-Piallat ML:
Arterial smooth muscle cell heterogeneity: Implications for
atherosclerosis and restenosis development. Arterioscler Thromb
Vasc Biol. 23:1510–1520. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dzau VJ, Braun-Dullaeus RC and Sedding DG:
Vascular proliferation and atherosclerosis: New perspectives and
therapeutic strategies. Nat Med. 8:1249–1256. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sherr CJ and Roberts JM: CDK inhibitors:
Positive and negative regulators of G1-phase progression. Genes
Dev. 13:1501–1512. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Malumbers M and Barbacid M: To cycle or
not to cycle: A critical decision in cancer. Nat Rev Cancer.
1:222–231. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burke JR, Hura GL and Rubin SM: Structures
of inactive retinoblastoma protein reveal multiple mechanisms for
cell cycle control. Genes Dev. 26:1156–1166. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Witkiewicz AK, Knudsen KE, Dicker AP and
Knudsen ES: The meaning of p16(ink4a) expression in tumors:
Functional significance, clinical associations and future
developments. Cell Cycle. 10:2497–2503. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rodriguez-Menocal L, Pham SM, Mateu D,
St-Pierre M, Wei Y, Pestana I, Aitouche A and Vazquez-Padron RI:
Aging increases p16 INK4a expression in vascular smooth-muscle
cells. Biosci Rep. 30:11–18. 2019. View Article : Google Scholar
|
|
11
|
Gizard F, Amant C, Barbier O, Bellosta S,
Robillard R, Percevault F, Sevestre H, Krimpenfort P, Corsini A,
Rochette J, et al: PPAR alpha inhibits vascular smooth muscle cell
proliferation underlying intimal hyperplasia by inducing the tumor
suppressor p16INK4a. J Clin Invest. 115:3228–3238. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Holdt LM and Teupser D: Recent studies of
the human chromosome 9p21 locus, which is associated with
atherosclerosis in human populations. Arterioscler Thromb Vasc
Biol. 32:196–206. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Holdt LM, Sass K, Gäbel G, Bergert H,
Thiery J and Teupser D: Expression of Chr9p21 genes CDKN2B
(p15(INK4b)), CDKN2A (p16(INK4a), p14(ARF)) and MTAP in human
atherosclerotic plaque. Atherosclerosis. 214:264–270. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yao Q, Xu H, Zhang QQ, Zhou H and Qu LH:
MicroRNA-21 promotes cell proliferation and down-regulates the
expression of programmed cell death 4 (PDCD4) in HeLa cervical
carcinoma cells. Biochem Biophys Res Commun. 388:539–542. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ou H, Shen YH, Utama B, Wang J, Wang X,
Coselli J and Wang XL: Effect of nuclear actin on endothelial
nitric oxide synthase expression. Arterioscler Thromb Vasc Biol.
25:2509–2514. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Motterle A, Pu X, Wood H, Xiao Q, Gor S,
Ng FL, Chan K, Cross F, Shohreh B, Poston RN, et al: Functional
analyses of coronary artery disease associated variation on
chromosome 9p21 in vascular smooth muscle cells. Hum Mol Genet.
21:4021–4029. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ghiorzo P, Villaggio B, Sementa AR,
Hansson J, Platz A, Nicoló G, Spina B, Canepa M, Palmer JM, Hayward
NK and Bianchi-Scarrà G: Expression and localization of mutant p16
proteins in melanocytic lesions from familial melanoma patients.
Hum Pathol. 35:25–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
LaPak KM and Burd CE: The molecular
balancing act of p16(INK4a) in cancer and aging. Mol Cancer Res.
12:167–183. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhuang J, Peng W, Li H, Wang W, Wei Y, Li
W and Xu Y: Methylation of p15INK4b and expression of ANRIL on
chromosome 9p21 are associated with coronary artery disease. PLoS
One. 7:e471932012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miki K, Shimizu E, Yano S, Tani K and Sone
S: Demethylation by 5-aza-2′-deoxycytidine (5-azadC) of p16INK4A
gene results in downregulation of vascular endothelial growth
factor expression in human lung cancer cell lines. Oncol Res.
12:335–342. 2001.PubMed/NCBI
|
|
21
|
Elledge SJ: Cell cycle checkpoints:
Preventing an identity crisis. Science. 274:1664–1672. 1996.
View Article : Google Scholar : PubMed/NCBI
|