Introduction
Acute myocardial infarction (AMI) is a major public
health problem. In 2010, age-standardized AMI incidence in all ages
was 195.3 per 100,000 in males and 115.0 in females (1). In Europe, ~1.8 million individuals
succumb to coronary heart disease each year (2). Reperfusion of the ischemic myocardium
is necessary to salvage tissue and limit cardiomyocyte death. While
reperfusion is beneficial, it also induces new pathophysiological
changes through the release of oxygen free radicals, cytokines and
other pro-inflammatory factors that initiate an inflammatory
cascade. This inflammation typically enhances damage to myocytes
and induces myocardial dysfunction (3–5).
Platelets serve key roles in myocardial reperfusion injury,
including platelet adhesion, aggregation, activation and inhibiting
the platelet activation, which reduces myocardial infarct size
(6). Furthermore, they induce new
thrombus formation and microembolization at sites of
atherosclerotic plaques in coronary arteries, and facilitate
interactions between the endothelium and leukocytes, consequently
prompting microcirculation disturbance and inflammation (7–9).
Suppression of platelet activation using Tirofiban to inhibit
glycoprotein IIb/IIIa may alleviate ischemia-reperfusion (I/R)
injury (10,11).
The Traditional Chinese Medicine Salvia miltiorrhiza
is widely used in the treatment of coronary artery disease and
cerebrovascular diseases and as a remedy to improve
microcirculation (12,13). Salvianolic acid A (SAA), as a
water-soluble polyphenolic compound, is a primary constituent of
Salvia miltiorrhiza and is extracted from Salvia miltiorrhiza Bunge
(14). A previous study by our group
revealed that SAA dose-dependently inhibited platelet aggregation
induced by adenosine diphosphate (ADP), thrombin, collagen and
U46619, and inhibited arterial thrombosis via the inhibition of
phosphoinositide 3-kinase in vitro (15). In the present study, the
cardioprotective effects of SAA against platelet activation and
inflammation during myocardial I/R injury were investigated by
ligating the left anterior descending branch of coronary artery.
Results indicated that the cardioprotective effects of SAA were
comparable to those of the positive control agent, Tirofiban.
Materials and methods
Animals
A total of 150, male Sprague-Dawley rats (aged 6–8
weeks and weighing 250–300 g) were purchased from the Zhejiang
Laboratory Animal Centre, China [certificate no. SCXL (Zhe)
2008–0033]. Animals were acclimated for at least 1 week at room
temperature (18–25°C), 55±5% humidity and a 12-h light/dark cycle.
The animals were given free access to a standard diet and tap
water. All experimental procedures were approved by the Ethics
Review of Animal Use Application of Fifth Affiliated Hospital of
Wenzhou Medical University (Zhejiang, China).
Animal groups and drug
pretreatment
All animals were randomly assigned to one of four
groups. Each group was divided into n1 and n2 subgroups and exposed
to reperfusion for 3 and 24 h, respectively. The model control
group (I/R; n1=10, n2=30) underwent I/R and received an intravenous
administration of glucose (5%), while the SAA group (I/R + SAA,
n1=10, n2=30) underwent I/R and received an intravenous
administration of SAA (10 mg/kg; Zhengda Qingchunbao Pharmaceutical
Co., Ltd., Zhejiang, China) dissolved in 5% glucose. The Tirofiban
group (I/R + Tirofiban, n1=10, n2=30) underwent I/R and received an
intravenous administration of Tirofiban (60 µg/kg; Wuhan Grand
Pharmaceutical Co., Ltd., Wuhan, China) dissolved in 5% glucose.
The sham group (sham; n1=10, n2=20) received an intravenous
administration of glucose (5%) alone. All drugs were administered
intravenously 10 min before reperfusion was initiated.
Myocardial I/R model protocol
Rats were anesthetized with an intraperitoneal
injection of 4% chloral hydrate (400 mg/kg). Following intubation,
a left thoracotomy was performed. The left anterior descending
coronary artery (LAD) was surgically occluded for 45 min through
ligation of a silk suture, and coronary occlusion was confirmed by
elevation of the ST segment (>0.1 mV) on an electrocardiogram
(MedLab-U/4C50; Nanjing Medease Science and Technology Co., Ltd.,
Nanjing, China). Reperfusion of the coronary artery was initiated
by release of the ligation tie. Following the procedure, the chest
was closed and rats in each group; n1 and n2 were monitored in the
animal facility for 3 and 24 h, respectively, anesthetized with an
intraperitoneal injection of 4% chloral hydrate (400 mg/kg), prior
to sacrifice with 10% potassium chloride solution (400 mg/kg; 1–2
ml) administered to the inferior vena cava. All groups underwent
the same surgical procedure, though the LAD suture was not tied for
rats in the sham group.
Measurement of myocardial infarct
size
Myocardial infarct size was evaluated by Evans blue
and 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) staining (both
from Sigma-Aldrich, Inc.; Merck KGaA, Darmstadt, Germany), as
described previously (16). Briefly,
after 24 h of reperfusion, rats were sacrificed and the LAD was
occluded with a silk suture in the same location as for the I/R
procedure. The abdomen was opened and 5 ml of 1% Evans blue dye was
injected into the aorta. The heart was immediately excised and the
right ventricle and left and right atria were removed. The left
ventricle was transversally cut into 1 mm thick slices and
incubated in 1% TTC at 37°C for 10 min. Evans blue was used to
stain the area outside of the risk area (RA), and the area
unstained by TTC represented the ischemic area. The infarct area
(ischemic area), RA and left ventricle wall size were also measured
digitally using Image J software (version 1.38; National Institutes
of Health, Bethesda, MA, USA).
Determination of platelet maximum
aggregation rate
Blood was collected from the abdominal aorta in an
anticoagulant solution containing 3.8% sodium citrate (Sinopharm
Chemical Reagent Co., Ltd., Shanghai, China), 1:9 citrate: whole
blood) after 3 h of reperfusion. The platelet-rich plasma (PRP)
fraction was obtained by centrifugation at 174 × g at 25°C for 10
min, and the remaining blood was further centrifuged at 1,570 × g
at 25°C for 10 min to prepare the platelet-poor plasma (PPP)
fraction. The platelet concentration, according to a previously
described method, using 1×20 l Coulter Isoton® II Dilutent (cat.
no. 8546719; Beckman Coulter, Brea, CA, USA) (15), was adjusted to 250×106
platelets/ml and incubated at room temperature for 30 min to allow
the platelets to clot. The platelet agonist ADP disodium (5 µM
final concentration; Helena Laboratories, Beaumont, TX, USA) was
used to stimulate platelet aggregation as a positive control. The
level of platelet aggregation was measured using an aggregometer
(AggRAM; cat. no. 8JF52001; Helena Laboratories, Beaumont, Texas,
USA).
Enzyme-linked immunosorbent assay
(ELISA)
After 3 and 24 h of reperfusion, blood samples were
collected from the abdominal aorta using 3.8% sodium citrate as the
anticoagulant, and samples were centrifuged at 25°C and 3,500 × g
for 15 min to isolate the plasma. Quantikine ELISA kits (96Test;
Abcam, Cambridge, UK) were used to measure the plasma levels of
creatine kinase isoenzyme MB (CK-MB; cat. no. XF020852B), cardiac
troponin T (cTnT; cat. no. XF03363B), p-selectin (cat. no.
XF03259B), interleukin-1β (IL-1β; cat. no. XF01588B) and tumor
necrosis factor-α (TNF-α; cat. no. XF01721B), according to the
manufacturer's protocol.
Histological analysis by light
microscopy
After 24 h of reperfusion, myocardial samples from
the RA were rinsed with PBS (pH 7.4) and fixed in 4%
paraformaldehyde and 25°C. After 24 h fixation, tissues were
dehydrated in graded alcohol (75, 85 and 95% twice and then 100%
twice) at a temperature of 25°C, embedded in paraffin and cut into
3–5 µm thin sections. The tissue sections were then stained with
hematoxylin and eosin and histologically examined under a light
microscope.
Measurement of myocardial nitric oxide
(NO) content
The myocardial NO concentration was measured as
described previously (17). Briefly,
after 3 h of reperfusion, myocardial samples from the RA were
rinsed and homogenized in deionized water (1:10 w/v) prior to
centrifugation at 3,000 × g at 25°C for 5 min. The concentration of
NO in the supernatant was determined using an NO detection kit
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China),
according to the manufacturer's instructions.
Statistical analysis
All data are presented as the mean ± standard error
of the mean and were analyzed by one-way analysis of variance,
followed by a least-significant-difference test for multiple
comparisons. Survival rate was analyzed by Kaplan-Meier analysis
with SPSS, version 19.0 software (IBM Corp., Armonk, NY, USA).
P<0.05 was determined to represent statistically significant
differences following a log-rank test.
Results
SAA treatment reduces myocardial
injury and mortality following myocardial I/R
After 24 h of reperfusion, the RAs of the SAA,
Tirofiban and control I/R groups were all of a similar size. The
infarct areas were significantly reduced in both the SAA and
Tirofiban groups compared to the I/R group (both P<0.05;
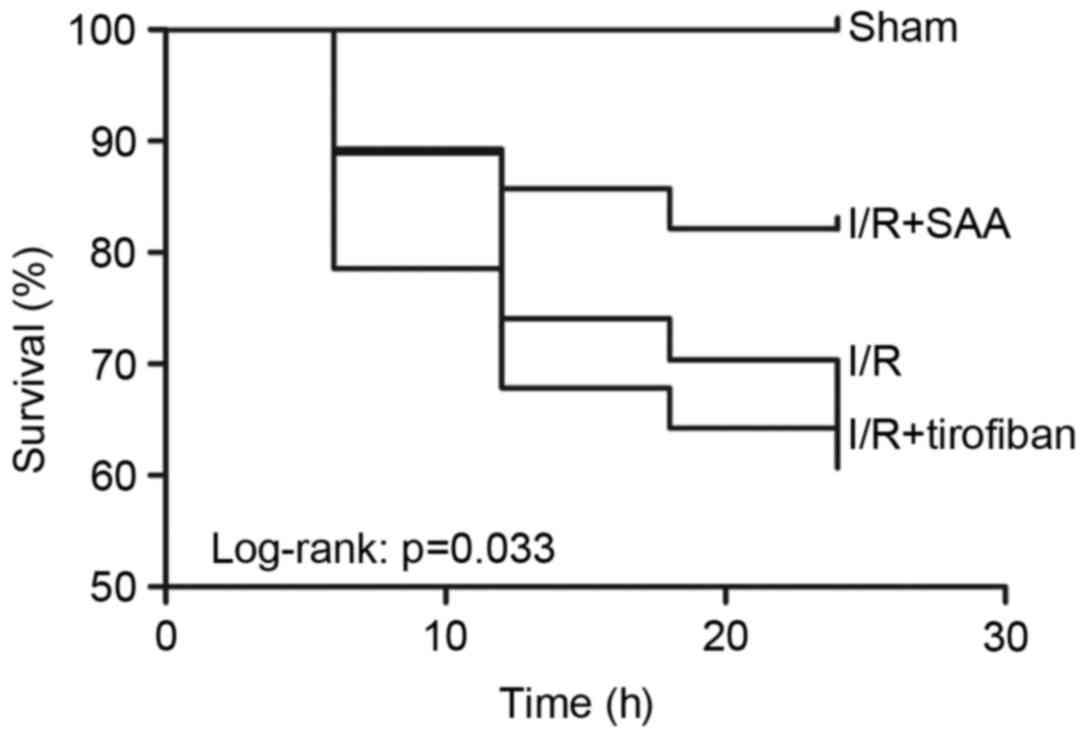
Table I; Fig. 1). Furthermore, survival rates of rats
in the sham, I/R, SAA and Tirofiban groups were 100, 61, 82 and
63%, respectively (log-rank test P=0.033 SAA group vs. I/R group;
Fig. 2), indicating that rats in the
SAA group had an improved survival rate relative to those in the
I/R and Tirofiban groups. In addition, after 3 and 24 h of
reperfusion, levels of cTnT and CK-MB in the serum were
significantly increased in the I/R group, relative to the sham
group (both P<0.05). This effect was significantly reversed by
pretreatment with SAA or Tirofiban after 24 h reperfusion (all
P<0.05; Fig. 3).
 | Table I.Effect of SAA and Tirofiban on
myocardial I/R injury in rats. |
Table I.
Effect of SAA and Tirofiban on
myocardial I/R injury in rats.
| Group | Dosage | Risk area, % | Infarct area, % |
|---|
| Sham | – | NA | NA |
| I/R | – | 30±2 | 29±5 |
| SAA | 10 mg/kg | 29±4 | 19±11a |
| Tirofiban | 60 µg/kg | 29±3 | 19±4a |
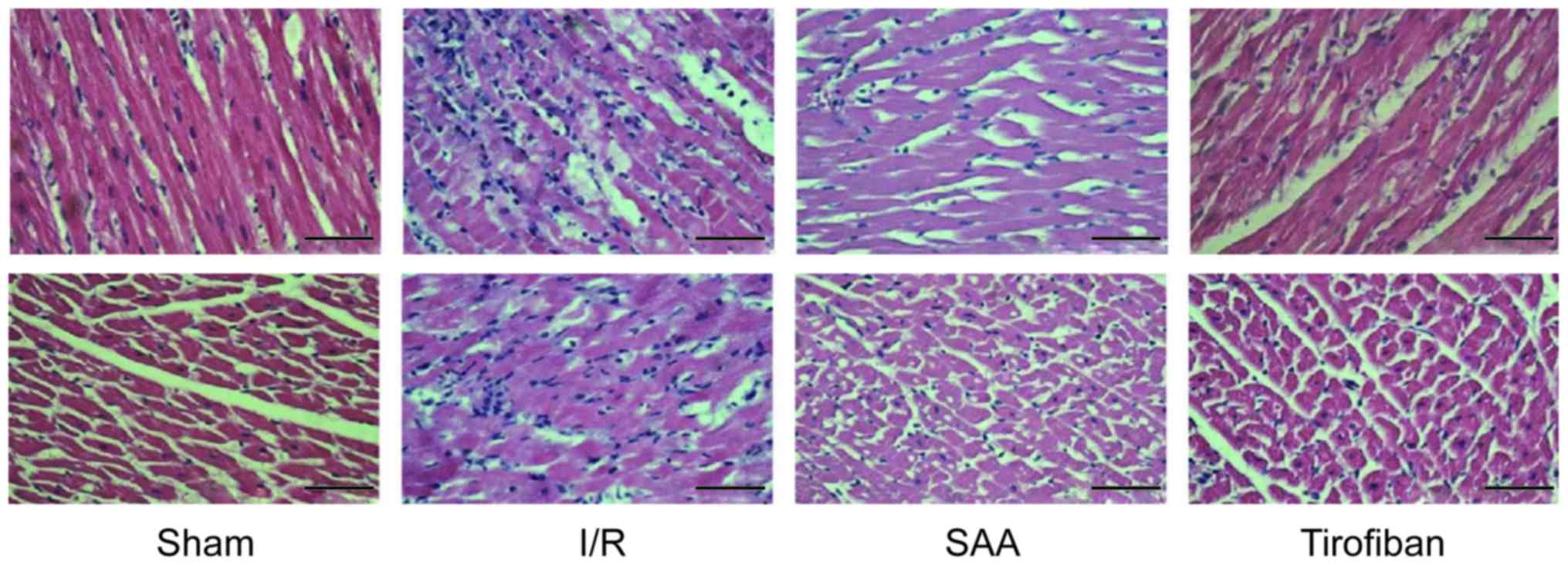
Under light microscopy, the myocardial samples of
the I/R group exhibited a disordered myocardial cell arrangement
and robust inflammatory cell infiltration, relative to the Sham
group. In turn, myocardial injury and inflammatory exudation were
attenuated in both the SAA and Tirofiban groups (Fig. 4).
SAA treatment inhibits platelet
aggregation during myocardial I/R
After 3 h of reperfusion, the maximum rate of
platelet aggregation in the I/R group was similar to that in the
sham group (P=0.195). This rate was significantly reduced by
pretreatment with SAA or Tirofiban (both P<0.05 vs. I/R group).
The platelet aggregation rates of the SAA and Tirofiban groups did
not differ significantly (P=0.59; Fig.
5).
SAA treatment reduces serum levels of
p-selectin
After 3 or 24 h of reperfusion, levels of p-selectin
in the serum were significantly increased in the I/R group,
compared with the sham group (P<0.05). This effect was
significantly reversed by pretreatment with SAA or Tirofiban
(P<0.05 vs. I/R). Serum levels of p-selectin did not differ
significantly between the SAA and Tirofiban groups (P>0.05;
Fig. 6).
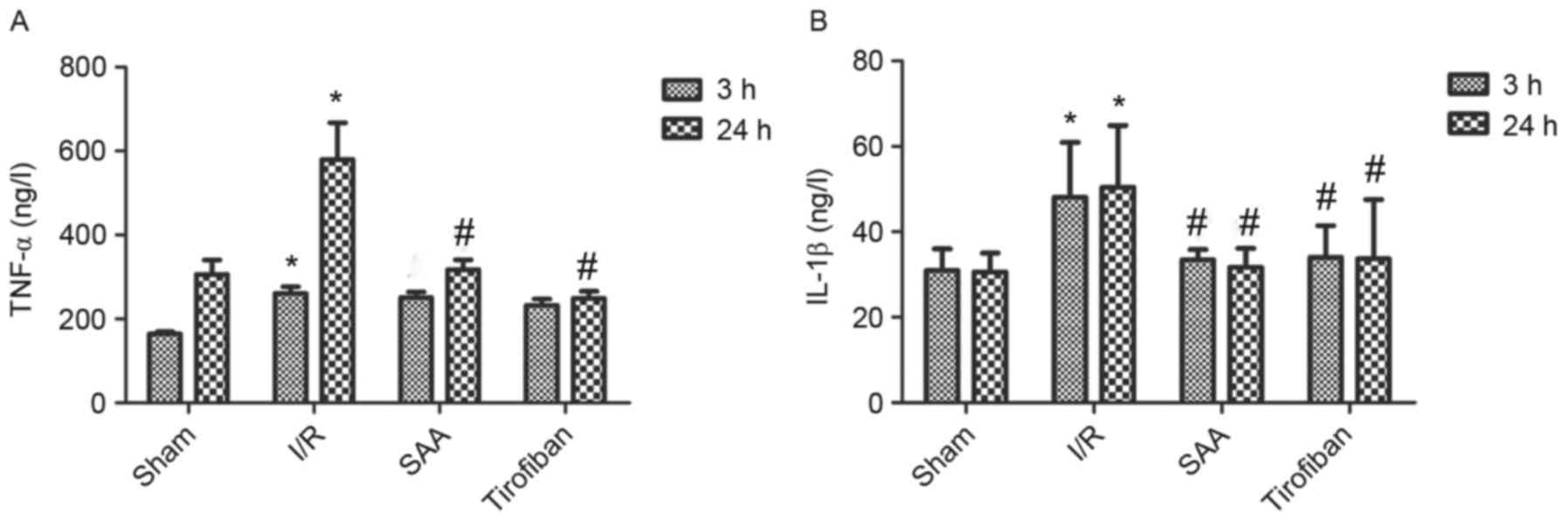
SAA treatment reduces serum levels of
TNF-α and IL-1β
After 24 h of reperfusion, levels of TNF-α in the
serum significantly increased in the I/R group compared to the sham
group (both P<0.05). In turn, pretreatment with SAA or Tirofiban
significantly reduced serum levels of TNF-α after 24 h of
reperfusion (both P<0.05; Fig.
7A).
Similarly, after 3 or 24 h of reperfusion, levels of
IL-1β in the serum were significantly increased in the I/R group,
relative to the sham group (both P<0.05). In turn, pretreatment
with SAA significantly reduced the serum levels of IL-1β levels
(both P<0.05 compared to I/R) such that the serum levels of
IL-1β did not differ significantly between the SAA and Tirofiban
groups (P>0.05; SAA vs Tirofiban group for 3 or 24 h; Fig. 7B).
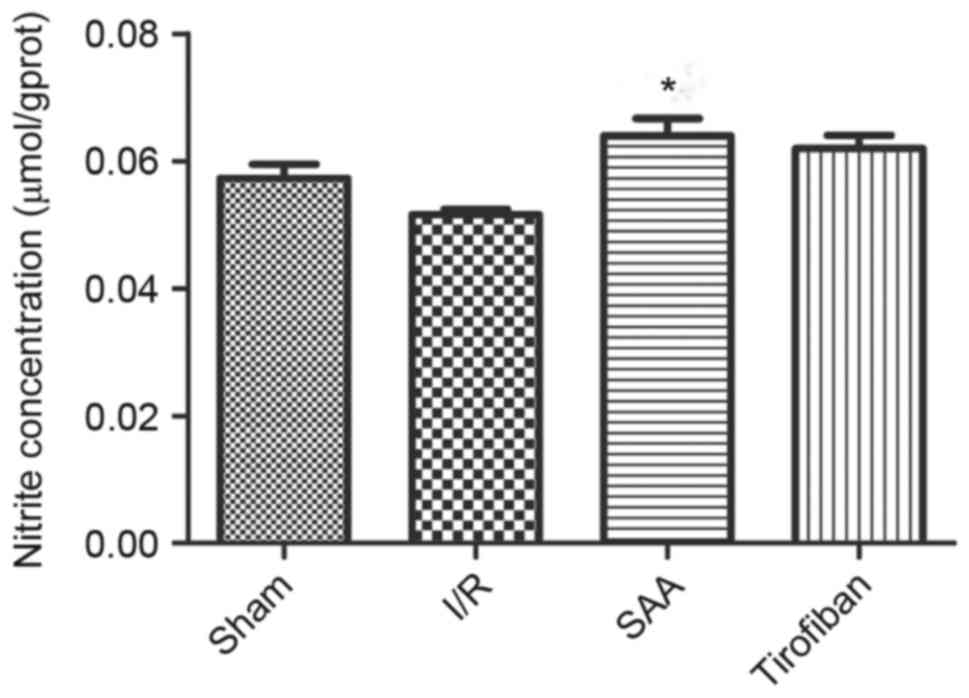
SAA treatment increases the NO
concentration in myocardial tissue
After 3 h of reperfusion, concentrations of NO in
the RA were similar between the I/R and sham groups (P>0.05).
Relative to the I/R the SAA group, but not the Tirofiban group,
exhibited significantly increased concentrations of NO in the RA
(P<0.05; Fig. 8).
Discussion
The present study demonstrated that pretreatment
with SAA significantly ameliorated cardiac injury and improved the
survival rate of rats exposed to I/R. SAA reduced serum levels of
cTnT and CK-MB and myocardial infarct size. Furthermore, SAA
inhibited ADP-induced platelet aggregation, decreased IL-1β and
TNF-α activity, reduced swelling of ischemic myocardial cells and
inflammatory cell infiltration, and increased NO synthesis in the
area at risk. These observations indicate that SAA may be a
potential therapeutic agent in the treatment of myocardial I/R
injury.
Platelets serve critical roles in I/R injury.
Following reperfusion, platelet contact with subendothelial
collagen stimulates platelet adhesion and activation (18). Activated platelets generate a variety
of factors that induce a feed forward loop to reinforce the
activation process. For instance, thromboxane A2 enhances platelet
activation and adhesion, while ADP and thrombin recruit circulating
platelets and promote platelet aggregation. Concomitant with these
events, tissue factor XIII is activated to promote the formation of
clots (both large thrombi and small platelet microthrombi). In the
present study, SAA exhibited strong antiplatelet activity, as
observed in an in vitro platelet aggregation assay. The
in vitro experiments of the present study demonstrated that
treatment with SAA significantly decreased ADP-induced platelet
aggregation, eliciting an inhibitory effect similar to Tirofiban.
Therefore, SAA may reduce coronary thrombus by inhibiting platelet
activation and improving coronary blood flow.
The inflammatory response and platelet aggregation
have been implicated in myocardial I/R injury (19). Within min of reperfusion, an
inflammatory cascade is triggered and numerous pro-inflammatory
cytokines are released into the region, including TNF-α, IL-1β,
IL-6 and IL-8. These pro-inflammatory cytokines, particularly
TNF-α, function as key mediators in cardiac dysfunction, serving to
activate endothelial cells and neutrophils and aggravate myocardial
I/R injury (20). In addition to
regulating thrombosis, SAA modulates the inflammatory process
(21). During adhesion, platelets
are activated and release a variety of potent chemotactic factors,
as previously described (22).
Moreover, platelets may modulate the chemotactic properties of
other cells through platelet-leukocyte (23) and platelet-endothelium interactions
(24).
P-selectin is present on activated platelets and
mediates loose contact between circulating platelets and the
vascular endothelium. Once platelets are activated during ischemia
and reperfusion, p-selectin may be cleaved from the membrane to
generate a soluble form that is readily detected in the plasma
(25). The present study observed
that levels of p-selectin were significantly elevated in the
myocardium after 3 and 24 h of reperfusion when compared to the
sham group, indicating that platelet activation occurs as a result
of I/R. Furthermore, it was observed that SAA decreased levels of
serum p-selectin and plasma IL-1β at 3 and 24 h after reperfusion,
and reduced plasma levels of TNF-α at 24 h after reperfusion.
Reduced cytokine levels are both a cause and consequence of reduced
inflammatory cell infiltration. These data suggest that SAA
functions to reduce platelet activation and inflammation.
NO is a diatomic free-radical gas that serves key
roles in a numerous of biological processes, including inhibited
platelet aggregation via a cyclic GMP-dependent mechanism and
relaxed vascular endothelium as an endothelium derived relaxation
factor (16). Maintaining a basal
level of NO is critical for various physiological processes,
including vascular smooth muscle cell relaxation, prevention of
neutrophil and platelet adhesion to the endothelium, and
maintenance of an anti-apoptotic environment in the vessel wall.
I/R alters the levels of NO to further aggravate I/R injury
(26). Furthermore, it has been
documented that Tirofiban increased NO production, and that SAA
alleviated impaired expression of endothelial nitric oxide synthase
and NO formation in response to I/R (10,27).
Similarly, results of the present study indicated that SAA
protected endothelial cells by improving NO production within the
vessel wall.
SAA has been demonstrated to prevent I/R-induced
myocardial damage by reducing cardiomyocyte necrosis and apoptosis
(28,29). The present study revealed that SAA
and Tirofiban share similarities in terms of inhibiting the
ADP-induced platelet aggregation rate, reducing plasma levels of
p-selectin, reducing inflammatory factors and increasing the
generation of NO to reduce myocardial infarct size. These
similarities suggest that SAA may protect myocardial cells
principally through antiplatelet activity. Notably, Tirofiban does
not increase the survival rate of rats following exposure to I/R,
potentially due to severe hemorrhage complications associated with
its use (30–32).
In conclusion, the present study demonstrated that
SAA protected the myocardium against I/R injury and improved
survival rate by reducing platelet activation and inflammation. The
protective effects of SAA may be principally related to the
antiplatelet effects of SAA. Thus, SAA may be viable as a novel
antiplatelet drug for use in both ischemic heart disease and
cardiac surgeries associated with I/R injury.
Acknowledgements
The present study was supported by the Technology
Bureau of Lishui (grant no. 20110410).
References
|
1
|
Moran AE, Forouzanfar MH, Roth GA, Mensah
GA, Ezzati M, Flaxman A, Murray CJ and Naghavi M: The global burden
of ischemic heart disease in 1990 and 2010: The Global Burden of
Disease 2010 study. Circulation. 129:1493–1501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fröhlich GM, Meier P, White SK, Yellon DM
and Hausenloy DJ: Myocardial reperfusion injury: Looking beyond
primary PCI. Eur Heart J. 34:1714–1722. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Becker LC and Ambrosio G: Myocardial
consequences of reperfusion. Prog Cardiovasc Dis. 30:23–44. 1987.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Maxwell SR and Lip GY: Reperfusion injury:
A review of the pathophysiology, clinical manifestations and
therapeutic options. Int J Cardiol. 58:95–117. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Piper HM, Meuter K and Schäfer C: Cellular
mechanisms of ischemia-reperfusion injury. Ann Thorac Surg.
75:S644–S648. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pachel C, Mathes D, Arias-Loza AP,
Heitzmann W, Nordbeck P, Deppermann C, Lorenz V, Hofmann U,
Nieswandt B and Frantz S: Inhibition of platelet GPVI protects
against myocardial ischemia-reperfusion injury. Arterioscler Thromb
Vasc Biol. 36:629–635. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gawaz M, Langer H and May AE: Platelets in
inflammation and atherogenesis. J Clin Invest. 115:3378–3384. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klinger MH and Jelkmann W: Role of blood
platelets in infection and inflammation. J Interferon Cytokine Res.
22:913–922. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Skyschally A, Erbel R and Heusch G:
Coronary microembolization. Circ J. 67:279–286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu X and Tao GZ: Effects of tirofiban on
the reperfusion-related no-reflow in rats with acute myocardial
infarction. J Geriatr Cardiol. 10:52–58. 2013.PubMed/NCBI
|
|
11
|
Howard JP, Jones DA, Gallagher S, Rathod
K, Antoniou S, Wright P, Knight C, Mathur A, Weerackody R and Wragg
A: Glycoprotein IIb/IIIa inhibitors use and outcome after
percutaneous coronary intervention for non-ST elevation myocardial
infarction. Biomed Res Int. 2014:6439812014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cheng TO: Cardiovascular effects of
Danshen. Int J Cardiol. 121:9–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han JY, Fan JY, Horie Y, Miura S, Cui DH,
Ishii H, Hibi T, Tsuneki H and Kimura I: Ameliorating effects of
compounds derived from Salvia miltiorrhiza root extract on
microcirculatory disturbance and target organ injury by ischemia
and reperfusion. Pharmacol Ther. 117:280–295. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lian-Niang L, Rui T and Wei-Ming C:
Salvianolic acid A, a new depside from roots of Salvia
miltiorrhiza. Planta Med. 50:227–228. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Huang ZS, Zeng CL, Zhu LJ, Jiang L, Li N
and Hu H: Salvianolic acid A inhibits platelet activation and
arterial thrombosis via inhibition of phosphoinositide 3-kinase. J
Thromb Haemost. 8:1383–1393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Moncada S, Palmer RM and Higgs EA: Nitric
oxide: Physiology, pathophysiology, and pharmacology. Pharmacol
Rev. 43:109–142. 1991.PubMed/NCBI
|
|
17
|
Liu X and Tao GZ: Effects of tirofiban on
the reperfusion-related no-reflow in rats with acute myocardial
infarction. J Geriatr Cardiol. 10:52–58. 2013.PubMed/NCBI
|
|
18
|
Ruggeri ZM: Platelets in atherothrombosis.
Nat Med. 8:1227–1234. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hansen PR: Inflammatory alterations in the
myocardial microcirculation. J Mol Cell Cardiol. 30:2555–2559.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Vinten-Johansen J, Jiang R, Reeves JG,
Mykytenko J, Deneve J and Jobe LJ: Inflammation, proinflammatory
mediators and myocardial ischemia-reperfusion injury. Hematol Oncol
Clin North Am. 21:123–145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li J, Gu T, Fu X and Zhao R: Effect of
salvianolic acid A and C compatibility on inflammatory cytokines in
rats with unilateral ureteral obstruction. J Tradit Chin Med.
35:564–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rendu F and Brohard-Bohn B: The platelet
release reaction: Granules' constituents, secretion and functions.
Platelets. 12:261–273. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Neumann FJ, Marx N, Gawaz M, Brand K, Ott
I, Rokitta C, Sticherling C, Meinl C, May A and Schömig A:
Induction of cytokine expression in leukocytes by binding of
thrombin-stimulated platelets. Circulation. 95:2387–2394. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gawaz M, Neumann FJ, Dickfeld T, Koch W,
Laugwitz KL, Adelsberger H, Langenbrink K, Page S, Neumeier D,
Schömig A and Brand K: Activated platelets induce monocyte
chemotactic protein-1 secretion and surface expression of
intercellular adhesion molecule-1 on endothelial cells.
Circulation. 98:1164–1171. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Massberg S, Enders G, Leiderer R,
Eisenmenger S, Vestweber D, Krombach F and Messmer K:
Platelet-endothelial cell interactions during ischemia/reperfusion:
The role of P-selectin. Blood. 92:507–515. 1998.PubMed/NCBI
|
|
26
|
Rafikov R, Fonseca FV, Kumar S, Pardo D,
Darragh C, Elms S, Fulton D and Black SM: eNOS activation and NO
function: Structural motifs responsible for the posttranslational
control of endothelial nitric oxide synthase activity. J
Endocrinol. 210:271–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang D, Xie P and Liu Z:
Ischemia/reperfusion-induced MKP-3 impairs endothelial NO formation
via inactivation of ERK1/2 pathway. PLoS One. 7:e420762012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pan H, Li D, Fang F, Chen D, Qi L, Zhang
R, Xu T and Sun H: Salvianolic acid A demonstrates cardioprotective
effects in rat hearts and cardiomyocytes after ischemia/reperfusion
injury. J Cardiovasc Pharmacol. 58:535–542. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fan H, Yang L, Fu F, Xu H, Meng Q, Zhu H,
Teng L, Yang M, Zhang L, Zhang Z and Liu K: Cardioprotective
effects of salvianolic acid A on myocardial ischemia-reperfusion
injury in vivo and in vitro. Evid Based Complement Alternat Med.
2012:5089382012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aguirre FV, Topol EJ, Ferguson JJ,
Anderson K, Blankenship JC, Heuser RR, Sigmon K, Taylor M, Gottlieb
R, Hanovich G, et al: Bleeding complications with the chimeric
antibody to platelet glycoprotein IIb/IIIa integrin in patients
undergoing percutaneous coronary intervention. EPIC Investigators.
Circulation. 91:2882–2890. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kellert L, Hametner C, Rohde S, Bendszus
M, Hacke W, Ringleb P and Stampfl S: Endovascular stroke therapy:
Tirofiban is associated with risk of fatal intracerebral hemorrhage
and poor outcome. Stroke. 44:1453–1455. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ilhan E, Güvenc TS, Güzelburc O, Altay S,
Özer N, Soylu O, Hasdemir H and Ergelen M: A fatal complication of
tirofiban in an octogenarian: Diffuse alveolar hemorrhage. J
Cardiol Cases. 2:e48–e51. 2010. View Article : Google Scholar
|