Introduction
There is a close relationship between bone formation
and angiogenesis. The germinal center of the bone is located around
blood vessels during embryonic osteogenesis. During development and
elongation of the bone, angiogenesis factors in the epiphyseal
growth plate induce blood vessel growth into the metaphysis and
cartilage region, thus promoting the formation of new bone. In
addition, angiogenesis is required for endochondral ossification
during fracture healing (1). In
this process, avascular cartilage tissue is gradually replaced by
bone tissue. Many factors influence fracture healing by affecting
angiogenesis at the fracture site, including fibroblast growth
factor (FGF), transforming growth factor (TGF), platelet-derived
growth factor (PDGF), prostaglandin E (PGE), vascular endothelial
growth factor (VEGF), and tumor necrosis factor (TNF). Of these
factors, VEGF is particularly important in angiogenesis, acting on
vascular endothelial cells to promote proliferation and
angiogenesis (2,3). Various types of cells secrete VEGF,
including fibroblasts, smooth muscle cells, hypertrophic
chondrocytes and osteoblasts. Hypertrophic chondrocytes secrete
VEGF, and induced metaphysis blood vessel growth into the cartilage
region, thus promoting ossification. Secretion of VEGF from
osteoblasts is critical for bone metabolism, and osteoblasts
secrete VEGF during fracture healing (3).
Despite the importance of osteoblast VEGF release to
long bone development and fracture healing, the molecular signal
transduction mechanisms for the induction and control of VEGF
release are unknown. VEGF release is induced by many factors in
vivo, including hypoxia, steroids and cytokines like
insulin-like growth factor-1 (IGF-1), keratinocyte growth factor,
epidermal growth factor (EGF), TNF-α, TGF, inter-leukin-1 (IL-1),
IL-6. Intracellular signaling cascades involving tyrosine kinases
also greatly impact VEGF release and signal transduction.
Extracellular signal-regulated kinases 1/2 (ERK1/2) play a central
role in regulating VEGF secretion (4). First, ERK1/2 signals directly to the
VFGF promoter to control expression of VEGF protein under normoxia.
Under hypoxia, hypoxia inducible factor-1 (HIF-1) is activated by
ERK1/2 to promote the expression of VEGF protein. ERK1/2 is a
serine and threonine kinase that interacts with a myriad of
signaling pathways (5). In turn,
many signaling molecules regulate ERK1/2 activity, including
activator protein 1 (AP-1), angiotensin-II (Ang-II), and negative
regulatory factor (Nef) (6).
We previously reported that G protein-coupled
receptor kinase interacting protein 1 (GIT1) is an important
regulator of ERK1/2 activity by acting as a specific shuttle
protein that associates ERK1/2 with upstream kinases and downstream
targets (7). We further
demonstrated that GIT1 knockout mice had both greatly reduced
ERK1/2 activity and VEGF expression. In addition, GIT1 knockout
impeded formation of the rat pulmonary artery, indicating that the
protein is involved in angiogenesis (8). The yeast two-hybrid method
demonstrated that GIT1 can combine with G protein coupled receptor
kinase 2 (GRK2). This interaction regulates cytoskeletal structure,
membrane transport, other signaling molecules, and cell polarity
(9). While GIT1 itself has no
catalytic activity, GIT1 activation by tyrosine phosphorylation
triggers a series of intracellular signal transduction events. We
have previously reported that GIT1 was tyrosine phosphorylated by
Src, leading to increased ERK1/2 activity (10). Indeed, protein tyrosine
phosphorylation is vital for GIT1 signaling. Webb et al
(11) described several important
tyrosine phosphorylation sites in GIT1 by mass spectrometry
analysis. We also found that GIT1Y392 was phosphorylated by Src,
and that Src-mediated tyrosine phosphorylation activated
intracellular PLC-γ, leading to podosome formation in smooth muscle
cells (12). Phosphorylation of
GIT1Y321 is key to enhancing ERK1/2 activity (7,13).
In this study, we clarified the mechanisms of
GIT1Y321 phosphorylation during PDGF-induced secretion of VEGF from
osteoblasts. We then examined the role of this signaling cascade in
bone fracture healing.
Materials and methods
Materials and reagents
Twenty 1-2-day-old Sprague Dawley (SD) rats (both
male and female) and 30 SD male rats weighing about 200–300 g were
obtained from the Department of Animal Science, Nanjing Medical
University. The drugs and reagents (and their suppliers) were as
follows: Dulbecco’s modified Eagle’s medium (DMEM) and fetal bovine
serum (FBS) were from Gibco (USA); EDTA-trypsin, collagenase II,
and Src inhibitor PP2 and MEK inhibitor PD98059 were from Sigma
(USA); GTI1 antibody, Flag antibody, PECAM-1 antibody were
purchased from Cell Signaling Technology (USA) while the ERK1/2
antibody, pERK1/2 antibody, pERK2 antibody, and VEGF antibody were
purchased from Santa Cruz Biotechnology, Inc. (USA); tubulin
antibody and 4G10 antibody were from Cell Signaling (USA). Finally,
VEGF primers were custom synthesized by Invitrogen (USA).
Lentivirus grain package and
titration
Various restriction enzymes, T4 ligase, pyrobest DNA
polymerase, and a DNA fragment purification kit were purchased from
Gibco. A site-directed mutagenesis kit was purchased from Takara
Bio, Inc. (Japan). A mouse testis cDNA library, lentiviral vector
plasmids pLJM-GFP, lentiviral packaging plasmids Delta891, pVSVG,
and 293T cell packaging virus were from the Laboratory of Cell
Biology, Nanjing Medical University. The pCMV-Tag2B-GIT1WT,
pCMV-Tag2B-GIT1Y293F, pCMV-Tag2B-GIT1Y321F and pCMV-Tag2B-GIT1Y392F
vectors were a kind gift of Professor Bradford C. Berk (University
of Rochester, NY). The GIT1, GIT1Y293F and GIT1Y321F vectors were
constructed into pLJM to form pLJM-GIT1, pLJM-GIT1Y293F and
pLJM-GIT1Y321F. These plasmids and supporting original viral
packaging (delta891 and pVSVG) were transfected into 293T cells.
The original medium was replaced by complete medium after 6 h
transfection and was incubated for 48 h to allow for viral
replication. The supernatant containing lentiviral particles was
collected and concentrated. The virus titer was determined by a
previously described method and stored at −80°C (14).
Isolation and culture of osteoblasts and
group
Primary cultures of neonatal rat calvarial (NRC)
cells were obtained from the calvaria of 1-2-day-old SD rat pups by
the method described by Frick and Bushinsky with some modifications
(15). All procedures were
performed in accordance with Nanjing Medical University Animal Care
Committee guidelines. Briefly, neonatal rats were sacrificed by 75%
ethanol soak for 20 min, and the calvaria were immediately
dissected and placed in chilled DMEM. The calvaria were cleaned of
all loose tissues, including the periosteum and dura mater, and
washed in a dilute solution of phosphate-buffered saline (PBS).
Cells were then released by five sequential 10-min digestions with
buffer containing 0.1% collagenase and 0.2% hyaluronidase.
Fractions 2–5 were collected, pelleted by centrifugation at 1,500
rpm for 3 min, resuspended in DMEM, and plated in T-75 flasks.
Cells were grown in DMEM supplemented with 10% FBS, 100 IU/ml
penicillin, and 100 IU/ml streptomycin and incubated at 37°C in a
5% CO2 atmosphere. Confluent cells were passaged in
medium with 0.25% trypsin and EDTA and plated onto petri dishes and
allowed to grow to confluence. Culture plates were divided into a
non-pretreated group and pretreated group. Osteoblast cultures in
the non-pretreated group were transfected with different virus
solutions, starved for 6 h in basal medium, then stimulated for 0
(baseline), 0.5, 2, 6 or 8 h with 10 ng/ml PDGF. Osteoblast
cultures in the pretreated group were starved 6 h after infection,
then pretreated with the MEK inhibitor PD98059 (10 μM) or
the Src inhibitor PP2 (10 μM) for 30 min, followed by PDGF
treatment at the same concentration and treatment durations as in
the non-pretreated group. Following PDGF treatment, cells were
harvested for detection of protein and mRNA expression.
Western blot analysis assays for
expression of pERK1/2, ERK1/2, GIT1 and tyrosine phosphorylation
GIT1 in osteoblasts, VEGF and GIT1 from callus tissue
Cells from each treatment group were lysed and
proteins were isolated. Protein concentrations were determined by
the BCA protein assay. Separated proteins were transferred to
polyvinylidene fluoride (PVDF) membranes at 100 V. Membranes were
blocked for 2 h in 5% bovine serum albumin (BSA) in PBS and then
incubated overnight at 4°C in primary antibody. Antibody-treated
membranes were washed and incubated in the appropriate secondary
antibody (1:5,000) at 37°C for 1 h. Antibody staining was
visualized using enhanced chemiluminescence.
Immunoprecipitation assays to detect the
association of GIT1 and GIT1Y321F to ERK1/2
Protein samples (100 μg) from each group were
mixed with 1 μg anti-Flag antibody and incubated overnight
at 4°C with slow shaking. Protein G/A agarose beads were added and
the mixture incubated for 2 h at 4°C. The mixture was centrifuged
at 5,000 rpm for 3 min at 4°C. The supernatant was discarded and
the beads washed in 1 ml PBS, then centrifuged again at 5,000 rpm
for 3 min at 4°C. Washing was repeated two more times. Sufficient
sample buffer was added in the precipitate followed by boiling at
100°C for 5 min. The beads were then centrifuged at 5,000 rpm for 5
min at 4°C. After cooling, the supernatant was loaded onto a gel
for electrophoretic separation and western blotting as described
above.
Bone fracture modeling in rats
Rats (200–300 g) were injected intraperitoneally
with 10% chloral hydrate. The dorsal side of one leg was shaved and
sterilized with an iodine solution. An open fracture was created in
the tibia using a modification of the method (16). Thirty rats with tibia fractures
were divided randomly into three groups of 10. The first group was
injected with GIT1WT lentivirus at the fracture site, the second
group was injected with mutant GIT1Y293F virus, and the third group
was injected with mutant GIT1Y321F virus at the fracture site. The
three groups were housed separately during recovery without any
limitation on activity. Starting at 1–3 days after surgery, rats
were treated daily with 4,000 M gentamicin i.m. to prevent
infection. Rats were sacrificed at 5, 7, 10 and 14 days after
fracture. The tibia fracture site was X-rayed before sacrificing
the animals. Following sacrifice, we examined the range of calli
derived and randomly selected rats for immunohistochemical analysis
and western blotting.
RT-PCR
Callus at the site of fracture were collected to
extract total-RNA for amplification. Total-RNA was amplified by two
pairs of primers: sense, 5′-TGCGATGCGGGGGCTGC-3′ and antisense,
5′-TTTCCTGGTGAGAGTCT-3′. The PCR reaction was run in a 50 μl
volume. The thermocycle settings used were 36 cycles of 94°C for 30
sec, 55°C for 30 sec, and 72°C for 30 sec. After the reaction, 4
μl of the reaction solution was combined with 1 μl
bromophenol blue loading buffer and run on 1.5% agarose gels (85 V)
and bands were analyzed using a UVP GDS 8000 gel imaging analysis
system camera (UVP, Cambridge, UK).
Immunohistochemistry
Tissue samples from the fracture site were isolated
at various times following the injury and consecutive paraffin
sections were prepared. For immunohistochemical staining, sections
were first dewaxed in xylene. Dewaxed sections were soaked for 10
min in 3% H2O2 solution in methanol to
eliminate endogenous catalase. Sections were then washed 3 times
with PBS wash, treated with 10% goat serum for 10 min and incubated
in rat anti-mouse VEGF antibody overnight in a wet box. Labeled
sections were washed 3 times in PBS (2 min each), treated with
anti-rat biotinylated secondary antibody for 10 min, washed as
before in PBS, incubated in HRP-streptavidin for 10 min, washed in
PBS, and finally treated with DAB color liquid at ambient
temperature. For quantification, we first selected the middle
section of the callus. Multiple sections were used to count the
number of VEGF-positive cells under light microscopy.
Immunofluorescence detection of PECAM-1
expression in callus tissue
The methods for PECAM-1 staining were as above
except that the primary antibody (anti-PECAM-1) was labeled with a
rhodamine-conjugated secondary goat anti-mouse antibody for 1 h at
37°C. Sections were counterstained with DAPI nuclear stain for 5–10
min and mounted in 80% glycerol/PBS. Rhodamine and DAPI
fluorescence were imaged using a Nikon 80i fluorescence microscope.
Photomicrographs were processed using Photoshop CS.
Statistical methods
The SPSS 11.0 statistical package was used for all
statistical analysis. Measures are expressed as means ± SD and
analyzed by ANOVA with P<0.05 considered statistically
significant.
Results
PDGF increased ERK1/2 activation, GIT1
phosphorylation, the association of GIT1 with ERK1/2, and VEGF mRNA
expression
Our previous studies revealed that EGF, angiotensin
II, and VEGF stimulate different cell types to activate ERK1/2
activity, increase tyrosine phosphorylation of GIT1, and promote
protein-protein interactions between GIT1 and ERK1/2. In this
study, osteoblasts were treated with PDGF (10 ng/ml) and ERK1/2
activation, VEGF mRNA expression, and GIT1-ERK1/2 interaction were
examined. Indeed, ERK1/2 activation (pERK1/2) was significantly
increased after only 0.5 h exposure to PDGF but decreased with
longer stimulation times (Fig.
1A). In contrast, PDGF increased GIT1 tyrosine phosphorylation
in osteoblasts and this response was observed for PDGF treatments
up to 6 h and began to decline thereafter) (Fig. 1B). In osteoblasts treated with
PDGF for 0.5 h, GIT1 binding to ERK1/2 was enhanced significantly,
and like ERK1/2 phosphorylation, decreased back to baseline as the
PDGF treatment duration was extended (Fig. 1C). The expression of VEGF mRNA in
osteoblasts increased during PDGF stimulation, peaking at 8 h (the
longest treatment time assessed) (Fig. 1D). To clarify the relationship
between ERK1/2 activation, GIT1 phosphorylation, GIT1-ERK binding,
and VEGF mRNA expression, we performed these same western blotting
experiments on lysates from osteoblasts pretreated with the ERK
pathway inhibitor PD98059 and the Src inhibitor PP2. Pretreatment
with PD98059 inhibited PDGF-induced ERK1/2 activity and
simultaneously inhibited expression of VEGF mRNA in osteoblasts
(Fig. 2A and D). Pretreatment
with PP2 inhibited PDGF-induced tyrosine phosphorylation of GIT1
(Fig. 2B), the interaction of
GIT1 with ERK1/2 (Fig. 2C), and
significantly reduced VEGF mRNA expression in osteoblasts (Fig. 2D). These results indicate that
ERK1/2 phospho-activation, GIT1 phosphorylation, GIT1-ERK binding,
and VEGF mRNA expression in response to PDGF depend on Src and
MEK1/2 activation.
Interaction of GIT1 with ERK1/2 and VEGF
expression in osteoblasts depends on GIT1 tyrosine 321
phosphorylation
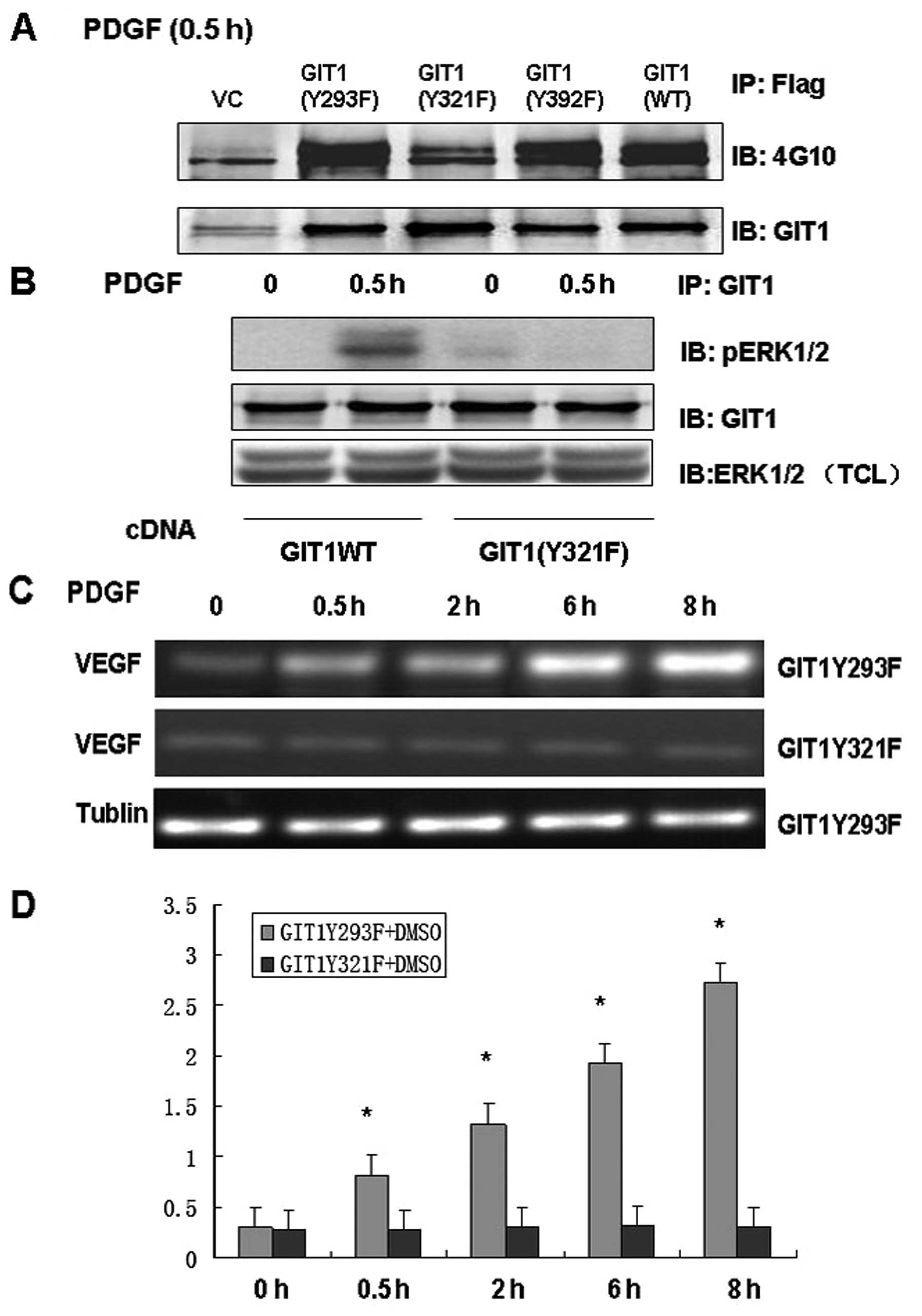
Our previous studies demonstrated that tyrosine
phosphorylation of GIT1 altered ERK1/2 activity and that GIT1
tyrosine 321 played an important role. To further clarify the role
of Y321 phosphorylation in GIT1-ERK1/2 interaction and VEGF mRNA
expression, we infected osteoblasts with mutant or wild type GIT1
and analyzed the responses to PDGF stimulation. We constructed
lentiviral vectors including GIT1WT, pCMV-Tag2B-GIT1Y293F,
pCMV-Tag2B-GIT1Y321F and pCMV-Tag2B-GIT1Y392F. In osteoblasts
treated with PDGF, infection with GIT1WT, GIT1Y293F and GIT1Y392F
significantly increased tyrosine phosphorylation, while
phosphorylation was completely inhibited in cells infected with a
virus containing GIT1Y321F (Fig.
3A). This suggests that Y321 is the target of PDGF-mediated
phosphorylation. Furthermore, immunoprecipitation demonstrated that
GIT1Y321F inhibited the interaction of GIT1 with ERK1/2, and that
the PDGF-induced increase in pERK1/2 activity was effectively
blocked in osteoblasts expressing mutant GIT1Y321F (Fig. 3B and C). These results indicate
that GIT1Y321 is the key tyrosine phosphorylation site that
regulates both the interaction between ERK1/2 and GIT1 and
PDGF-induced ERK1/2 activity. In order to determine the role of
Y321 phosphorylation in VEGF mRNA expression, we treated
osteoblasts expressing GIT1Y293F and GIT1Y321F with PDGF and
analyzed VEGF mRNA expression. Infection with GIT1Y293F had no
effect on PDGF-induced VEGF mRNA expression, while GIT1Y321F
infection significantly inhibited VEGF mRNA expression in
osteoblasts (Fig. 3D). Thus, Y321
is the key phosphorylation site mediating PDGF-induced ERK1/2
activity, ERK1/2-GIT1 interactions and VEGF mRNA expression in
osteoblasts.
Phosphorylation of GIT1Y321 regulates
VEGF expression, angiogenesis, and healing at the fracture
callus
We demonstrated that PDFG-induced VEGF expression in
cultured osteoblasts depended on phosphorylation of GIT1Y321,
ERK1/2 activation, and GIT1-ERK1/2 interaction. VEGF acts on
endothelial cells to trigger angiogenesis, a process that is
critical from ossification and fracture healing. We examined the
effects of GIT1WT, GIT1Y293F and GIT1Y321F lentiviral infection in
a rat tibia fracture model. A group of rats were anesthetized and
injured by a single fracture to the tibia. One group was injected
with a lentivirus containing GIT1WT and the others with GIT1Y293F
and GIT1Y321F lentivirus respectively. VEGF expression was
determined at the fracture site (Fig.
4A). At 5, 7, 10 and 14 days after lentiviral infection, we
detected VEGF protein expression. Expression of VEGF was
significantly elevated at 5, 7, 10 and 14 days after infection with
GIT1WT and GIT1Y293F mutants, while VEGF expression in GIT1Y321F
lentiviral infected bone was barely above baseline (Fig. 4B and C). To confirm that VEGF
protein expression was inhibited by GIT1Y321F infection, callus
sections were stained by immunohistochemistry. As shown in Fig. 5A, there was significantly higher
VEGF immuno-reactivity in calli sections infected with GIT1WT
lentivirus compared to GIT1Y321F lentivirus in 7 and 14 days. VEGF
is a major inducer of angiogenesis, so reduced expression would
tend to suppress angiogenesis. To confirm that reduced VEGF
expression did indeed result in lower angiogenesis at the fracture
site, we stained calli sections with an antibody against the
angiogenic marker protein PECAM-1. As shown in Fig. 5B, 7 and 14 days after fracture,
angiogenesis was significantly lower at fracture sites injected
with GIT1Y321F lentiviral than at sites injected with GIT1WT
lentivirus. Previous studies demonstrated that reduced angiogenesis
will impede fracture healing. As predicted the healing of the
tibial fracture was greatly reduced on Day 14 by GIT1Y321F
lentiviral infection (Fig. 5D).
In contrast, fractures infected with GIT1WT were fully healed on
Day 14, indicating that tyrosine 321 phosphorylation is critical
for bone healing by upregulating the expression of the angiogenic
factor VEGF.
Discussion
Fracture healing is a complex physiological process
involving the coordinated expression of immune, angiogenic, and
ossification factors. VEGF is a disulfide-linked homodimer
glycoprotein that promotes endothelial cell proliferation and
angiogenesis during development. At a fracture site, VEGF also
promotes vascular permeability, cartilage cell differentiation,
osteoblast differentiation, and osteoclast recruitment, all vital
processes for new bone formation (17). Thus, VEGF is a major regulator of
fracture healing. Promotion of angiogenesis facilitates bone
healing, while VEGF indirectly induces osteoblast proliferation,
migration, and differentiation by stimulating endothelial cells to
secrete several osteo anabolic factors, including endothelin-I and
insulin-like growth factor-I (17). Vascularization by VEGF promotes
cartilage absorption, ossification, and subperiosteal bone
formation (18). Niida et
al (19) have shown that VEGF
promotes osteoclast differentiation and bone resorption similar to
that evoked by macrophage colony-stimulating factor (M-CSF).
VEGF is a multifunctional cytokine secreted by
platelets, megakaryocytes, endothelial cells, osteoblasts, and
tumor cells in response to a multitude of extracellular signals.
Deckers et al (20)
confirmed that proliferation of osteoblasts could regulate VEGF
release and that bone morphogenic proteins stimulated osteoblasts
to secrete VEGF. Induction of VEGF by IGF-I in osteoblast-like
cells was mediated by the PI3K signaling pathway. Transforming
growth factor-β (TGF-β) stimulated heat shock protein 27 (HSP27)
induction via p44/p42 mitogen-activated protein (MAP) kinase, p38
MAP kinase and stress-activated protein kinase/c-Jun N-terminal
kinase in osteoblast-like MC3T3-E1 cells, and that the release of
VEGF was induced by TGF-β in these cells (21). Stimulation of EGF receptors
regulated the production of VEGF through the ERK1/2 pathway
(22). Application of the ERK1/2
inhibitor 6-thioguanine inhibited the secretion of VEGF, as did
application of the MEK1 inhibitor PD98059 (23), underscoring the importance of
MEK1/ERK1/2 signaling in the regulation of VEGF. The VEGF promoter
region contains binding sites for the transcription factor Sp1, and
Sp1 activity is controlled by extracellular signals through ERK1/2.
Indeed, ERK1/2 activity and phosphorylation are correlated with Sp1
activity to regulate the expression of VEGF (24).
We have previously reported that GIT1 regulates
ERK1/2 activity through direct protein-protein interactions, likely
by acting as a specific shuttle protein in the MEK1/ERK1/2
signaling pathway (25). The GIT1
protein is an important signaling protein in osteoblasts. The
interaction of GIT1 with signaling molecules is mediated by
multiple interacting domains, including an N-terminal ARFGAP
domain, an ankyrin repeats (ANK) domain, a PIX-binding domain (Spa2
homology domain 1, SHD) and a C-terminal paxillin-binding domain
(26). The ARF-GAP domain of GIT1
regulates ARF1, ARF5, ARF6 and other signaling molecules to control
formation of vesicles from the Golgi complex, in addition to roles
in endosome-mediated intermembrane and membrane transport, cell
proliferation and migration, and synaptogenesis (27). At the SHD domain of GIT1, a
complex set of protein interactions occurs to form large oligomeric
protein complexes with a multitude of functions. The main binding
partner for this domain is p21-activated kinase interacting
exchange factor (PIX). We have previously reported that GIT1,
through the SHD domain, allows the GIT1 CC2 domain to combine with
MEK1 and ERK1/2 to facilitate ERK1/2 activation (28). We also found that GIT1 was
tyrosine phosphorylated by Src, while the Src inhibitor PP2
inhibited tyrosine phosphorylation of GIT1 in osteoblasts in
vitro (7,13).
Tyrosine phosphorylation is essential to GIT1
function. Mass spectrometry found that the SHD and CC2 regions have
three major tyrosine phosphorylation sites at 293, 321 and 392
(11). Segura et al
(29) demonstrated that
phosphorylation at GIT1 tyrosine 392 could promote protein-protein
interaction with Grb4 and modulate spine morphogenesis and synapse
formation. We also reported that GIT1 tyrosine 392 is critical for
PDBU-induced podosome formation by regulating PLC activation. Here,
we confirm that GIT1Y321 phosphorylation is a key signal for the
activation ERK1/2 and show that this signaling pathway is a major
regulator of VEGF expression and fracture healing.
The results of this study indicate that GIT1Y321
promotes VEGF release to facilitate fracture healing through the
MEK/ERK signaling pathway. These results suggest that VEGF
expression through ERK1/2 signaling could be manipulated to enhance
fracture healing, but the details of these signaling pathway,
specifically how pY321 activates ERK and how ERK evokes VEGF
release, require further study. The GIT1 is a central mediator of
VEGF-regulated healing of nonunion bone fractures that may be
exploited clinically in the future.
Acknowledgements
This study was supported by the
National Natural and Science Foundation (81071481).
References
|
1.
|
G AugustinA AntabakS DavilaThe periosteum.
Part 1: anatomy, histology and molecular
biologyInjury3811151130200717889870
|
|
2.
|
ML BrandiP Collin-OsdobyVascular biology
and the skeletonJ Bone Miner
Res21183192200610.1359/JBMR.05091716418774
|
|
3.
|
H EckardtM DingM LindRecombinant human
vascular endothelial growth factor enhances bone healing in an
experimental nonunion modelJ Bone Joint Surg
Br8714341438200510.1302/0301-620X.87B10.1622616189323
|
|
4.
|
DE RichardE BerraE Gothiep42/44
mitogen-activated protein kinases phosphorylate hypoxia-inducible
factor1α (HIF-1) and enhance the transcriptional activity of HIF-1J
Biol Chem2743263132637199910551817
|
|
5.
|
G PagesJ MilaniniDE RichardSignaling
angiogenesis via p42/p44 MAP kinase cascadeJ Ann NY Acad
Sci902187200200010.1111/j.1749-6632.2000.tb06313.x10865838
|
|
6.
|
V WitteB LaffertP GintschelInduction of
HIV transcription by Nef involves Lck activation and protein kinase
C theta raft recruitment leading to activation of ERK1/2 but not NF
kappa BJ
Immumol18184258432200810.4049/jimmunol.181.12.842519050260
|
|
7.
|
G YinJ HaendelerC YanGIT1 functions as a
scaffold for MEK1-extracellular signal-regulated kinase 1 and 2
activation by angiotensin II and epidermal growth factorMol Cell
Biol24875885200410.1128/MCB.24.2.875-885.200414701758
|
|
8.
|
J PangR HoefenGS PryhuberG-protein-coupled
receptor kinase interacting protein-1 is required for pulmonary
vascular
developmentCirculation11915241532200910.1161/CIRCULATIONAHA.108.82399719273721
|
|
9.
|
CE TurnerMC BrownJA PerrottaPaxillin LD4
motif binds PAK and PIX through a novel 95-kD ankyrin repeat,
ARF-GAP protein: a role in cytoskeletal remodelingJ Cell
Biol145851863199910.1083/jcb.145.4.85110330411
|
|
10.
|
G YinC YanBC BerkAng II signaling pathways
mediated by tyrosineInt J Biochem Cell
Biol35780783200310.1016/S1357-2725(02)00300-X12676164
|
|
11.
|
DJ WebbMW MayhewM KovalenkoIdentifucation
of phosphorylation sites in GIT1J Cell
Sci11928472850200610.1242/jcs.0304416825424
|
|
12.
|
J WangG YinP MenonPhosphorylation of G
protein-coupled receptor kinase 2-interacting protein 1 tyrosine
321 is required for phospholipase C-gamma activation and podosome
formation in vascular smooth muscle cellsArterioscler Thromb Vasc
Biol3019761982201010.1161/ATVBAHA.110.212415
|
|
13.
|
G YinQ ZhengC YanGIT1 is a scaffold for
ERK1/2 activation in focal adhesionsJ Biol
Chem2802770527712200510.1074/jbc.M50227120015923189
|
|
14.
|
G TiscorniaO SingerIM VermaProduction and
purification of lentiviral vectorsNat
Protoc1241245200610.1038/nprot.2006.3717406239
|
|
15.
|
KK FrickDA BushinskyChronic metabolic
acidosis reversibly inhibits extracellular matrix gene expression
in mouse osteoblastsAm J Physiol Renal
Physiol275F840F84719989815143
|
|
16.
|
S SakanoY ZhuL SandellCartilage-derived
retinoic acid-sensitive protein and type II collagen expression
during fracture healing are potential targets for Sox9 regulationJ
Bone Miner
Res1418911901199910.1359/jbmr.1999.14.11.189110571689
|
|
17.
|
H MayerH BertramW LindenmaierVascular
endothelial growth factor (VEGF-A) expression in human mesenchyreal
stem cells: autocrine and paracrine role on osteoblastic and
endothelial differentiationJ Cell
Biochem95827839200510.1002/jcb.2046215838884
|
|
18.
|
SE AldridgeTW LennardJR WilliamsMA
BirchVascular endothelial growth factor receptors in osteoclast
diferentiation and functionBiochem Biophys Res
Commun335793798200510.1016/j.bbrc.2005.07.14516105658
|
|
19.
|
S NiidaT KondoS HiratsukaVEGF receptor 1
signaling is essential for osteoclast development and bone marrow
formation in colony-stimulating factor 1-deficient miceProc Natl
Acad Sci USA1021401614021200510.1073/pnas.050354410216172397
|
|
20.
|
MM DeckersRL van BezooijenG van der
HorstBone morphogenetic proteins stimulate angiogenesis through
osteoblast-derived vascular endothelial growth factor
AEndocrinology14315451553200210.1210/endo.143.4.8719
|
|
21.
|
K KatoH TokudaS AdachiRole of heat shock
protein 27 in transforming growth factor-β stimulated vascular
endothelial growth factor release in osteoblastsInt J Mol
Med274234282011
|
|
22.
|
D FeldserF AganiNV IyerReciprocal positive
regulation of hypoxia-inducible factor 1alpha and insulin-like
growth factor 2Cancer Res5939153918199910463582
|
|
23.
|
S SalcedaI BeckV SrinivasJ CaroComplex
role of protein phosphorylation in gene activation by hypoxiaKidney
Int51556559199710.1038/ki.1997.789027738
|
|
24.
|
J WuS BrandtSM HuderLingand and
cell-specific effects of signal transduction pathway inhibitors on
progestin-induced vascular endothelial growth factor levels in
human breast cancer cellsMol
Endocrinol19312326200510.1210/me.2004-0252
|
|
25.
|
KP HofmannP ScheererPW HildebrandA G
protein-coupled receptor at work the rhodopsin modelTrends Biochen
Sci341102211027200919836958
|
|
26.
|
A ClaingW ChenWE
MillerBeta-Arrestin-mediated ADP-ribosylation factor 6 activation
and beta 2-adrenergic receptor endocytosisJ Biol
Chen2764250942513200110.1074/jbc.M10839920011533043
|
|
27.
|
J OverbaughAD MillerMV EidenReceptors and
entry cofactors for retroviruses include single and multiple
transmembrane-spanning proteins as well as newly described
glycophosphatidylinositol-anchored and secreted proteinsMicrobiol
Mol Biol Rev65371389200110.1128/MMBR.65.3.371-389.2001
|
|
28.
|
J FernandezI YamanWC MerrickRegulation of
internal ribosome entry site-mediated translation by eukaryotic
initiation factor-2alpha phosphorylation and translation of a small
upstream open reading frameJ Biol
Chem27720502058200210.1074/jbc.M109199200
|
|
29.
|
I SeguraCL EssmannS WeingesA
Acker-PalmerGrb4 and GIT1 transduce ephrinB reversesignals
modulating spine morphogenesis and synapse formationNat
Neurosci10301310200710.1038/nn185817310244
|