Introduction
Human hepatocellular carcinoma (HCC) is the fifth
most common malignancy worldwide, and is the third leading cause of
cancer-related mortality. The incidence and mortality of HCC is
inhomogenous in undeveloped and developed countries, and the
majority of HCC occurs in Asia and Africa. However, its incidence
has increased rapidly in the past two decades in Western Europe
(1,2). Despite the magnitude of the disease,
existing treatments are of limited efficacy. Less than 30% of the
patients are suitable for curative treatment, and the recurrence is
a frequent issue following tumor ablation (3). Accordingly, new novel therapeutic
approaches for HCC are urgently required.
Among the molecular mechanisms of HCC, several
signaling pathways play a vital role in hepatocarcinogenesis
(4), and the phosphoinositide
3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) signaling
is aberrantly activated in HCC. The PI3K/AKT/mTOR signaling pathway
has been the focus of research in recent years due to its potential
role in cancer (5,6). The PI3K/AKT/mTOR signaling pathway
acts as a convergence for numerous upstream signals and stimulates
the activity of a multitude of downstream effectors, thereby
mediating cellular survival, growth (5). Therefore, it is not surprising that
aberrant and deregulation activation of this signaling is a common
molecular event in several types of malignancy (6). These insights have resulted in the
development of novel therapies targeting constituents of this
pathway (single or multiple PI3K/mTOR inhibitors) and currently
several inhibitors are in clinical trials (7–9).
Therefore, targeting the PI3K/AKT/mTOR signaling has become the
novel therapeutic approach for human HCC treatment (9).
Autophagy is a cellular lysosomal degradation
pathway that is essential for regulation of cell survival and death
to maintain cellular homeostasis (10,11). One of the key regulators of
autophagy is mTOR, which is the major inhibitory signal that shuts
off autophagy in the presence of growth factors and abundant
nutrients (11). Accordingly,
inhibition of mTOR signaling (e.g., by the mTOR inhibitor
rapamycin) induces autophagy (12). Autophagy can be either a
pro-survival or death mechanism depending on the circumstances
(10,11), thereby generating a variable
impact on the outcome of cancer therapy. In the present study, we
investigated whether inhibition of autophagy would enhance the
apoptosis of HCC cells.
We focused on the role of NVP-BEZ235, the effective
PI3K/mTOR dual inhibitor on cancer therapy (13–15) on the induction of apoptosis in
human HCC cell lines and explored the impact of autophagy induction
on its anticancer activity against HCC. We also examined if
combination of autophagy inhibitors with NVP-BEZ235 could enhance
the apoptosis of HCC cells.
Materials and methods
Materials
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT) and 3-methyladenine (3-MA) were purchased from Sigma.
Atg5 siRNA plasmid was purchased from Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA (sc-29918). Dulbecco’s modified Eagle’s
medium (DMEM) and fetal bovine serum (FBS) were purchased from
Gibco. The antibodies anti-caspase-3, anti-cleaved caspase-3,
anti-PARP, anti-LC3, anti-Atg5, anti-AKT, anti-p-AKT, anti-p70S6K,
anti-p-p70S6K and anti-p62 were purchased from Santa Cruz
Biotechnology, Inc. NVP-BEZ235 was purchased from LC
Laboratories.
Cell culture
The human Hep3B and PLC/PRF/5 cells were cultured in
DMEM with 10% FBS, under standard culture conditions (37°C and 5%
CO2).
Cell viability assays
Hep3B and PLC/PRF/5 cells were cultured in 96-well
plates at a density of 1×104 cells/well in 100 μl
of complete medium. Each group was repeated in nine separate wells.
MTT reagent [12 μl, 5 mg/ml in phosphate-buffered saline
(PBS)] was added to each well for 4–6 h. Following treatment, each
well was dissolved in 150 μl DMSO. Absorbance was recorded
at a wavelength of 490 nm.
Western blot analysis
Hep3B and PLC/PRF/5 cells were washed with cold PBS
and then 300 μl radioimmunoprecipitation (RIPA) buffer [50
mM Tris-HCl (pH 6.8), 0.1% SDS, 150 mM NaCl, 1 mM EDTA, 0.1 mM
Na3VO4, 1 mM sodium fluoride (NaF), 1% Triton
X-100, 1% NP-40, 1 mM dithiothreitol, and 1 mM PMSF, 1 μg/ml
aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A]
were added to each group. Cell lysates were then shaken in a cold
room (4°C) for 30 min and centrifuged at 14,000 × g for 10 min.
Protein concentrations in the supernatants were detected using the
BCA protein assay. For western blot analysis, 50 μg proteins
were separated by 12% (w/v) SDS-polyacrylamide gel electrophoresis.
After running gels, proteins were transferred onto PVDF membranes
and were then blocked with 5% (w/v) skim milk in buffer [10 mM
Tris-HCl (pH 7.6), 100 mM NaCl, and 0.1% (v/v) Tween-20] for 2 h at
room temperature (25°C); the primary antibodies were added
overnight on the shaker in a cold room. Then, PVDF membranes were
incubated with secondary antibodies (Sigma) for 1 h at room
temperature. The semi-quantitation of proteins was surveyed with a
Tanon GIS gel imager system.
Atg5 siRNA transfection
Prior to transfection, 50% Hep3B and PLC/PRF/5 cells
were grown in each dish. These cells were transfected with 50
nmol/l of siRNA using Lipofectamine RNAiMAX (Invitrogen, Carlsbad,
CA, USA) according to the manufacturer’s protocol. Cells were
harvested for western blotting at 24–48 h post transfection.
Mitochondrial membrane potential (MMP)
analysis
To detect the change of MMP, we used the JC-1
staining through flow cytometry. The assay was performed according
to the manufacturer’s instructions (Invitrogen). Briefly, Hep3B and
PLC/PRF/5 cells were trypsinized, washed with PBS twice, and
resuspended in PBS at a concentration of 1–1.5×106
cells/ml. Hep3B and PLC/PRF/5 cells were then stained with 3
μl of JC-1 (1 mg/ml) and incubated in the dark at 37°C for 1
h. The JC-1 positive cells were subsequently detected by a
FACSCalibur flow cytometer.
Statistical analysis
The data were analyzed by t-test. P<0.05 was
considered to represent a statistically significant difference.
Data are representative of three independent experiments performed
in triplicate.
Results
NVP-BEZ235 (NVP) inhibits growth and the
PI3K/mTOR pathway of the HCC cell lines Hep3B and PLC/PRF/5
Overexpression of PI3K/AKT/mTOR signaling has been
reported in several types of cancer, including HCC (5,9).
Targeting the PI3K/AKT/mTOR signal pathway may be the novel
therapeutic approach for HCC. NVP-BEZ235 is a novel, orally
bioavailable dual PI3K/mTOR inhibitor that has exhibited promising
activity in preclinical models (13,14). First, we used MTT to detect the
effect of NVP on the two HCC cell lines, Hep3B and PLC/PRF/5. NVP
decreased the cell viability of the two cell lines in a
dose-dependent manner (Fig. 1A and
B). We also detected the expression of PI3K/mTOR pathway
proteins in the two cell lines treated with NVP. NVP downregulated
the expression of p-AKT and p-p70S6K (Fig. 1C and D).
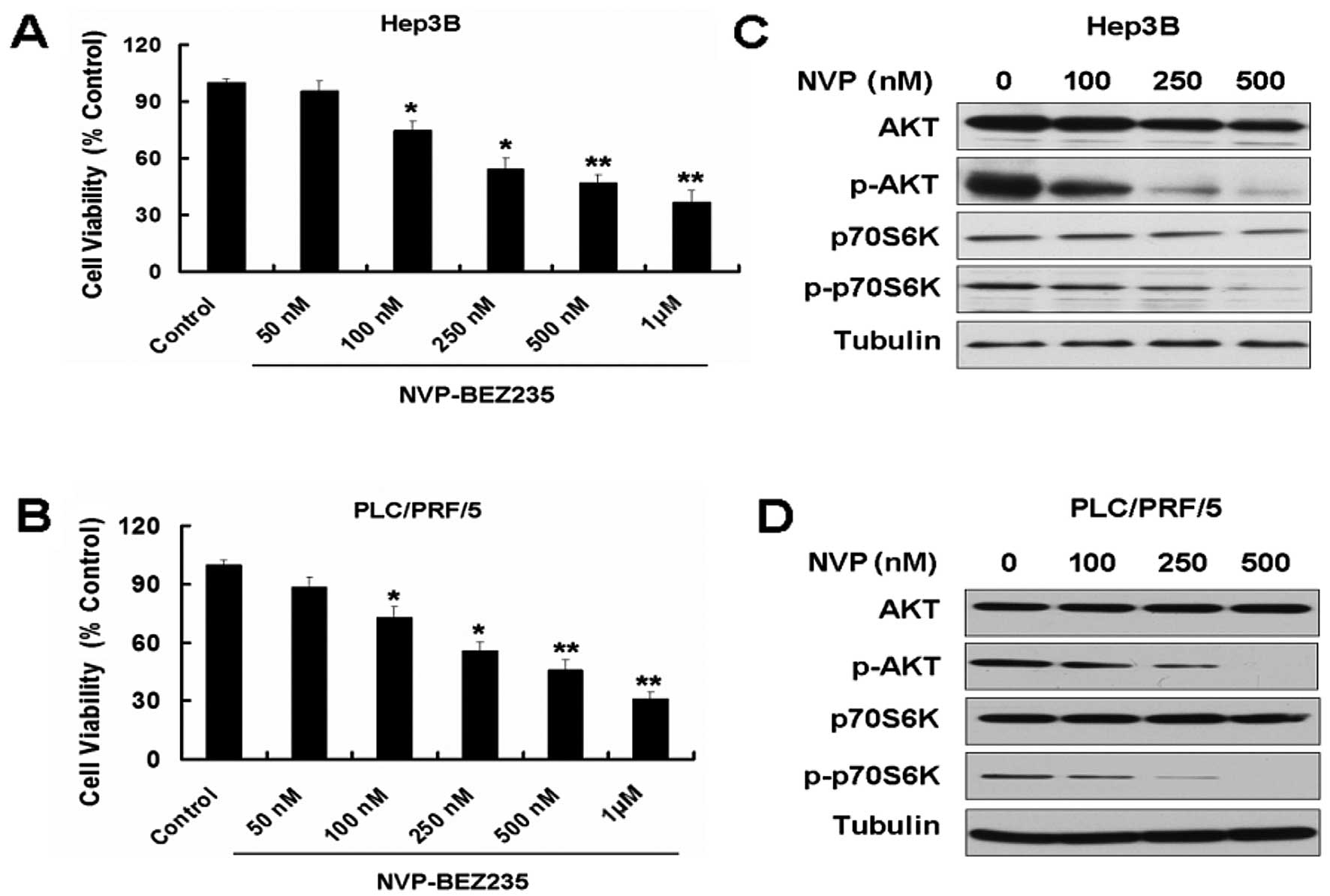 | Figure 1NVP-BEZ235 (NVP) inhibits growth and
the PI3K/mTOR pathway of the hepatocellular carcinoma (HCC) cell
lines Hep3B and PLC/PRF/5. (A) Hep3B cells were treated with 50,
100, 250, 500 and 1,000 nM NVP for 24 h. Cell viability was
determined by MTT assay. Data are presented as the means ± SD, n=9.
(B) PLC/PRF/5 cells were treated with 50, 100, 250, 500 and 1,000
nM NVP for 24 h. Cell viability was determined by MTT assay. Data
are presented as the means ± SD, n=9. *P<0.05 vs.
control group; **P<0.01 vs. control group. (C)
Western blot analysis for the expression of AKT, p-AKT, p70S6K and
p-p70S6K in Hep3B cells treated with 100, 250 and 500 nM NVP for 24
h. (D) Western blot analysis for the expression of AKT, p-AKT,
p70S6K and p-p70S6K in PLC/PRF/5 cells treated with 100, 250 and
500 nM NVP for 24 h. |
NVP induces apoptosis in Hep3B and
PLC/PRF/5 cells
Based on the above results, we further detected the
apoptosis-related proteins in Hep3B and PLC/PRF/5 treated with NVP.
We detected the apoptotic relative proteins caspase-3 and PARP. NVP
increased the expression of cleaved caspase-3 and cleaved PARP in
Hep3B (Fig. 2A and B). Similar
results can be seen in another HCC cell line, PLC/PRF/5 (Fig. 2C and D). We next measured the MMP
in Hep3B and PLC/PRF/5 cells. NVP clearly induced the loss of the
MMP in Hep3B and PLC/PRF/5 cells (Fig. 2E and F). These results indicate
that NVP induces apoptosis in Hep3B and PLC/PRF/5 cells, possibly
through the mitochondrial apoptotic pathway.
NVP induces autophagy in Hep3B and
PLC/PRF/5 cells
Aside from the effect on tumor cell growth, the
PI3K/AKT/mTOR pathway is also involved in autophagy. We further
detected the autophagy-related proteins in Hep3B and PLC/PRF/5
cells treated with NVP. Microtubule-associated protein LC3 (the
mammalian equivalent of yeast ATG8) and p62 are the two markers for
autophagy. There are two cellular forms of LC3, LC3-I and LC3-II.
LC3-I converts to LC3-II when autophagy occurs and the amount of
LC3-II becomes a marker for the formation of autophagosomes.
Following the occurrence of autophagy, the expression of p62
decreased. We used western blotting to detect the expression of the
two proteins in Hep3B and PLC/PRF/5 cells treated with NVP. NVP
enhanced the expression of LC3-II and decreased the expression of
p62 in Hep3B and PLC/PRF/5 cells, indicating that NVP induces
autophagy in Hep3B and PLC/PRF/5 cells (Fig. 3).
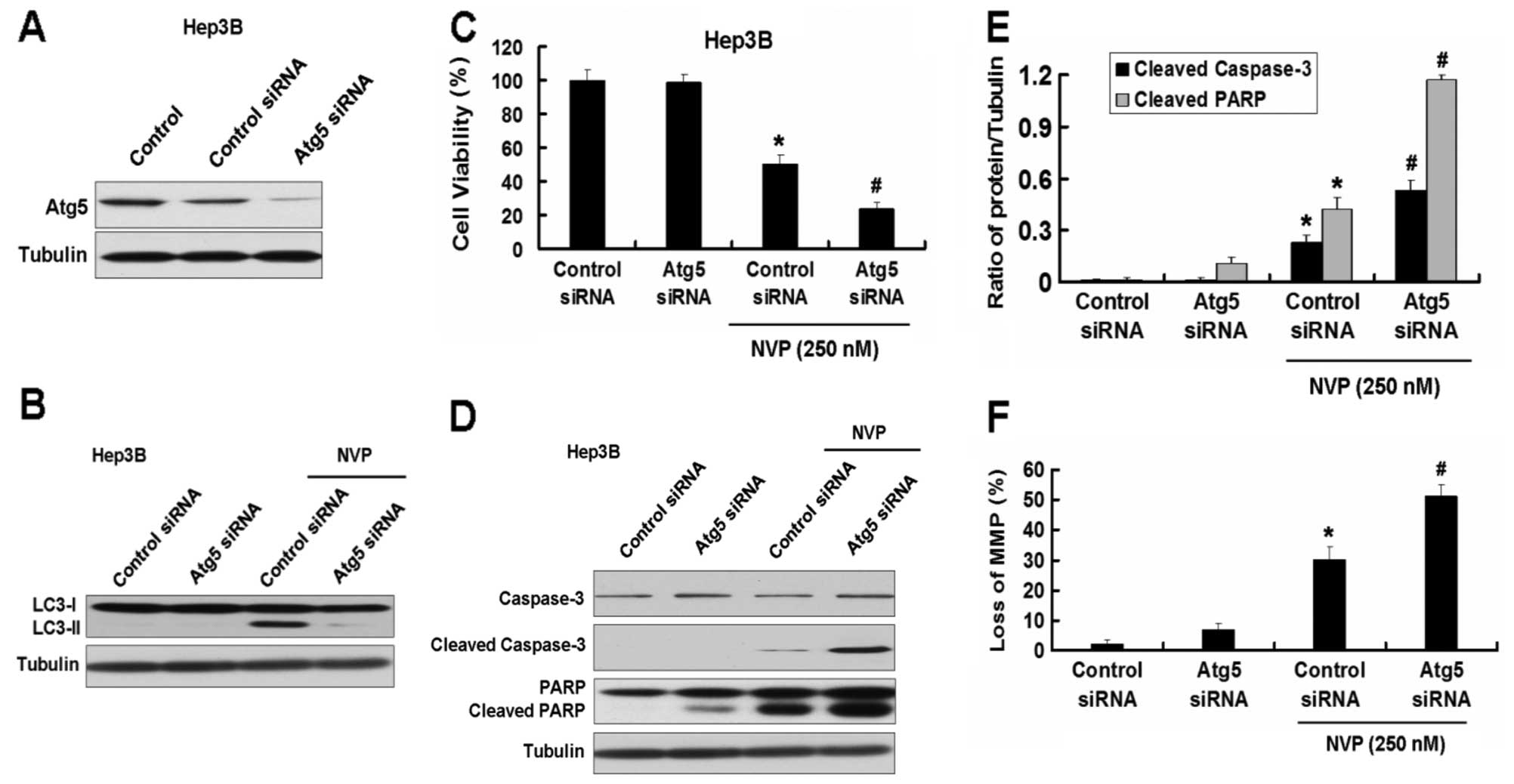
Inhibition of autophagy by Atg5 siRNA
enhances apoptosis induced by NVP in Hep3B and PLC/PRF/5 cells
As shown in the results above, NVP induces apoptosis
and autophagy in HCC cell lines. Autophagy is always considered a
protection process when cells are under low nutrition. However, the
role of autophagy in apoptosis induced by chemicals is uncertain.
We first used the Atg5 siRNA plasmid to explore the role of
autophagy in apoptosis induced by NVP. Atg5 siRNA inhibited the
expression of Atg5 and the increase of LC3-II induced by NVP in
Hep3B cells (Fig. 4A and B).
Inhibition of autophagy by Atg5 siRNA intensified the growth
inhibition of Hep3B cells induced by NVP (Fig. 4C). Meanwhile, Atg5 siRNA further
increased the apoptotic relative proteins cleaved caspase-3 and
cleaved PARP induced by NVP in Hep3B cells (Fig. 4D and E). Atg5 siRNA also increased
the loss of MMP in Hep3B cells induced by NVP (Fig. 5F). Similar results were found in
the other HCC cell line, PLC/PRF/5 (Fig. 5).
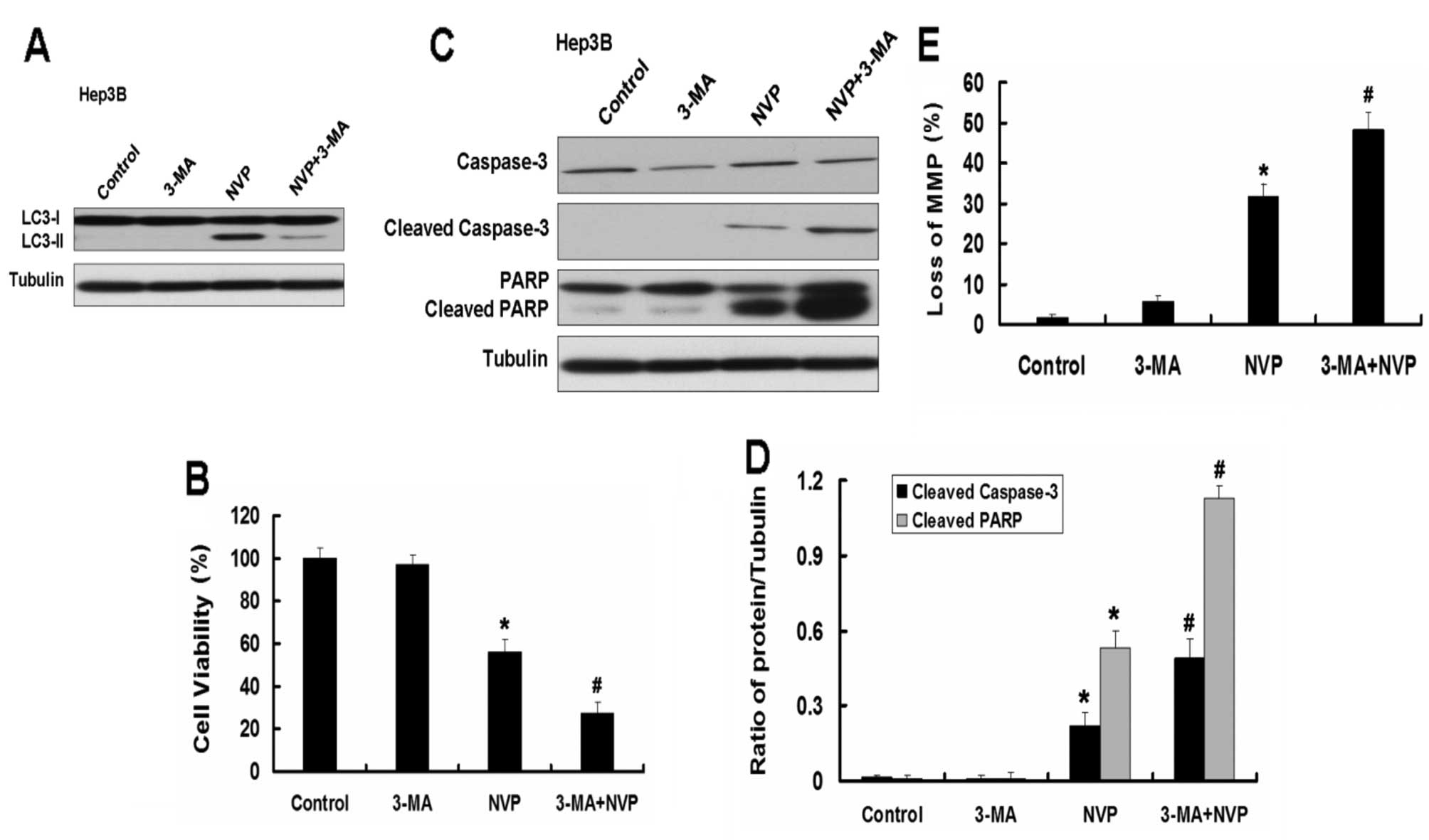
Inhibition of autophagy by autophagy
inhibitor 3-MA enhances apoptosis induced by NVP in Hep3B and
PLC/PRF/5 cells
We further explored the role of autophagy in
apoptosis induced by NVP. We used the autophagy inhibitor 3-MA to
analyze the effect of autophagy on apoptosis in Hep3B and PLC/PRF/5
cells induced by NVP. The autophagy inhibitor 3-MA inhibits the
autophagy induced by NVP (Fig.
6A). As the Atg5 siRNA, 3-MA further augmented the decrease of
cell viability induced by NVP in Hep3B cells. Combination of NVP
and 3-MA further increased the expression of apoptotic relative
proteins cleaved caspase-3 and cleaved PARP and the loss of MMP
induced by NVP in Hep3B cells. Similar results were observed in
PLC/PRF/5 cells (Fig. 7).
These results indicate that autophagy plays a key
role in apoptosis induced by NVP. Combination of NVP and autophagy
inhibition may be a novel strategy for the treatment of HCC.
Discussion
HCC is a fatal cancer for men and women and is
caused by complicated risk factors (16). Due to frequent de novo and
acquired resistance of HCC to chemotherapy, the effective options
for therapy of HCC are limited (17,18). Consequently, there is a strong
interest in identifying novel molecular targets for therapy of
HCC.
Taking into account the role of PI3K/AKT/mTOR
signaling in HCC cellular survival, negating apoptosis (19) would indicate that a marked
proapoptotic response to the inhibition of this signaling pathway
may be expected. However, as previously found (20), apoptosis is not necessarily the
primary response to PI3K/AKT/mTOR inhibition, particularly in
cancer cells where marked apoptosis suppression may be the
consequence of multiple genetic alterations. The current study
suggests that the PI3K/AKT/mTOR inhibitors induce productive
autophagy in cancer cells (21,22). Jung et al (23) reported that mTOR is a master
regulator of autophagy through the phosphorylation of its
downstream gene ULK. Thus, it is not surprising that mTOR blockade
would lead to autophagy induction.
Autophagy induced by the blockade of the
PI3K/AKT/mTOR pathway has been identified in several studies as a
mechanism of cell death, whereas other articles have provided data
showing the role of this process in therapeutic resistance. Recent
examples of the former include the finding that PI3K/AKT/mTOR
pathway inhibition increases radiosensitivity by augmenting
autophagic response (24), and
that combining the dual PI3K/mTOR inhibitor NVP-BEZ235 with the
mTORC1 inhibitor temsirolimus leads to cell death secondary to
massive autophagic response (25). On the contrary, autophagy blockade
has been indicated as enhancing the proapoptotic effects of
PI3K/mTOR inhibitors in preclinical cancer models (21,22).
Apoptosis and autophagy are significantly involved
in cancer. Both apoptosis and autophagy play important roles in the
development, cellular homeostasis and, particularly, in the
oncogenesis of mammals. The mechanisms of apoptosis and autophagy
are different and involve fundamentally distinct sets of regulatory
and executioner molecules (26–28). The crosstalk between apoptosis and
autophagy is therefore complex, sometimes even contradictory, in
nature, yet it is critical to the overall fate of the cell
(29). In some cellular settings,
autophagy serves as a cell survival pathway to suppress apoptosis
(30). On the other hand,
autophagy could lead to cell death, either in collaboration with
apoptosis or as a back-up mechanism when apoptosis is defective
(29). Recent studies have shown
that autophagy may play an important role in the regulation of
cancer development and progression. Whether autophagy represents a
mechanism for resisting apoptosis or a mechanism for initiating a
nonapoptotic form of programmed cell death remains unclear
(31–33).
Thus, we hypothesize that inhibition of autophagy
may be an effective way of improving antitumor strategies by
enhancing apoptosis. In this study, we found that the combined
treatment of NVP-BEZ235 and autophagy inhibition led to marked cell
growth inhibition and increase of apoptosis. These results
indicated that inhibition of autophagy likely impairs the
proliferation mechanism of tumor cells, consequently leading to
tumor cell growth inhibition. Furthermore, inhibition of autophagy
increased the apoptotic marker caspase-3 activation, and thus led
to the enhancement of tumor cell apoptosis. Indeed, previous
studies have shown that inhibition of autophagy enhanced
inhibitor-induced apoptosis (34–35). These findings are consistent with
our hypothesis and indicate that autophagy inhibition could enhance
the apoptosis of HCC cells and finally increase the growth
inhibition of HCC cells. Thus, induction of autophagy by NVP-BEZ235
may be a survival mechanism that counteracts its anticancer
effects. Based on these data, we suggest a strategy to enhance the
anticancer efficacy of BEZ235 by blockade of autophagy.
In this study, we provided significant data
indicating that inhibition of autophagy could enhance tumor cell
growth inhibition and apoptosis of HCC cells induced by PI3K/mTOR
inhibitor NVP-BEZ235. Thus, activation of autophagy may be involved
in the resistance to apoptosis in HCC cells. Autophagy may help
tumor cells mitigate metabolic stress and promote cell survival
during apoptosis. In conclusion, we showed that inhibition of
autophagy may be a novel way to increase the efficacy of antitumor
strategies in the treatment of HCC.
Acknowledgements
This study was supported by the Basic
Research Foundation of Jilin University, China (no. 201103048).
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Siegel R, Naishadham D and Jemal A: Cancer
statistics, 2012. CA Cancer J Clin. 62:10–29. 2012. View Article : Google Scholar
|
|
3
|
Verslype C, Van Cutsem E, Dicato M, et al:
The management of hepatocellular carcinoma. Current expert opinion
and recommendations derived from the 10th World Congress on
Gastrointestinal Cancer, Barcelona, 2008. Ann Oncol. 20(Suppl 7):
vii1–vii6. 2009. View Article : Google Scholar
|
|
4
|
Sia D and Villanueva A: Signaling pathways
in hepatocellular carcinoma. Oncology. 81(Suppl 1): S18–S23. 2011.
View Article : Google Scholar
|
|
5
|
Vivanco I and Sawyers CL: The
phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev
Cancer. 2:489–501. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Luo J, Manning BD and Cantley LC:
Targeting the PI3K-Akt pathway in human cancer: rationale and
promise. Cancer Cell. 4:257–262. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sabatini DM: mTOR and cancer: insights
into a complex relationship. Nat Rev Cancer. 6:729–734. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kong D and Yamori T: Advances in
development of phosphatidylinositol 3-kinase inhibitors. Curr Med
Chem. 16:2839–2854. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pang RW and Poon RT: From molecular
biology to targeted therapies for hepatocellular carcinoma: the
future is now. Oncology. 72(Suppl 1): S30–S44. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rubinsztein DC, Gestwicki JE, Murphy LO
and Klionsky DJ: Potential therapeutic applications of autophagy.
Nat Rev Drug Discov. 6:304–312. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Masuda M, Shimomura M, Kobayashi K, et al:
Growth inhibition by NVP-BEZ235, a dual PI3K/mTOR inhibitor, in
hepatocellular carcinoma cell lines. Oncol Rep. 26:1273–1279.
2011.PubMed/NCBI
|
|
14
|
Thomas HE, Mercer CA, Carnevalli LS, et
al: mTOR inhibitors synergize on regression, reversal of gene
expression, and autophagy in hepatocellular carcinoma. Sci Transl
Med. 4:139–184. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Herrera VA, Zeindl-Eberhart E, Jung A, et
al: The dual PI3K/mTOR inhibitor BEZ235 is effective in lung cancer
cell lines. Anticancer Res. 31:849–854. 2011.PubMed/NCBI
|
|
16
|
Shire AM and Roberts LR: Prevention of
hepatocellular carcinoma: progress and challenges. Minerva
Gastroenterol Dietol. 58:49–64. 2012.PubMed/NCBI
|
|
17
|
Sandhu DS, Tharayil VS, Lai JP and Roberts
LR: Treatment options for hepatocellular carcinoma. Expert Rev
Gastroenterol Hepatol. 2:81–92. 2008. View Article : Google Scholar
|
|
18
|
Roberts LR and Gores GJ: Emerging drugs
for hepatocellular carcinoma. Expert Opin Emerg Drugs. 11:469–487.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tokunaga E, Oki E, Egashira A, et al:
Deregulation of the Akt pathway in human cancer. Curr Cancer Drug
Targets. 8:27–36. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zou CY, Smith KD, Zhu QS, et al: Dual
targeting of AKT and mammalian target of rapamycin: a potential
therapeutic approach for malignant peripheral nerve sheath tumor.
Mol Cancer Ther. 8:1157–1168. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu CX, Zhao L, Yue P, et al: Augmentation
of NVP-BEZ235’s anticancer activity against human lung cancer cells
by blockage of autophagy. Cancer Biol Ther. 12:549–555. 2011.
|
|
22
|
Mirzoeva OK, Hann B, Hom YK, et al:
Autophagy suppression promotes apoptotic cell death in response to
inhibition of the PI3K-mTOR pathway in pancreatic adenocarcinoma. J
Mol Med. 89:877–889. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jung CH, Jun CB, Ro SH, et al:
ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy
machinery. Mol Biol Cell. 20:1992–2003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fujiwara K, Iwado E, Mills GB, et al: Akt
inhibitor shows anticancer and radiosensitizing effects in
malignant glioma cells by inducing autophagy. Int J Oncol.
31:753–760. 2007.PubMed/NCBI
|
|
25
|
Yang S, Xiao X, Meng X and Leslie KK: A
mechanism for synergy with combined mTOR and PI3 kinase inhibitors.
PLoS One. 6:e263432011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Danial NN and Korsmeyer SJ: Cell death:
critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Levine B and Klionsky DJ: Development by
self-digestion: molecular mechanisms and biological functions of
autophagy. Dev Cell. 6:463–477. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mizushima N and Klionsky DJ: Protein
turnover via autophagy: implications for metabolism. Annu Rev Nutr.
27:19–40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eisenberg-Lerner A, Bialik S, Simon HU and
Kimchi A: Life and death partners: apoptosis, autophagy and the
cross-talk between them. Cell Death Differ. 16:966–975. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang Y, Xing D, Zhou F and Chen Q:
Mitochondrial autophagy protects against heat shock-induced
apoptosis through reducing cytosolic cytochrome c release and
downstream caspase-3 activation. Biochem Biophys Res Commun.
395:190–195. 2010. View Article : Google Scholar
|
|
31
|
Zhuang W, Qin Z and Liang Z: The role of
autophagy in sensitizing malignant glioma cells to radiation
therapy. Acta Biochim Biophys Sin. 41:341–351. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
White E and DiPaola RS: The double-edged
sword of autophagy modulation in cancer. Clin Cancer Res.
15:5308–5316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dikic I, Johansen T and Kirkin V:
Selective autophagy in cancer development and therapy. Cancer Res.
70:3431–3434. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nguyen TM, Subramanian IV, Xiao X, et al:
Endostatin induces autophagy in endothelial cells by modulating
Beclin 1 and beta-catenin levels. J Cell Mol Med. 13:3687–3698.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nguyen TM, Subramanian IV, Kelekar A and
Ramakrishnan S: Kringle 5 of human plasminogen, an angiogenesis
inhibitor, induces both autophagy and apoptotic death in
endothelial cells. Blood. 109:4793–4802. 2007. View Article : Google Scholar : PubMed/NCBI
|