Introduction
The abnormal proliferation and migration of vascular
smooth muscle cells (VSMCs) are major events in the development and
progression of atherosclerosis. During the early stages of
atherosclerosis, VSMCs migrate from the tunica media into the
tunica intima of the arterial wall, and this, along with
proliferation, cause intimal thickening and narrowing of the
arterial space (1).
Atherogenic lesions are characterized by the
accumulation of inflammatory cells and released cytokines (2). Tumor necrosis factor-α (TNF-α) is a
major inflammatory cytokine that plays an important role in the
initiation and development of atherosclerosis (3). TNF-α is secreted by activated
macrophages in atherosclerotic lesions and by VSMCs in the
neointima following balloon injury; this cytokine induces the
proliferation and migration of VSMCs (4–6).
For VSMC migration to occur, the proteolytic degradation or
remodeling of the extracellular matrix (ECM) is required. Matrix
metalloproteinases (MMPs) are a family of endopeptidases that
degrade ECM components, including type IV collagen, laminin and
elastin (7,8). Among the MMPs, MMP-9 plays a
critical role in VSMC migration and neointima formation, and TNF-α
is known to induce VSMC migration via the induction of MMP-9
expression (9,10). As previously demonstrated in an
animal model of restenosis, MMP-9-deficient mice exhibit reduced
neointima formation due to a defect in VSMC migration, suggesting
an important role for MMP-9 in the progression of atherosclerosis
(11). TNF-α also induces the
expression of cellular adhesion molecules, such as intercellular
adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1
(VCAM-1), and recruits monocytes to injury sites, thereby enhancing
the interaction between VSMCs and monocytes at inflammatory sites.
This process also plays an important role in the development and
progression of atherosclerosis (12). Therefore, the inhibition of
TNF-α-mediated VSMC proliferation and migration is considered an
important therapeutic strategy for atherosclerosis.
Cinnamon is a widely used food spice that is
obtained from the inner bark of Cinnamomum cassia.
Cinnamaldehyde, an active component of cinnamon, exhibits various
biological functions, such as anti-bacterial, anti-fungal,
anti-inflammatory and antitumor activities (13–16). Specifically, the natural
derivative, 2-hydroxycinnamaldehyde (HCA), and the synthetic
derivative, 2-benzoyloxycinnamaldehyde (BCA), have been shown to
effectively induce cell cycle arrest and the subsequent apoptosis
of various human cancer cells, including those from breast, colon,
leukemia, lung and oral cancers (16–19). Recently, we demonstrated that HCA
induced the activation of the cell death pathway in a
p53-independent manner and that autophagy was actively involved in
the HCA-induced apoptosis of oral cancer cells (20). To date, the majority of studies
evaluating cinnamaldehyde have focused on its antitumor activity.
However, Liao et al demonstrated that cinnamaldehyde
inhibited the adhesion of TNF-α-induced monocytes to endothelial
cells by suppressing the expression of the cell adhesion molecules,
VCAM-1 and ICMA-1 (21).
Furthermore, we recently demonstrated that BCA inhibited
LPS-induced inducible nitric oxide (NO) synthase (iNOS) expression
and subsequent NO production in vitro and in vivo
(15). These data demonstrate the
anti-inflammatory effects of cinnamaldehyde and suggest that
cinnamaldehyde and its derivatives may be possible candidates for
use in the treatment of inflammation-related diseases.
In the present study, we evaluated
2-methoxycinnamaldehyde (MCA), a natural cinnamaldehyde derivative,
to determine whether it would be useful as an anti-atherosclerotic
agent. Specifically, we evaluated the effects of MCA on the
proliferation and migration of human aortic smooth muscle cells
(HASMCs) that were exposed to TNF-α. The molecular mechanisms of
action of MCA as an anti-atherosclerotic agent were also assessed.
To the best of our knowledge, our data provide initial evidence
that MCA is a novel candidate for the treatment of
atherosclerosis.
Materials and methods
Materials
The HASMCs and smooth muscle cell medium (SMCM) were
purchased from ScienCell (Carlsbad, CA, USA). Recombinant human
TNF-α was purchased from R&D Systems (Minneapolis, MN, USA).
The antibodies against cyclin D1 (#2926), cyclin D3 (#2936),
cyclin-dependent kinase (CDK)4 (#2906), CDK6 (#3136), p15 (#4822),
p21 (#2946), p27 (#2552), p65 (#4764), phosphorylated (p-)p65
(#3033), IκBα (#4814), c-Jun N-terminal kinase (JNK; #9258), p-JNK
(#4668), p38 (#9212), p-p38 (#9211), extracellular signal-regulated
kinase (ERK; #4695) and p-ERK (#4370) were obtained from Cell
Signaling Technology (Danvers, MA, USA) and the antibodies against
TATA-binding protein (TBP; ab818) were purchased from Abcam
(Cambridge, MA, USA). Recombinant human platelet-derived growth
factor (PDGF) and antibodies against β-actin (A1978) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). HCA and MCA were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Unless
otherwise mentioned, all other chemicals were purchased from
Sigma-Aldrich.
Cell culture and treatment
The HASMCs were maintained at 37°C under 5%
CO2 in SMCM supplemented with the reagents provided with
the medium. The cells were treated with various concentrations
(1-50 µM) of MCA or HCA (dissolved in 0.1% DMSO) for the
indicated periods of time.
Cell proliferation and cytotoxicity
assays
The HASMCs were seeded on 12-well plates at a
density of 1.5×105 cells/ml. The cells were cultured
overnight and treated with various concentrations of MCA or HCA for
24 h. Cell proliferation was evaluated by MTT assay according to a
previously described method (22). Briefly, each well was washed twice
with PBS, and 0.5 ml of cell culture medium and 50 µl of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
solution (5 mg/ml in PBS) were added. After 3 h of incubation, the
medium was removed and 250 µl of acid-isopropanol (0.04
mol/l HCl in isopropanol) were added. The absorbance was measured
at 570 nm using a Microplate reader (iMarkTM; Bio-Rad, Hercules,
CA, USA).
For the cell cytotoxicity assay, the cells were
treated with various concentrations of MCA or HCA for 24 h. Cell
toxicity was evaluated by measuring the lactate dehydrogenase (LDH)
activity using the CytoTox 96® non-radioactive assay kit
(Promega, Madison, WI, USA) in accordance with the manufacturer's
instructions. In brief, the supernatant of the cell media was
transferred to a 96-well plate. An equal volume of CytoTox
96® reagent was then added and incubated for 30 min at
room temperature. After adding stop solution, the absorbance was
measured at 490 nm using a Microplate reader (iMarkTM;
Bio-Rad).
Western blot analysis
The HASMCs were exposed to TNF-α (10 ng/ml) alone or
together with MCA for 24 h. The cells were washed with
phosphate-buffered saline (PBS) and lysed in RIPA buffer (PBS
supplemented with 1% NP-40, 0.5% sodium deoxycholate, 1 mM PMSF, 1
µg/ml aprotinin, and 1 mM sodium orthovanadate). The cell
lysates were then incubated at 4°C for 30 min, followed by
centrifugation at 10,000 × g for 10 min. Alternatively, the
cytoplasmic and nuclear fractions were obtained using a subcellular
fractionation method that has been described previously (23). The protein samples were resolved
by sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred onto PVDF membranes. The blots were
blocked and then incubated with primary antibodies. The
immunoreactive bands were detected using the Immobilon™ Western
chemiluminescent HRP substrate (Millipore, Billerica, MA, USA).
Semi-quantitative RT-PCR
The cells were exposed to TNF-α alone or together
with various concentrations of MCA for 24 h. Total RNA was isolated
using TRIzol® reagent (Invitrogen, Carlsbad, CA, USA),
and semi-quantitative RT-PCR was conducted using the One-Step
RT-PCR PreMix kit (Intron Biotechnology, Seongnam, Korea) according
to the manufacturer's instructions. The specific primers used for
RT-PCR are shown in Table I.
RT-PCR was performed under the following conditions: 1 cycle of 30
min at 45°C, 1 cycle of 5 min at 94°C, and 25 to 30 cycles of 30
sec at 94°C, 30 sec at 55°C, and 40 sec at 72°C, with a final
extension at 72°C for 5 min. The PCR products were electrophoresed
on a 1.7% agarose gel and visualized by ethidium bromide
staining.
 | Table IPrimers used for semi-quantitative
RT-PCR. |
Table I
Primers used for semi-quantitative
RT-PCR.
| Gene | Primer sequences
(5′→3′) | Size (bp) |
|---|
| MMP-9 | F:
GGATGGGAAGTACTGGCGATTCT | 478 |
| R:
CACTTGGTCCACCTGGTTCAAC | |
| MMP-2 | F:
CTTCCAAGTCTGGAGCGATGT | 209 |
| R:
TCTCCCAAGGTCCATAGCTCA | |
| TIMP1 | F:
GCTGACATCCGGTTCGTCTAC | 272 |
| R:
CAAGCAATGAGTGCCACTCTG | |
| GAPDH | F:
CCAAGGTCATCCATGACAACTTTG | 464 |
| R:
GTCATACCAGGAAATGAGCTTGACA | |
Gelatin zymography
The cell culture supernatants were resuspended in
sample buffer (60 mM Tris-Cl, pH 6.8, 15% glycerol, 2% SDS and
0.001% bromophenol blue) and loaded, without boiling, onto a 0.1%
gelatin gel containing 10% acrylamide. Following electrophoresis,
the gels were washed twice with 0.25% Triton X-100 solution for 30
min/wash and then incubated in incubation buffer (50 mM Tris-Cl, pH
7.6, 5 mM CaCl2, 20 mM NaCl) at 37°C for 18–24 h to
allow for proteolysis of the gelatin. The gels were stained with
Coomassie Brilliant Blue R. Proteolysis can be detected as a white
zone in a dark blue field.
Luciferase reporter gene assay
A 0.71 kb segment at the 5′-flanking region of the
human MMP-9 gene, corresponding to GenBank®
accession number D10051 was amplified by PCR using genomic DNA from
293 cells (American Type Culture Collection, Manassas, VA, USA) as
a template. The luciferase reporter vector for the MMP-9 promoter
was created by inserting the MMP-9 promoter DNA fragment into the
5′ SacI and 3′ HindIII sites of the pGL3-Basic
vector. The constructs were confirmed by DNA sequencing. To assess
the effects of MCA on MMP-9 promoter activity, the cells were
co-transfected with pGL3-MMP-9-Luc and pCH110 using the Neon
Transfection system (Invitrogen) according to the manufacturer's
instructions. After 30 h, the cells were exposed to TNF-α and/or
various concentrations of MCA for 24 h. The cells were then lysed
and luciferase activity was measured using the Luciferase assay
system (Promega). The luciferase activity was normalized to the
β-galactosidase activity.
Immunofluorescence microscopy
The HASMCs were seeded at a density of
1.5×105 cells/ml. The cells were cultured overnight and
exposed to TNF-α and/or MCA for 3 h. The cells were washed twice
with PBS and fixed with 4% paraformaldehyde for 10 min. After being
washed twice with PBS, the cells were incubated with methanol for 2
min. The immunostaining was performed as previously described
(24). The fluorescence analysis
was performed by conventional fluorescence microscopy (Axio
Observer D1; Carl Zeiss, Oberkochen, Germany).
Cell migration assay
Cell migration assay was performed using the
Transwell® system (Corning Inc., Corning, NY, USA).
Briefly, the cells were seeded at a density of 3×104
cells/100 µl on a 0.1% gelatin-coated upper chamber. A 500
µl aliquot of serum-free SMCM with hPDGF (10 ng/ml) and/or
MCA was added to the lower compartment of the invasion chamber.
Following 24 h of incubation, the filter insert within the upper
chamber was removed. The cells on the upper side of the filter were
removed using cotton swabs, and the cells that had migrated to the
underside of the filter were stained with hematoxylin and eosin
(H&E). The migrated cells were observed under a light
microscope (Olympus CKX41; Olympus, Tokyo, Japan).
Statistical analysis
All experiments were performed at least 3 times and
the data are expressed as the means ± SD. ANOVA and the Student's
t-test were applied to determine the statistical significance.
P-values <0.01 were considered to indicate statistically
significant differences.
Results
Effect of MCA on HASMC proliferation and
cytotoxicity
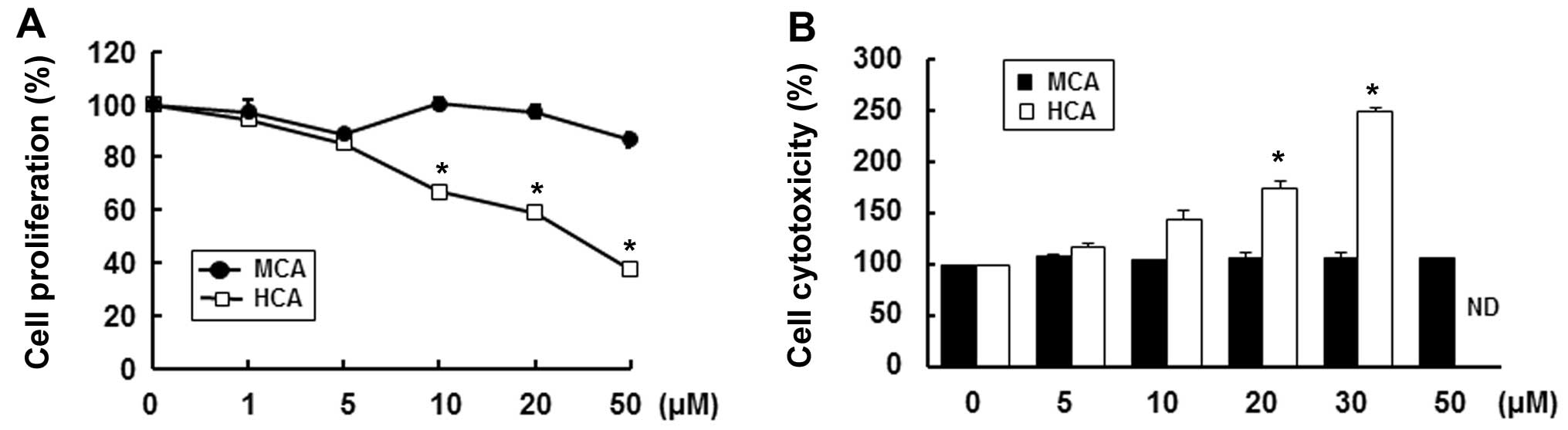
In an effort to find possible anti-atherosclerotic
agents, we first evaluated the effects of MCA and HCA, natural
derivatives of cinnamaldehyde, on HASMC proliferation (25). Instead of a hydrogen, HCA has a
hydroxy group and MCA has a methoxy group at the 2′ site of
cinnamaldehyde (Fig. 1). We, as
well as others have demonstrated that HCA markedly inhibits the
proliferation and induces the apoptosis of various human cancer
cells (16,18–20). Consistent with these studies, in
this study, HCA significantly decreased cell proliferation in a
dose-dependent manner. Treatment with 50 µM HCA reduced cell
proliferation by 62.7% compared with the untreated control cells
(Fig. 2A). Furthermore, HCA
markedly increased cytotoxicity in a dose-dependent manner at a
concentration of up to 30 µM (Fig. 2B). We could not obtain reasonable
cytotoxicity data for concentrations >50 µM as HCA was
highly toxic. On the other hand, MCA only slightly decreased cell
proliferation, by 13.3%, at a concentration of 50 µM and did
not result in any cytotoxicity up to a concentration of 50
µM (Fig. 2B). Therefore,
we used MCA for the subsequent experiments.
MCA inhibits TNF-α-induced HASMC
proliferation
We then assessed the effects of MCA on TNF-α-induced
cell proliferation. The HASMCs were exposed to 10 ng/ml TNF-α alone
or together with 50 µM MCA for the indicated periods of
time, and cell proliferation was monitored by MTT assay. TNF-α
increased cell proliferation by approximately 19.2% at day 3
compared with the untreated control cells, while MCA decreased cell
proliferation by approximately 31.1% (Fig. 3A). More importantly, MCA
completely abolished the TNF-α-induced increase in HASMC
proliferation (Fig. 3A). To
confirm the inhibitory effects of MCA on TNF-α-induced cell
proliferation, we then examined the expression levels of cell cycle
regulatory proteins. The cells were exposed to TNF-α alone or with
a combination of various concentrations of MCA for 24 h. TNF-α
increased the levels of cyclin D1 and cyclin D3 and markedly
increased the levels of CDK6 (Fig.
3B). Of note, MCA attenuated the TNF-α-induced increase in the
levels of cyclin D1, cyclin D3 and CDK6 in a dose-dependent manner
compared with the control levels of these proteins. Additionally,
the levels of the CDK inhibitor (CDKI) proteins, p21 and p27, were
increased by MCA. These results suggest that MCA effectively
suppresses TNF-α-induced cell proliferation through the regulation
of cell cycle regulatory proteins.
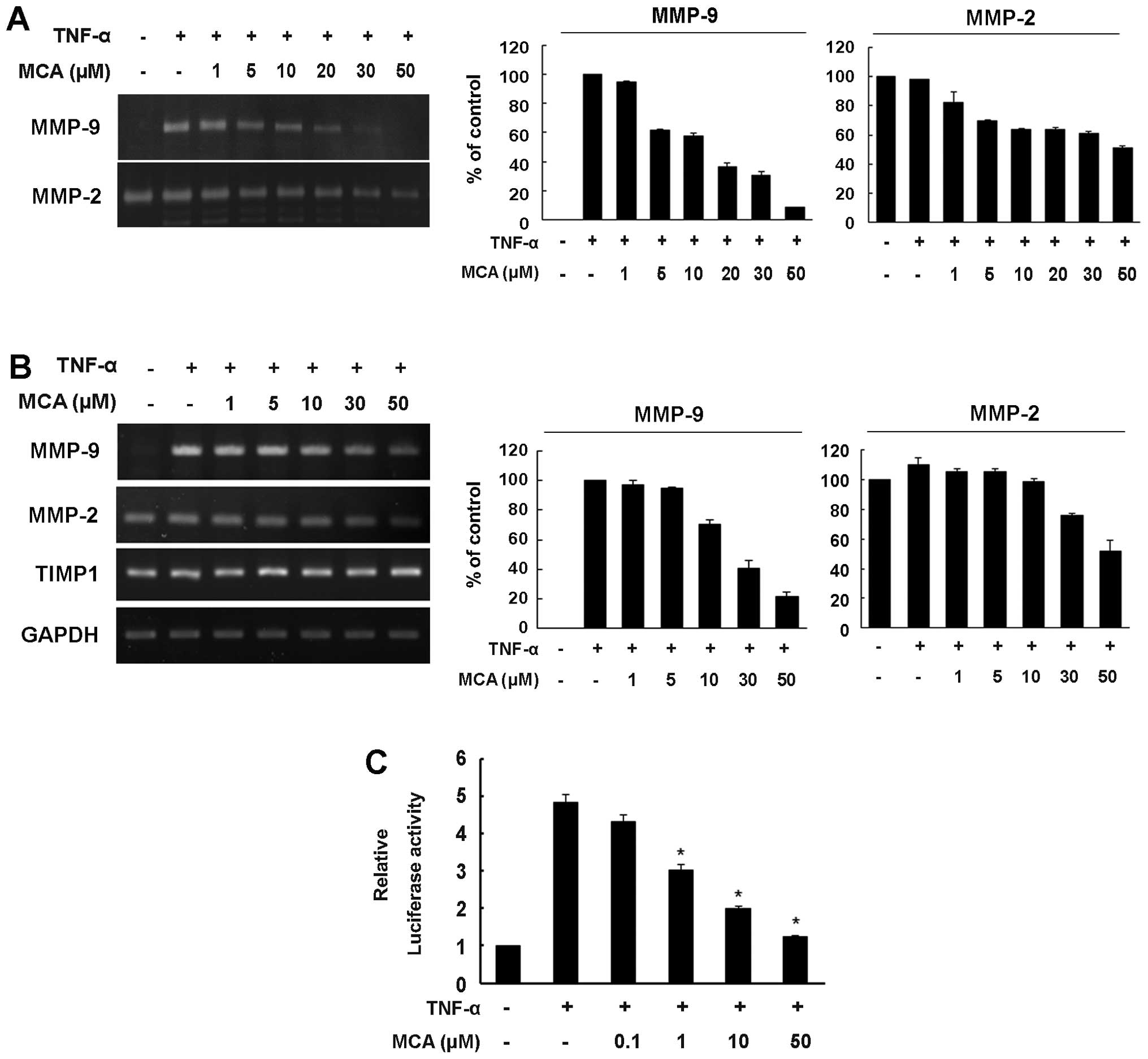
MCA inhibits the TNF-α-induced increase
in MMP-9 expression
MMP-9 plays an important role in VSMC proliferation
and migration, and the expression of MMP-9 can be induced by TNF-α
(9,10). Thus, to assess whether MCA affects
TNF-α-induced MMP-9 expression, a gelatin zymography assay was
performed. While no MMP-9 activity was detected in the medium from
the untreated control cells, exposure to TNF-α markedly enhanced
the secretion of MMP-9, and MCA significantly inhibited the
secretion of MMP-9 in a dose-dependent manner (Fig. 4A). Unlike MMP-9, MMP-2 exhibited
high proteolytic activity in the medium from the untreated control
cells. Although TNF-α did not affect the level of MMP-2, MCA
slightly reduced the level of MMP-2. Compared with the
TNF-α-exposed controls, MCA inhibited MMP-9 and MMP-2 secretion by
approximately 91.5 and 48.8%, respectively (Fig. 4A).
We then examined whether the MCA-dependent decrease
in MMP-9 secretion is caused by the transcriptional regulation of
the MMP-9 gene. The cells were exposed to TNF-α alone or
together with various concentrations of MCA for 24 h. Total RNA was
then isolated, and semi-quantitative RT-PCR was performed.
Consistent with the results from zymography assay, TNF-α markedly
increased the mRNA level of MMP-9, and the TNF-α-induced increase
in the MMP-9 mRNA levels was attenuated by MCA in a dose-dependent
manner, with a 78.7% inhibition observed in the cells treated with
MCA. The constitutive mRNA expression of MMP-2 was also
down-regulated, exhibiting a 48% inhibition in the MCA-treated
cells. However, MCA did not affect the transcription of tissue
inhibitor of metalloproteinases (TIMP)1 (Fig. 4B).
To further confirm the transcriptional regulation of
MMP-9 by MCA, a luciferase reporter gene assay was performed. The
cells were transfected with the pGL3-MMP-9-Luc reporter vector and
then exposed to TNF-α alone or together with various concentrations
of MCA. TNF-α enhanced MMP-9 promoter activity up to 5-fold
compared with the untreated controls (Fig. 4C). However, when the cells were
treated with MCA in the presence of TNF-α, MMP-9 promoter activity
was reduced in a dose-dependent manner, suggesting that MCA
inhibits TNF-α-induced MMP-9 transcriptional activity.
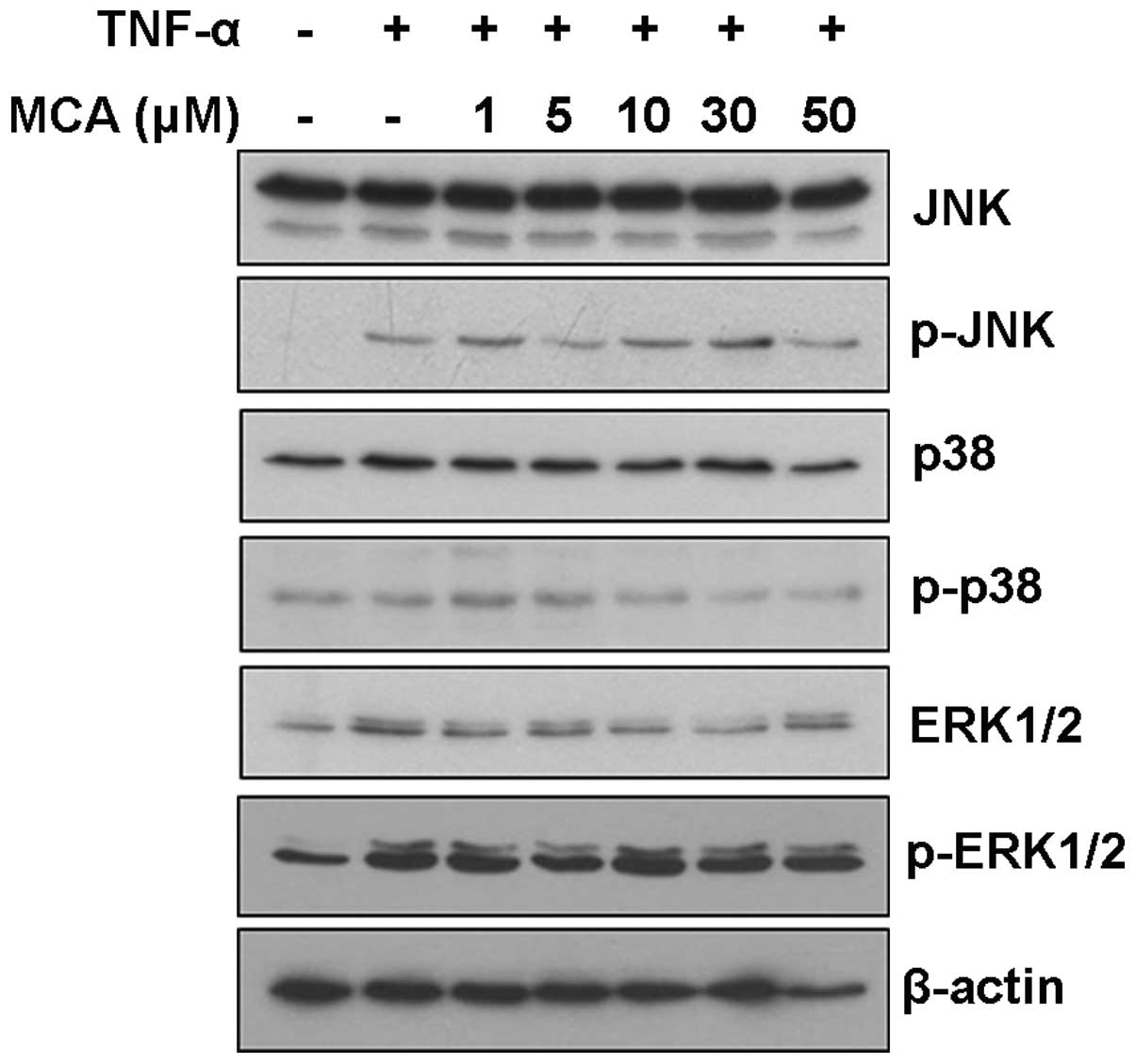
MCA inhibits TNF-α-induced NF-κB nuclear
translocation
To further understand the molecular mechanisms
through which MCA alters MMP-9 expression, the effects of MCA on
mitogen-activated protein kinase (MAPK) signaling pathways were
examined. Although TNF-α induced the activation of JNK and ERK, MCA
did not affect the JNK, p38 and ERK signaling pathways, suggesting
that MCA functions through another signaling pathway (Fig. 5).
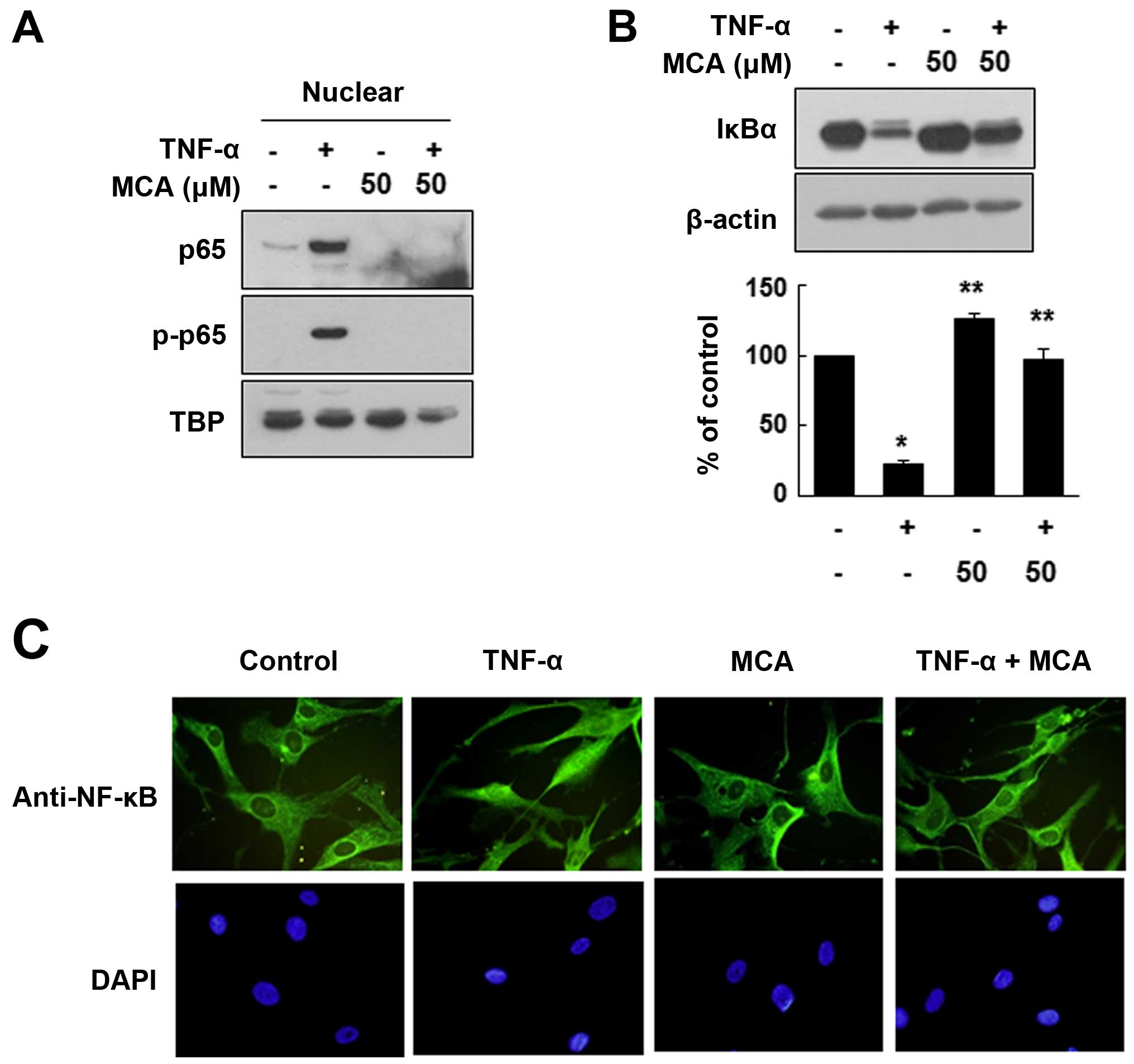
As MCA did not exert any significant effects on the
TNF-α-induced activation of MAPK signaling pathways, we examined
the activity of the transcription factor, NF-κB, a well-known
effector of TNF-α. As the nuclear translocation of NF-κB is an
important indicator of its activation, we evaluated whether MCA
affects NF-κB subcellular translocation. For this experiment, the
HASMCs were exposed to TNF-α and/or MCA for 24 h. The nuclear
proteins were separated from the cytosolic proteins through
fractionation, and the distribution of NF-κB was evaluated. As
expected, TNF-α markedly enhanced the nuclear localization of the
NF-κB p65 subunit. However, MCA significantly inhibited the
TNF-α-induced nuclear localization of p65. We also demonstrated
that the TNF-α-induced nuclear p65 was phosphorylated, which is
required for NF-κB activation (Fig.
6A). It is well known that the nuclear translocation of p65 is
caused by the degradation of IκBα. Thus, to confirm the effects of
MCA on the activation of NF-κB, the levels of IκBα were evaluated.
Exposure to TNF-α decreased the level of IκBα compared with the
untreated controls (Fig. 6B).
However, MCA effectively inhibited the TNF-α-induced IκBα
degradation. The effects of MCA on NF-κB activation were also
assessed by immunofluorescence staining. Under the control
conditions, NF-κB was mainly localized in the cytoplasm, and
exposure to TNF-α induced the nuclear translocation of NF-κB.
However, co-treatment with MCA and TNF-α resulted in the
cytoplasmic localization of NF-κB, suggesting that MCA inhibits the
TNF-α-induced NF-κB translocation (Fig. 6C).
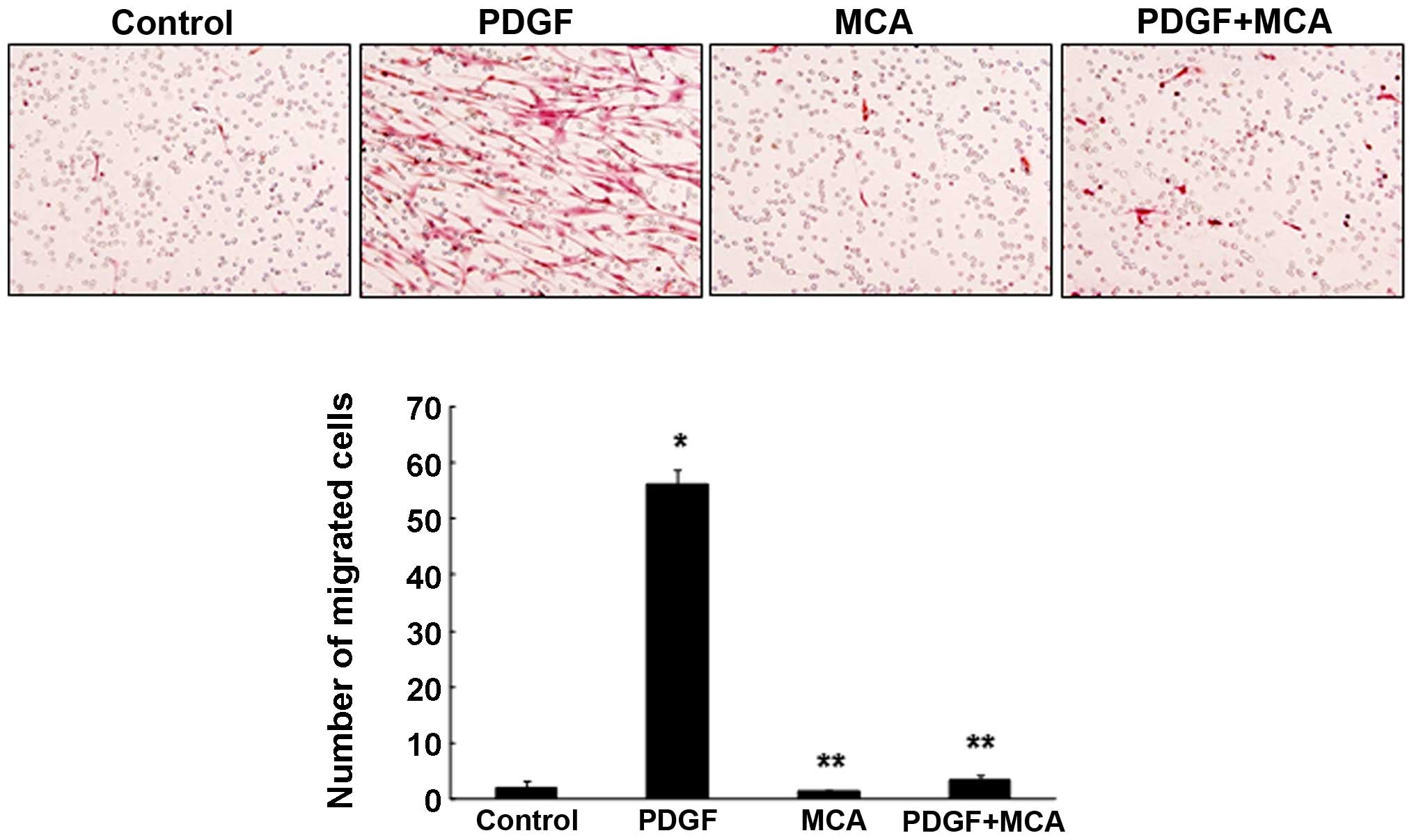
MCA inhibits cytokine-induced HASMC
migration
Our results demonstrated that MCA effectively
inhibited the TNF-α-induced HASMC proliferation and migration by
inhibiting the activation of the NF-κB signaling pathway. To
further evaluate the potential of MCA as an anti-atherosclerotic
agent, a migration assay was performed. For this experiment, HASMC
migration was induced by treating the cells with PDGF. PDGF is the
strongest chemoattractant that contributes to the progression of
atherosclerosis by inducing the proliferation and migration of
VSMCs (2,26). Treatment with PDGF (10 ng/ml)
markedly enhanced HASMC migration (Fig. 7). However, combined treatment with
PDGF and MCA significantly inhibited the PDGF-induced cell
migration, suggesting that MCA may be a potential therapeutic agent
for atherosclerosis. The TNF-α-induced cell migration was also
effectively inhibited by MCA (data not shown).
Discussion
Researches evaluating the pharmacological properties
of cinnamaldehyde and its derivatives have focused on its antitumor
activity. Among the known natural constituents isolated from
Cinnamomum cassia, HCA is the most widely studied due to its
antitumor activity (16,18–20). However, MCA, another constituent
of Cinnamomum cassia, has not attracted researchers'
interest due to its moderate cytotoxic effects on cancer cells.
The proliferation and migration of VSMCs are
critical events in the development of atherosclerotic lesions and
cytokines, such as TNF-α and PDGF, are intimately involved in
regulating these processes (4–6,26).
The first response upon vascular injury is increased SMC
proliferation, which continues for 1 to 3 days following injury. In
the second phase of lesion development, SMCs migrate from the
internal lamina to the intima, usually beginning at day 3 (1,2,4).
In this study, we evaluated the anti-atherosclerotic effects of MCA
on HASMCs. As expected, HCA exhibited strong cytotoxicity, while
MCA did not result in any cytotoxicity up to a concentration of 50
µM (Fig. 2B). Even 100
µM MCA did not result in any cytotoxicity (data not shown).
Furthermore, MCA effectively inhibited the TNF-α-induced HASMC
proliferation, primarily by decreasing the levels of cyclin D1/CDK6
and inducing the expression of the CDKIs, p21 and p27 (Fig. 3). These results suggest that MCA
may be a suitable candidate for the treatment of
atherosclerosis.
MMPs play an important role in ECM degradation and
remodeling. Among them, MMP-9 and MMP-2 actively contribute to the
pathogenesis of atherosclerosis by facilitating the migration of
smooth muscle cells into the intima (11,27,28). Furthermore, experiments with
knock-out mice showed that MMP-9 is critical for the development of
arterial lesions due to its role in regulating VSMC migration and
proliferation (11,28,29). Although MMP-9 and MMP-2 have
similar substrate specificities, their expression patterns are
differentially regulated. MMP-2 is constitutively expressed in
smooth muscle cells, and its expression is not affected by
cytokines. By contrast, the basal level of MMP-9 is very low, and
its expression can be induced by TNF-α (30,31). In this study, the effects of MCA
on TNF-α-induced MMP-9 expression were assessed, and the data
demonstrated that MCA significantly inhibited TNF-α-induced MMP-9
secretion via the suppression of MMP-9 transcription (Fig. 4). Although the effect was small,
MCA also inhibited the constitutive expression of MMP-2 at the
transcriptional level.
Previous studies have reported that the induction of
MMP-9 expression by TNF-α is regulated by the ERK and JNK signaling
pathways, and by the subsequent activation of NF-κB and activator
protein 1 (AP-1) in VSMCs (32–34). To understand the precise molecular
mechanisms of action for MCA, MAPK activity was examined.
Consistent with previous studies, the levels of p-JNK and p-ERK1/2
were elevated by exposure to TNF-α. However, treatment with MCA did
not affect the levels of phosphorylated MAPKs, suggesting that MCA
inhibits MMP-9 expression through a different signaling pathway
(Fig. 5). Furthermore, our data
clearly demonstrated that MCA inhibited the TNF-α-induced increase
in MMP-9 expression through the inhibition of IκBα degradation and,
therefore, of subsequent NF-κB activation (Fig. 6).
PDGF is a well-known mitogen and chemoattractant for
VSMCs. Similar to TNF-α, PDGF potently stimulates VSMC
proliferation and migration and plays an important role in the
development of atherosclerosis (2,26).
To confirm the anti-atherosclerotic effects of MCA, we also
examined the effect of MCA on PDGF-induced HASMC migration; the
data clearly demonstrated that MCA potently inhibited, not only
TNF-α- (data not shown), but also PDGF-induced cell migration
(Fig. 7).
In this study, we demonstrated that MCA effectively
inhibited TNF-α-induced HASMC proliferation by reducing the levels
of cyclin D1/CDK6 and increasing the levels of the CDKIs, p21 and
p27. Furthermore, we demonstrated that MCA potently inhibited the
TNF-α-induced increase in MMP-9 expression at the transcriptional
level by inhibiting the nuclear translocation of NF-κB via a
MAPK-independent signaling pathway. To the best of our knowledge,
this is the first study showing that MCA, a natural constituent of
Cinnamomum cassia, effectively inhibits TNF-α-induced HASMC
proliferation and migration, and this study suggests that MCA may
be a potential candidate for the treatment of atherosclerosis.
Acknowledgments
The present study was supported by the MRC program
of MOST/KOSEF (no. R13-2005-013-01000-0).
References
|
1
|
Ross R: The pathogenesis of
atherosclerosis: A perspective for the 1990s. Nature. 362:801–809.
1993. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abedi H and Zachary I: Signalling
mechanisms in the regulation of vascular cell migration. Cardiovasc
Res. 30:544–556. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McKellar GE, McCarey DW, Sattar N and
McInnes IB: Role for TNF in atherosclerosis? Lessons from
autoimmune disease. Nat Rev Cardiol. 6:410–417. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hansson GK: Inflammation, atherosclerosis,
and coronary artery disease. N Engl J Med. 352:1685–1695. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hoefer IE, van Royen N, Rectenwald JE,
Bray EJ, Abouhamze Z, Moldawer LL, Voskuil M, Piek JJ, Buschmann IR
and Ozaki CK: Direct evidence for tumor necrosis factor-α signaling
in arteriogenesis. Circulation. 105:1639–1641. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Clausell N, de Lima VC, Molossi S, Liu P,
Turley E, Gotlieb AI, Adelman AG and Rabinovitch M: Expression of
tumour necrosis factor alpha and accumulation of fibronectin in
coronary artery restenotic lesions retrieved by atherectomy. Br
Heart J. 73:534–539. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Woessner JF Jr: MMPs and TIMPs - an
historical perspective. Mol Biotechnol. 22:33–49. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Siefert SA and Sarkar R: Matrix
metalloproteinases in vascular physiology and disease. Vascular.
20:210–216. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li H, Liang J, Castrillon DH, DePinho RA,
Olson EN and Liu ZP: FoxO4 regulates tumor necrosis factor
alpha-directed smooth muscle cell migration by activating matrix
metalloproteinase 9 gene transcription. Mol Cell Biol.
27:2676–2686. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xiong W, MacTaggart J, Knispel R, Worth J,
Persidsky Y and Baxter BT: Blocking TNF-alpha attenuates aneurysm
formation in a murine model. J Immunol. 183:2741–2746. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Johnson C and Galis ZS: Matrix
metalloproteinase-2 and -9 differentially regulate smooth muscle
cell migration and cell-mediated collagen organization.
Arterioscler Thromb Vasc Biol. 24:54–60. 2004. View Article : Google Scholar
|
|
12
|
Huo Y and Ley K: Adhesion molecules and
atherogenesis. Acta Physiol Scand. 173:35–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kwon JA, Yu CB and Park HD: Bacteriocidal
effects and inhibition of cell separation of cinnamic aldehyde on
Bacillus cereus. Lett Appl Microbiol. 37:61–65. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cheng SS, Liu JY, Chang EH and Chang ST:
Antifungal activity of cinnamaldehyde and eugenol congeners against
wood-rot fungi. Bioresour Technol. 99:5145–5149. 2008. View Article : Google Scholar
|
|
15
|
Kwon JY, Hong SH, Park SD, Ahn SG, Yoon
JH, Kwon BM and Kim SA: 2′-Benzoyloxycinnamaldehyde inhibits nitric
oxide production in lipopolysaccharide-stimulated RAW 264.7 cells
via regulation of AP-1 pathway. Eur J Pharmacol. 696:179–186. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee CW, Hong DH, Han SB, Park SH, Kim HK,
Kwon BM and Kim HM: Inhibition of human tumor growth by 2′-hydroxy-
and 2′-benzoyloxycinnamaldehydes. Planta Med. 65:263–266. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Han DC, Lee MY, Shin KD, Jeon SB, Kim JM,
Son KH, Kim HC, Kim HM and Kwon BM: 2′-benzoyloxycinnamaldehyde
induces apoptosis in human carcinoma via reactive oxygen species. J
Biol Chem. 279:6911–6920. 2004. View Article : Google Scholar
|
|
18
|
Lee CW, Lee SH, Lee JW, Ban JO, Lee SY,
Yoo HS, Jung JK, Moon DC, Oh KW and Hong JT:
2-hydroxycinnamaldehyde inhibits SW620 colon cancer cell growth
through AP-1 inactivation. J Pharmacol Sci. 104:19–28. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim SA, Sung YK, Kwon BM, Yoon JH, Lee H,
Ahn SG and Hong SH: 2′-Hydroxycinnamaldehyde shows antitumor
activity against oral cancer in vitro and in vivo in a rat tumor
model. Anticancer Res. 30:489–494. 2010.PubMed/NCBI
|
|
20
|
Ahn SG, Jin YH, Yoon JH and Kim SA: The
anticancer mechanism of 2′-hydroxycinnamaldehyde in human head and
neck cancer cells. Int J Oncol. 47:1793–1800. 2015.PubMed/NCBI
|
|
21
|
Liao BC, Hsieh CW, Liu YC, Tzeng TT, Sun
YW and Wung BS: Cinnamaldehyde inhibits the tumor necrosis
factor-α-induced expression of cell adhesion molecules in
endothelial cells by suppressing NF-kappaB activation: Effects upon
IkappaB and Nrf2. Toxicol Appl Pharmacol. 229:161–171. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kim SA, Kim YC, Kim SW, Lee SH, Min JJ,
Ahn SG and Yoon JH: Antitumor activity of novel indirubin
derivatives in rat tumor model. Clin Cancer Res. 13:253–259. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jin YH, Ahn SG and Kim SA: BAG3 affects
the nucleocytoplasmic shuttling of HSF1 upon heat stress. Biochem
Biophys Res Commun. 464:561–567. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim EJ, Park WH, Ahn SG, Yoon JH, Kim SW
and Kim SA: 5′-nitro-indirubinoxime inhibits inflammatory response
in TNF-alpha stimulated human umbilical vein endothelial cells.
Atherosclerosis. 211:77–83. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ngoc TM, Nhiem NX, Khoi NM, Son DC, Hung
TV and Van Kiem P: A new coumarin and cytotoxic activities of
constituents from Cinnamomum cassia. Nat Prod Commun. 9:487–488.
2014.PubMed/NCBI
|
|
26
|
Kim HJ, Cha BY, Choi B, Lim JS, Woo JT and
Kim JS: Glyceollins inhibit platelet-derived growth factor-mediated
human arterial smooth muscle cell proliferation and migration. Br J
Nutr. 107:24–35. 2012. View Article : Google Scholar
|
|
27
|
Bendeck MP, Zempo N, Clowes AW, Galardy RE
and Reidy MA: Smooth muscle cell migration and matrix
metalloproteinase expression after arterial injury in the rat. Circ
Res. 75:539–545. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Galis ZS, Johnson C, Godin D, Magid R,
Shipley JM, Senior RM and Ivan E: Targeted disruption of the matrix
metalloproteinase-9 gene impairs smooth muscle cell migration and
geometrical arterial remodeling. Circ Res. 91:852–859. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cho A and Reidy MA: Matrix
metalloproteinase-9 is necessary for the regulation of smooth
muscle cell replication and migration after arterial injury. Circ
Res. 91:845–851. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Galis ZS, Muszynski M, Sukhova GK,
Simon-Morrissey E, Unemori EN, Lark MW, Amento E and Libby P:
Cytokine-stimulated human vascular smooth muscle cells synthesize a
complement of enzymes required for extracellular matrix digestion.
Circ Res. 75:181–189. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fabunmi RP, Baker AH, Murray EJ, Booth RF
and Newby AC: Divergent regulation by growth factors and cytokines
of 95 kDa and 72 kDa gelatinases and tissue inhibitors or
metalloproteinases-1, -2, and -3 in rabbit aortic smooth muscle
cells. Biochem J. 315:335–342. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Moon SK, Cho GO, Jung SY, Gal SW, Kwon TK,
Lee YC, Madamanchi NR and Kim CH: Quercetin exerts multiple
inhibitory effects on vascular smooth muscle cells: Role of ERK1/2,
cell-cycle regulation, and matrix metalloproteinase-9. Biochem
Biophys Res Commun. 301:1069–1078. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Karki R, Jeon ER and Kim DW: Nelumbo
nucifera leaf extract inhibits neointimal hyperplasia through
modulation of smooth muscle cell proliferation and migration.
Nutrition. 29:268–275. 2013. View Article : Google Scholar
|
|
34
|
Suh SJ, Kwak CH, Chung TW, Park SJ,
Cheeeei M, Park SS, Seo CS, Son JK, Chang YC, Park YG, et al:
Pimaric acid from Aralia cordata has an inhibitory effect on
TNF-α-induced MMP-9 production and HASMC migration via
down-regulated NF-κB and AP-1. Chem Biol Interact. 199:112–119.
2012. View Article : Google Scholar : PubMed/NCBI
|