Introduction
Cervical cancer is ranked as the second cause of
cancer deaths in women worldwide (1–3).
Human papillomaviruses (HPV) are considered as the causative agents
of most cervical cancers (1–3). The
binding of HPV E6 and E7 oncoproteins to tumor-suppressor genes
p53 and RB could result in impaired
tumor-suppressor-gene function and eventual cell immortalization
(2). Although recent studies have
identified other equally important molecular events for malignant
transformation of cervical epithelial cells, including the
activation of certain tumor-promoting genes and pathways (1), E6 and E7 oncoproteins are widely
recognized as the driving force in carcinogenesis of cervical
epithelium. However, it is not clear whether and how E6 and E7
could promote the progression of cervical cancer after the tumor
formation.
The immortalized cells resulting from transfection
with HPV DNA have been found to express significantly higher levels
of Sine oculis homeobox homolog 1 (SIX1), suggesting that HPV might
be able to promote tumor cell proliferation by increasing SIX1
expression (4). SIX1 gene is one
of homeobox genes which code for transcription factors functioning
as master regulators in proliferation, apoptosis, migration and
invasion (5). Similar to many cell
cycle proteins, the expression of SIX1 is regulated in a cell
cycle-specific manner, where it is transcriptionally upregulated at
the G1/S boundary, highly expressed in S and G2/M phases, then
degraded via the ubiquitin-proteasome pathway (6). SIX1 is necessary in early
embryogenesis, but its expression is lost in most well
differentiated tissues. However, SIX1 has been found to be
overexpressed in a variety of malignancies, showing the ability to
promote tumor growth and metastasis (7). Moreover, SIX1 was reported to abolish
the G2 cell cycle checkpoint and upregulate cyclin A1 to promote
cell proliferation in breast cancer (8,9).
These results implied that SIX1 might play an important role in the
proliferation of tumor cells and the development of tumor.
The ability of tumor cells to sustain the
uncontrolled proliferation might be influenced by the factors that
could modulate DNA replication, cell cycle, apoptosis-resistance,
metabolic capacity, etc. In this study we investigated the
mechanism underlying the effect of SIX1 on the proliferation of
cervical cancer cells. The resultant data showed that SIX1
functioned as a master regulator in DNA replication. SIX1
upregulated multiple genes affecting the initiation of DNA
replication, accelerated G1 to S phase progression, and promoted
tumor cell proliferation. These results implied that SIX1 might
play a crucial role in tumorigenesis and progression of cervical
cancer, and also suggested that SIX1 might be a therapeutic target
in cervical cancer treatment.
Materials and methods
Cells and transfection
Human cervical cancer cell lines C33a
(HPV-negative), Siha and Caski (HPV-positive) were purchased from
the American Type Culture Collection (Manassas, VA). Cells were
transduced with CMV-Fluc-IRES-RFP lentiviral particles (GeneChem,
Shanghai, China). The cells with stable transfection
(RFP+) were isolated by FACS. RFP/luciferase-expressing
cells were used in living imaging. In cell cycle analysis by flow
cytometry, RFP-free cells were used.
The expression vectors for E6 and E7 were
constructed as previously described (10). Briefly, the cDNAs coding for E6 and
E7 were amplified from total RNA isolated from Caski cell lines
using following primers: E6, 5′-GCGGCCGCC ACCATGTTTCAGGACCACAG-3′
(sense) and 5′-CTGCGG CCGCGATTACAGCTGGGTTTTCTCT-3′ (antisense); E7,
5′-GCGGCCGCCACCATGGCATGGCATGGAGATAC ACCT-3′ (sense) and
5′-AGGCGGCCGCGATTATGGTTT CTGAGAACA-3′ (antisense). The cDNAs were
inserted into expression vector pcDNA3.1. C33a cells were
transfected with plasmid pcDNA3.1-SIX1 (a kind gift from Kong-Ming
Wu, Thomas Jefferson University, Philadelphia, PA), pcDNA3.1-E6,
pcDNA3.1-E7 or pcDNA3.1 using Lipofectamine 2000 (Invitrogen, Life
Technologies). ShSIX1(1) and
shSIX1(2) shRNA lentiviral
particles (GeneChem) were used to knockdown SIX1 expression,
targeting 5′-CCAGCTCAGAAGAGG AATT-3′ and 5′-CACGCCAGGAGCTCAAACT-3′,
respectively. Shcon, the shRNA not targeting any known gene, was
used as control. Caski cells were transduced with SIX1 shRNA
lentiviral particles, or control shRNA lentiviral particles. The
cells with stable transfection were selected with G418 (C33a) and
puromycin (Caski). The transfected cells were designated as
C33a-3.1, C33a-E6, C33a-E7, C33a-SIX1, Caski-shcon,
Caski-shSIX1(1) and
Caski-shSIX1(2), respectively.
Immunohistochemistry
A tissue microarray (TMA) (CIN481) was purchased
from Alenabio (Xi-an, China), containing cervical intraepithelial
neoplasia (CIN) samples. Tumor samples were acquired by surgery
from cancer patients without preoperative treatment, which was
approved by the Ethics Committee of the Medical Faculty of Tongji
Medical College. Informed consent was obtained from all subjects.
The tumor samples were processed into TMA at Outdo Biotech Company
(Shanghai, China). TMAs and tissue sections were subjected to
immunohistochemical analysis as described previously (11), using the Avidin-Biotin Complex
(ABC) Vectastain Kit (Zsgb-Bio, Beijing, China). Rabbit anti-human
SIX1 antibody (Sigma-Aldrich) and rabbit anti-human Ki67 antibody
(Santa Cruz Biotechnology) were used as primary antibodies. For
semi-quantitative evaluation of SIX1 expression in tissue, staining
intensity was graded (0, absence; 1, weak; 2, moderate; 3, strong),
and assessed using an immunoreactivity-scoring system, HSCORE
(12). The HSCORE was calculated
using the following formula: HSCORE = ∑Pi(i + 1), where i is the
staining intensity of tumor cells and Pi is the percentage of tumor
cells at each level of intensity. The percentage of Ki67-positive
tumor cells was measured as mean of five hot-spots on one slide
from each tumor. Hot-spots were defined as areas with the highest
Ki67 intensity. The slides were read by two pathologists and each
data point represents the mean of the scores from two
pathologists.
Assay of gene expression by real-time
RT-PCR
Total RNA was extracted from cells with TRIzol
reagent (Invitrogen). The relative quantity of mRNA was determined
by real-time RT-PCR. Gapdh and EEF1A1 were chosen as
reference genes, which were reported as the most reliable
combination in cervical cancer (13). The primer sequences are shown in
Table I. The expression of genes
was quantified using the comparative CT method. The
expression level of each mRNA was normalized to that of
GAPDH and EEF1A1 mRNAs, and expressed as n-fold
difference relative to the control.
 | Table IPrimers for real-time RT-PCR. |
Table I
Primers for real-time RT-PCR.
| Gene | Sense primers | Antisense
primers |
|---|
| GAPDH |
GACAGTCAGCCGCATCTTCT |
TTAAAAGCAGCCCTGGTGAC |
| EEF1A1 |
TGCGGTGGGTGTCATCAAA |
AAGAGTGGGGTGGCAGGTATTG |
| MCM2 |
TTGGCGTGAGTTGCGTATTC |
GAGACTGAAAACGATTACAAACATC |
| MCM3 |
CCACAGATGATCCCAACTTT |
GTCCCATGTAGAAGGTTGTC |
| MCM4 |
TGAACCTCTATACATGCAACGAC |
CAGGGTAACGGTCAAAGAAGATT |
| MCM5 |
TCACCAAGCAGAAATACCC |
GTCCATGAGTCCAGTGAG |
| MCM6 |
AGTGTGTGAGTCGATGAATA |
CATTAAAGAGGAGCGAGCTT |
| MCM7 |
GGAAATATCCCTCGTAGTATCAC |
CTGAGAGTAAACCCTGTACC |
| POLA1 |
AAAGATCCATTGGAGCTTCACC |
TCAGCACGTTTAAGAGGAACAG |
| POLA2 |
AGGAGCTAGAGACATTGTTTCCA |
CTCGCTTCTGAGAACCCTTTG |
| PRIM1 |
ACATTCGCTACCAATCCTTCAAC |
AGCTCCCAGCTTCACTGTATT |
| PRIM2 |
TCTTCGAGAACAGGAGATTGTTG |
CAGAGCATCAGCAAAAGGGAT |
| RFC1 |
TTGTCATGGGTCGTGATAGTGG |
CCTGGCATAGTCCGAATCAGAT |
| RFC2 |
GTGAGCAGGCTAGAGGTCTTT |
TGAGTTCCAACATGGCATCTTTG |
| RFC3 |
GTGGACAAGTATCGGCCCTG |
TGATGGTCCGTACACTAACAGAT |
| RFC4 |
CCGCTGACCAAGGATCGAG |
AGGGAACGGGTTTGGCTTTC |
| RFC5 |
GACATGCGTAGGGCTCTGAAC |
GTGCAGGTGTAGACAGTCTCC |
| POLD1 |
ATCCAGAACTTCGACCTTCCG |
ACGGCATTGAGCGTGTAGG |
| POLD2 |
ATGTTTTCTGAGCAGGCTGCC |
TGGGGGAGCAGGTTGTGCTC |
| POLD3 |
AAGCCATGCTAAAGGACAGTG |
CATTGGTGGTCAGCTCATTGTT |
| POLD4 |
ATCACTGATTCCTACCCGGTT |
AGAGATGCCAGAGACTGCACT |
| POLE |
TTGCGACCAGAAAGGGTTGT |
TGATTTGGCAAGTCCAGATCCT |
| POLE2 |
TGAGAAGCAACCCTTGTCATC |
TCATCAACAGACTGACTGCATTC |
| POLE3 |
GGCCCGAGGACCTAAACCT |
ATGTGGCGTACAGCACGAAG |
| POLE4 |
AAGGTCTTCCACTGTTTCTGTCTG |
CCTTTATTCTGGTCCTCATCATTC |
Western blot assay
Western blot assay was performed as described
previously (14). The antibodies
were purchased from Sigma-Aldrich, Biorbyt and Santa Cruz
Biotechnology.
Bioinformatic analysis
The normalized RNA-sequencing data of 116 cases of
cervical cancer samples was publicly available from TCGA database
(https://tcga-data.nci.nih.gov/tcga/tcgaHome2.jsp).
Differential gene expression based on SIX1 expression category was
determined using Bioconductor edgeR package (15) as described previously (16). Then, pathway analysis of
differentially expressed genes was performed using Onto-Express
from Onto-Tools package (17).
To further analyze the effect of SIX1 expression on
DNA replication and cell cycle pathways (gene sets), Gene set
enrichment analysis (GSEA) package was used as described previously
(18). For this analysis, SIX1
expression was set as continuous label. Heat map was generated by
GSEA.
BrdU incorporation
The BrdU colorimetric ELISA kit (Roche) was used to
detect 5′-bromo-2′-deoxyuridine (BrdU) incorporated into newly
synthesized DNA, according to the manufacturer’s protocol. Briefly,
tumor cells were incubated with BrdU solution for 2–16 h at 37°C,
then fixed and denatured and incubated with a peroxidase-conjugated
antibody against BrdU for 1 h at room temperature. The cells were
then incubated with the tetramethyl-benzidine (TMB) substrate
solution and absorbance was read at 450 nm using a multiskan
spectrum microplate reader (μQuant Bio-Tek Instruments, USA).
Flow cytometry analysis
Cells were harvested by trypsinization and fixed in
70% ice-cold ethanol overnight at −20°C, and then washed in PBS and
incubated in RNase A (10 U/ml) and propidium iodide (20 mg/ml) for
30 min at room temperature. The cells in different phases were
detected by flow cytometry on a FACS Calibur system
(Becton-Dickinson) and analyzed using FlowJo software. Serum
starvation was performed as described previously (19).
Tumor cell proliferation assay
Tumor cells were seeded at 2×103 per well
into 96-well plates. Using cell counting kit-8 (CCK8, Boster,
China), the proliferation of the cells at each time point was
measured with multiskan spectrum microplate reader (μQuant Bio-Tek
Instruments, USA) at 450 nm wavelength. Cell proliferation index
was expressed as fold change related to day 0.
Soft agar assay
Cells were suspended in 0.35% agar in DMEM (20% FBS)
and plated (1.5×103 per well) on a layer of 0.5% agar in
culture medium in 6-well plates. After two-week culture, colonies
were counted and photographed by phase-contrast microscopy (Nikon,
Japan). The size of 20 randomly chosen colonies per well was
measured. The average size of colony was calculated as (length +
width) × 0.5. The colony formation rate was calculated as the
number of colonies/1,500 ×100%.
Animal studies
Female NOD-SCID mice (4 weeks old) were purchased
from Beijing HFK Bio-Technology Co. Ltd. (Beijing, China). The
studies were approved by the Committee on Ethics of Animal
Experiments of Tongji Medical College. The mice were maintained in
the accredited animal facility of Tongji Medical College.
Intramuscular tumors were initiated by injecting 1×106
tumor cells in 50 μl of media into the hind limb of NOD-SCID mice.
The sizes of ectopic tumors were monitored by a caliper and
calculated by the formula: volume = (width)2 × length/2.
Orthotopic xenograft model of cervical cancer was performed as
previously described (20).
Briefly, intramuscular tumors were used as donor tumor fragments
for orthotopic transplantation into the cervix of NOD-SCID mice.
Tumor growth in cervix was dynamically monitored in living mice by
optical imaging of luciferase activity using the IVIS Spectrum
system (Caliper, Xenogen, USA). After anaesthetization with 3%
pentobarbital sodium, mice were imaged 10 min after intraperitoneal
injection of 100 mg/kg D-luciferin. Tumor size was measured by
three-dimensional reconstruction of image using Living Image
version 4.3.1 software.
Statistical analysis
Each experiment was repeated at least three times
independently. SPSS (version 13.0) software package was used for
statistical analysis. Results are expressed as mean value ±
standard error of the mean (SEM), and interpreted by one-way ANOVA.
Differences were considered to be statistically significant at
P<0.05.
Results
SIX1 expression is increased by HPV
oncoprotein in cervical cancer
To determine the role of SIX1 in cervical cancer, we
analyzed the expression of SIX1 in each stage of cancer
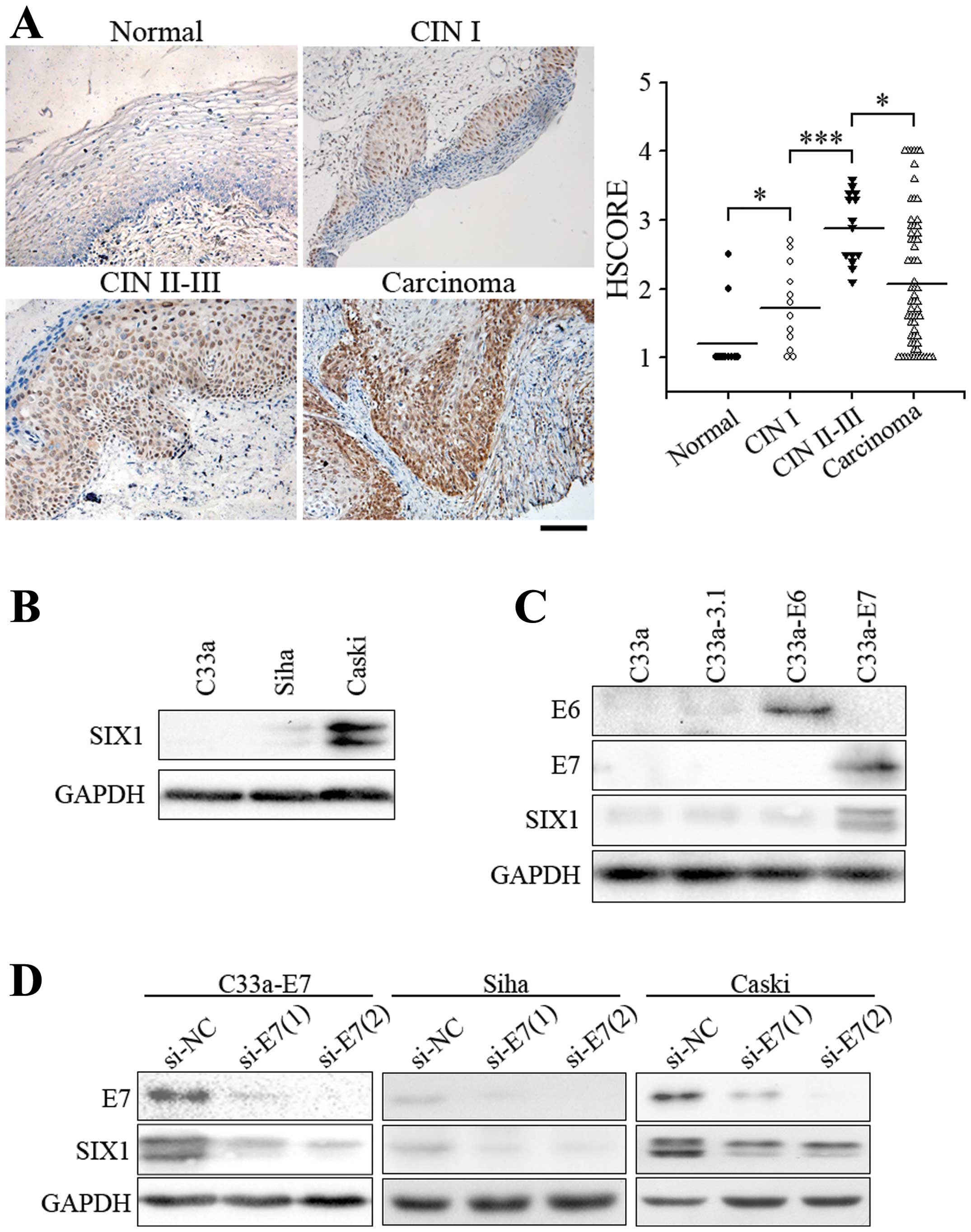
development. SIX1 expression was negligible in normal cervical
epithelium, but was significantly increased in low-grade cervical
intraepithelial neoplasia (CIN I, precancerous lesion) (Fig. 1A). Higher expression levels of SIX1
were observed in almost all cases of high-grade cervical
intraepithelial neoplasia (CIN II-III). SIX1-expressing cells
shared the same distribution area with the extent of
intraepithelial neoplasia (Fig.
1A). In three cervical squamous cell carcinoma cell lines, the
expression of SIX1 was very low in HPV-negative C33a cells,
slightly elevated in Siha cells which contain 1–2 copies of HPV
DNA, and much higher in Caski cells which contain 60–600 copies of
HPV DNA (Fig. 1B), suggesting that
HPV infection increased the expression of SIX1. We then further
tested whether SIX1 was induced by E6 or E7 oncoprotein, the
driving force of cervical cancer. Overexpression of E7, but not E6,
in HPV-negative C33a cells induced SIX1 expression (Fig. 1C). Consistently, when E7 were
knocked down in C33a-E7, Siha or Caski cells, the expression of
SIX1 was significantly decreased (Fig.
1D). These results suggested that HPV-E7 is one of the factors
that are required for inducing the expression of SIX1. Moreover,
the correlation of SIX1 expression to the progression of cervical
cancer suggested that SIX1 might play an important role in
tumorigenesis and progression of cervical cancer.
SIX1 may modulate the expression of
multiple genes to promote tumor cell proliferation
To identify the effect of SIX1 on the development of
cervical cancer, bioinformatic analysis was performed to predict
the effect of SIX1 on gene expression in cervical cancer. The
RNA-sequencing data of 116 cervical cancer specimens was obtained
from TCGA database. The specimens were divided into SIX1-low
expression group (n=58) and SIX1-high expression group (n=58).
Bioconductor edgeR package was used to identify the genes which
were significantly up-regulated in SIX1-high expression group.
Subsequent pathway analysis indicated five pathways which were
significantly enriched in the genes potentially upregulated by SIX1
(Fig. 2A). Two of the pathways
were correlated with cell proliferation: DNA replication and the
cell cycle. Further analysis by using GSEA package confirmed that
the enrichment of the upregulated genes related to DNA replication
was significantly correlated with the increase of SIX1 gene
expression (Fig. 2B, P<0.05,
FDR<0.25). The enrichment of the upregulated genes related to
cell cycle was correlated, although without statistical
significance, with the increase of SIX1 gene expression
(Fig. 2C). Importantly, the
analysis with both edgeR-Onto and GSEA methods showed that the
upregulated genes included many genes related to DNA replication
and the cell cycle, and that these genes could be considered as
candidate genes which might be up-regulated by SIX1 (Fig. 2D). These results implied that SIX1
may regulate the expression of the genes involved in DNA
replication to promote tumor cell proliferation.
SIX1 regulates the expression of genes
responsible for DNA replication and enhances DNA synthesis
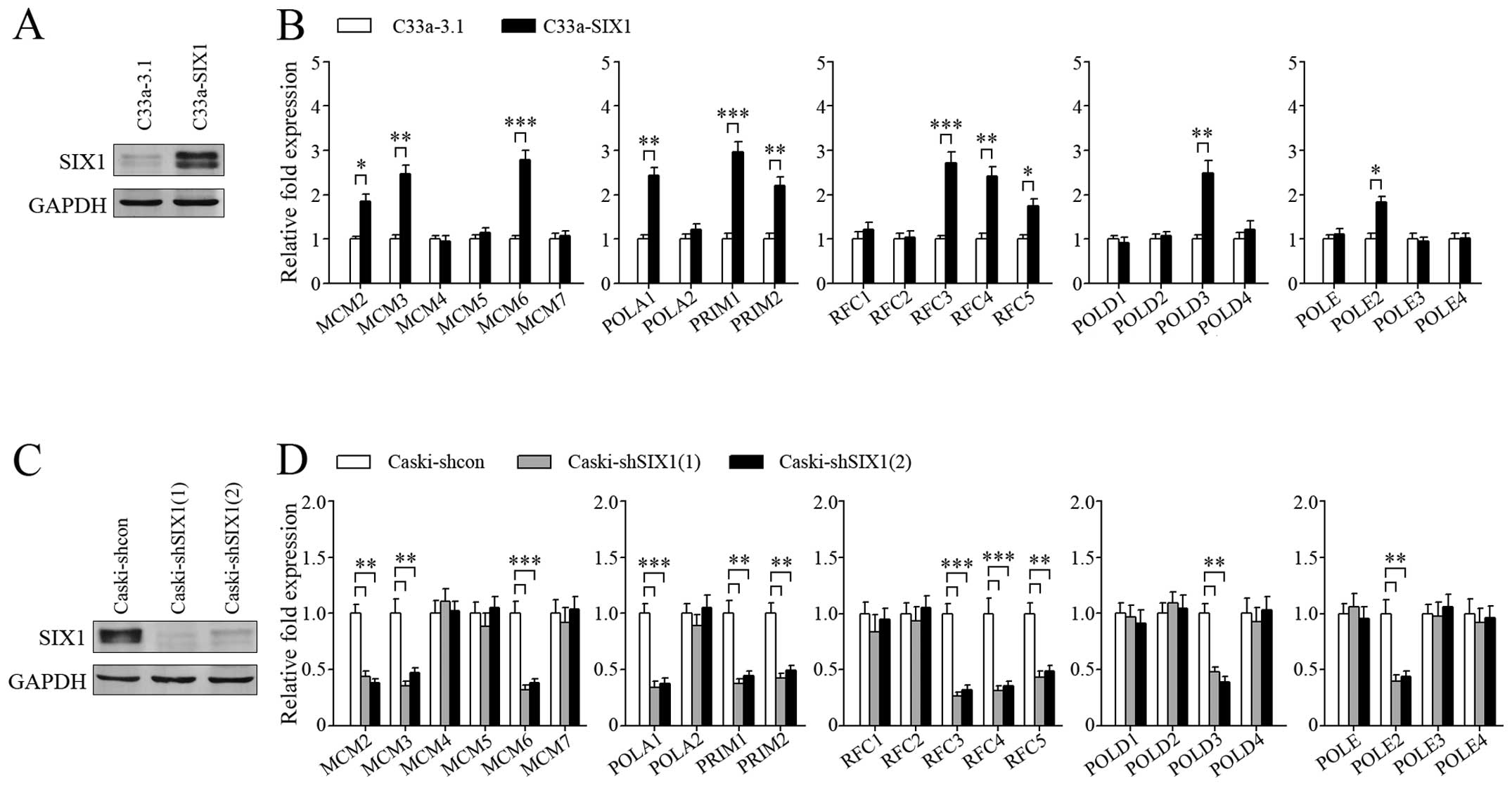
Based on the above results, we overexpressed SIX1 in
C33a cells (Fig. 3A) to
investigate the regulatory effect of SIX1 on DNA replication genes
(Fig. 3B). SIX1 expression was
knocked down in Caski cells (Fig.
3C) to further confirm the effect of SIX1 (Fig. 3D). The results showed that SIX1
could upregulate the expression of several genes coding for the
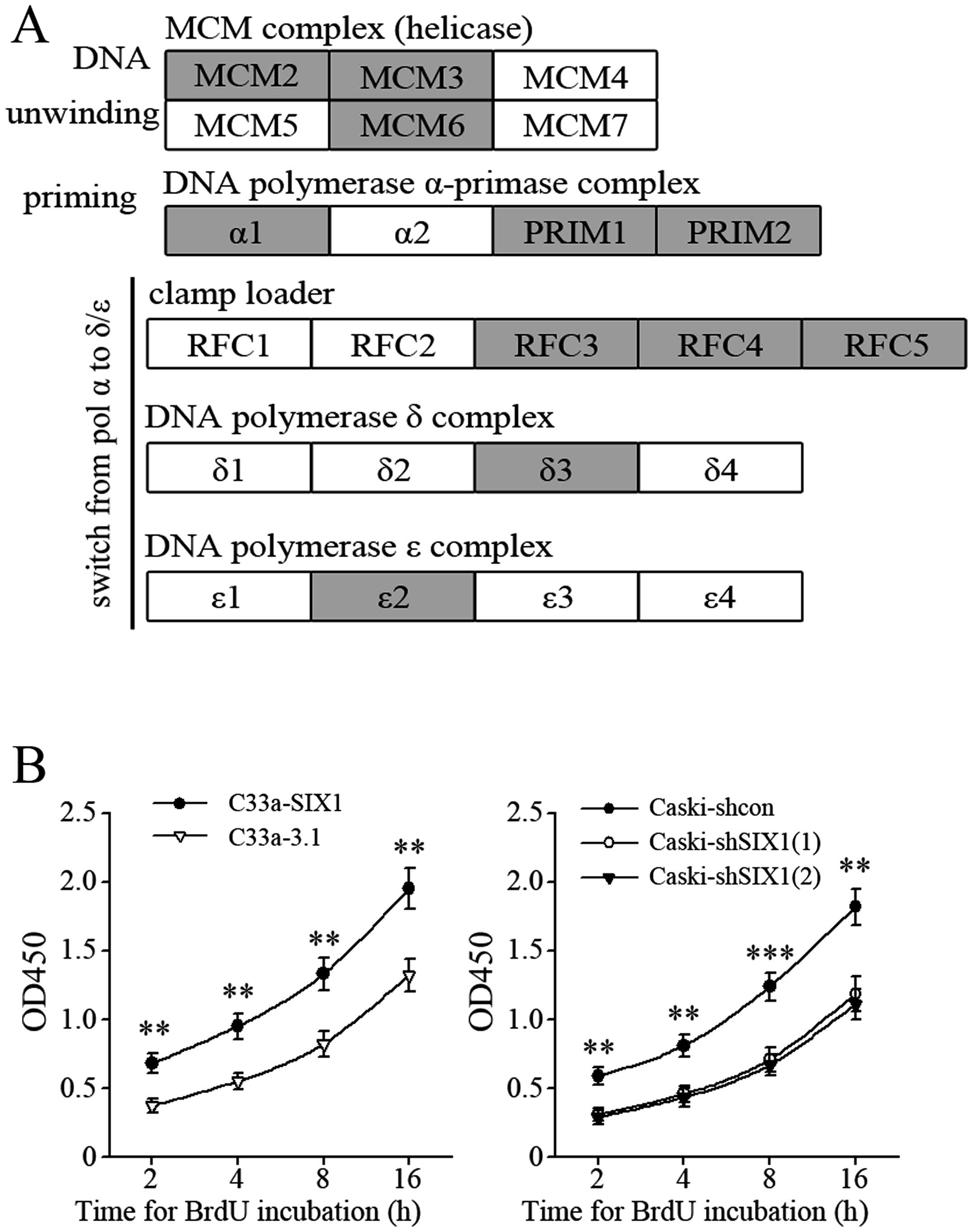
proteins in five complexes related to DNA replication, including
MCM complex (MCM2, MCM3, MCM6), DNA polymerase α-primase complex
(POLA1, PRIM1, PRIM2), clamp loader (RFC3, RFC4, RFC5), DNA
polymerase δ complex (POLD3) and DNA polymerase ɛ complex (POLE2).
These five complexes play critical roles in three important steps
of DNA replication initiation: DNA unwinding, synthesis of primer,
and switching from DNA polymerase α to polymerase δ or polymerase ɛ
(Fig. 4A). Consistently, BrdU
incorporation assay indicated that DNA synthesis was more active in
SIX1 highly expressed cervical cancer cells (Fig. 4B).
SIX1 modulates the cell cycle and
promotes G1 to S phase progression
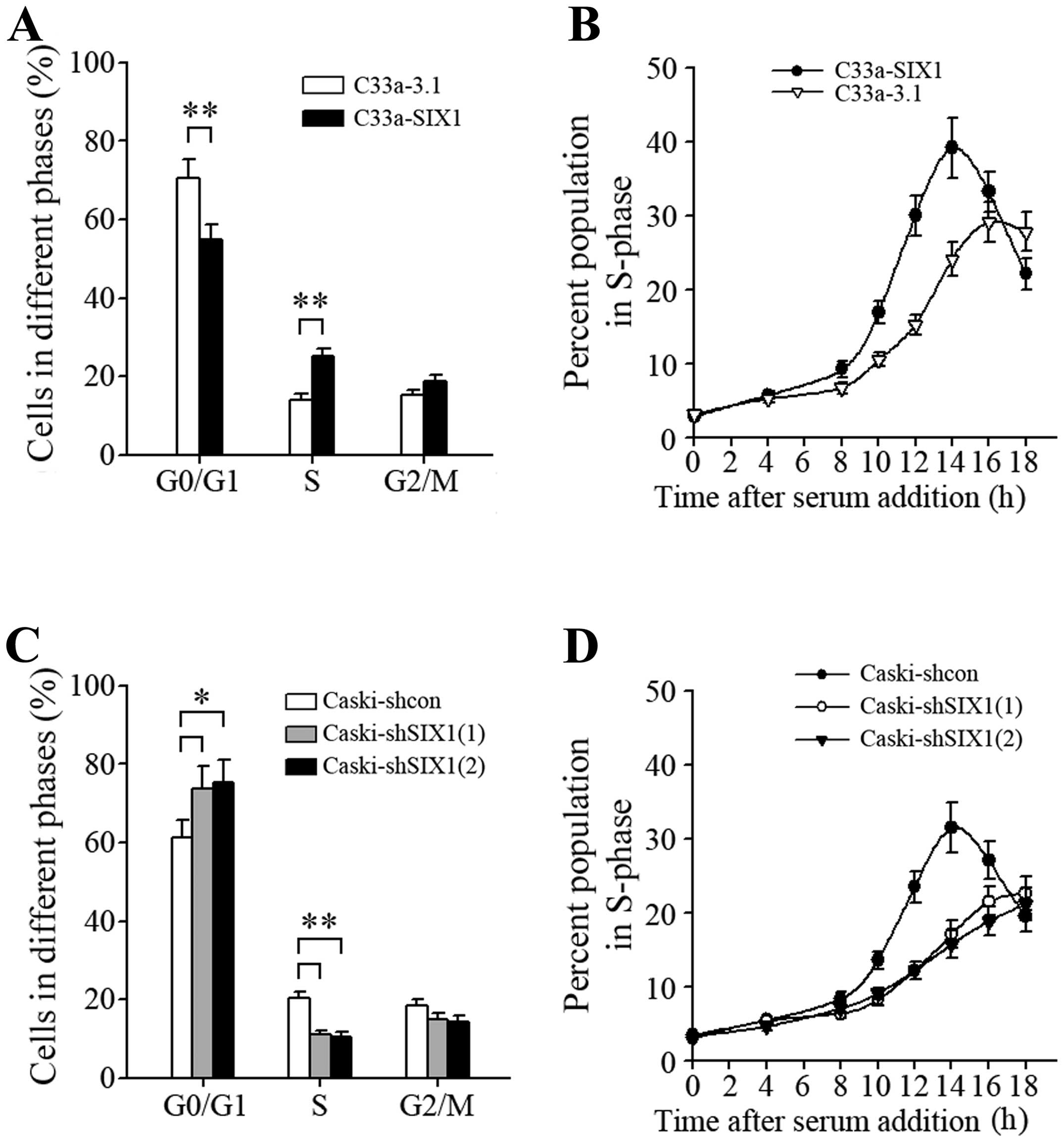
In accordance with the potential of SIX1 to promote
DNA replication, overexpression of SIX1 in C33a cells resulted in a
decrease of the percentage of cells in G0/G1 phase and a
concomitant increase of the percentage of cells in S phase
(Fig. 5A), indicating more active
cell proliferation. After synchronization by serum starvation and
following serum addition, C33a-SIX1 cells progressed into S phase
faster than corresponding control cells (Fig. 5B), and knockdown of SIX1 in Caski
cells resulted in the opposite effect (Fig. 5C and D). These results demonstrated
that SIX1 modulated the cell cycle and promoted G1 to S phase
progression.
SIX1 promotes tumor cell proliferation in
vitro
We next investigated whether SIX1 could promote
tumor cell proliferation. Overexpression of SIX1 in C33a cells
significantly, although slightly, promoted anchorage-dependent cell
growth, while knockdown of SIX1 in Caski cells decreased
anchorage-dependent cell growth (Fig.
6A). Intriguingly, in colony-forming assays in soft agar, the
tumor cells with highly expressed SIX1 showed remarkably larger
size colonies (Fig. 6B) and much
higher colony formation rate (Fig.
6C), indicating that SIX1 could more efficiently promote
anchorage-independent cell growth.
SIX1 promotes tumor growth in vivo
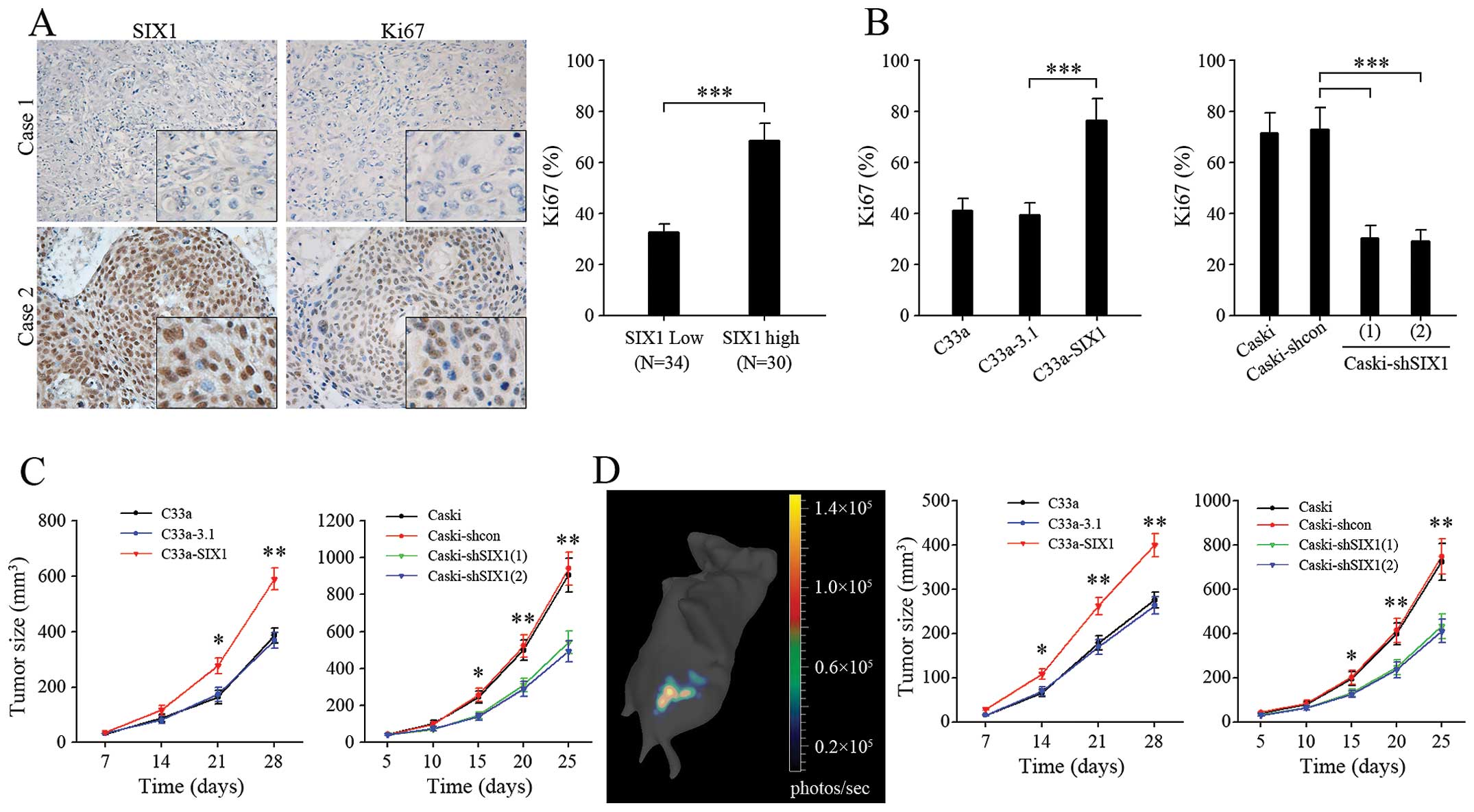
Expression of the proliferation marker Ki-67, was
significantly higher in SIX1 highly expressed cervical cancer
specimens (Fig. 7A). Comparing
with corresponding control groups, the percentage of Ki67-positive
tumor cells was increased in C33a-SIX1 tumors and decreased in
Caski-shSIX1 tumors (Fig. 7B),
indicating more active cell proliferation in SIX1 highly expressed
tumors. In line with this, in both ectopic (Fig. 7C) and orthotopic (Fig. 7D) xenograft models of cervical
cancer, overexpression of SIX1 in C33a cells significantly promoted
tumor growth, while knockdown of SIX1 in Caski cells decreased
tumor growth. Taken together, these results demonstrated that SIX1
could promote not only tumor cell proliferation in vitro,
but also tumor growth in vivo.
Discussion
Malignant proliferation is the fundamental trait of
tumor cells. The initiation of DNA replication represents a key
process for cell proliferation, and has a marked impact on
tumorigenesis and progression (19,21).
The factors that promote DNA replication are organized in
multiprotein complexes that coordinately help to recognize regions
of origin, unwind DNA and promote replisome formation (21). The data in this study showed that
SIX1 could modulate the expression of several genes related to DNA
replication, thus accelerating G1 to S phase progression, promoting
the proliferation of cervical cancer cells in vitro, and
promoting the growth of cervical cancer in vivo.
The expression of SIX1 was closely correlated with
the progression of cervical cancer as shown by our data. High
levels of SIX1 expression were observed in almost all regions that
developed intraepithelial neoplasia, suggesting that SIX1 might
play an important role in tumorigenesis and progression of cervical
cancer. Our data showed that the expression of SIX1 was positively
correlated with the copy number of HPV DNA in cervical squamous
cell carcinoma cells. These results were also supported by a report
that SIX1 was significantly increased in HPV-immortalized normal
human keratinocytes (4). Moreover,
we demonstrated that SIX1 expression in cervical cancer cells could
be modulated by HPV oncoprotein. HPV-E7, but not E6, was required
for sustaining the expression of SIX1 gene in cervical
cancer cells, including C33a-E7, Siha and Caski cells. These
results also implied that HPV-E7 oncoprotein might continuously
promote the progression of cervical cancer after the formation of
tumor by upregulating the expression of SIX1.
The general process of DNA replication initiation
involves several key steps: loading of helicase at replication
origins, unwinding of DNA by helicase, the synthesis of RNA primer
by primase, elongation of primer by DNA polymerase α, and then the
replacement of DNA polymerase α with polymerase δ or polymerase ɛ
for extending DNA strands (22,23).
SIX1 could modulate the expression of the genes that mainly affect
the initiation of DNA replication as shown by out data. The
upregulation of multiple genes by SIX1 might increase the
efficiency of DNA unwinding, synthesis of primer, and switching
from DNA polymerase α to polymerase δ or polymerase ɛ.
MCM complex serves as the primary helicase for
unwinding DNA during replication (21). Members of MCM family are proposed
as diagnostic or prognostic markers in various cancers due to their
increased proliferative potential (24). Studies have demonstrated that CDC6
is necessary for the loading of MCM complex at replication origins
(25). Our data in this study
showed that SIX1 upregulated the expression of MCM2, MCM3 and MCM6.
Bioinformatic analysis also suggested that SIX1 could upregulate
the expression of CDC6. Therefore, SIX1 might accelerate the
initiation of DNA replication by upregulating the expression of
these genes.
The synthesis of RNA primer by primase and the
following elongation of the primer by DNA polymerase α are required
for DNA replication in eukaryotes (23). The primase consists of two
subunits, p49 (PRIM1) and p58 (PRIM2), both of which were
upregulated by SIX1. Moreover, SIX1 could upregulate the expression
of polymerase α1 (POLA1). The increased expression of these genes
might make the initiation of DNA replication more efficient by
improving the efficiency of primer synthesis. After the synthesis
and elongation of primer, the synthesis of new DNA strands was
catalyzed by DNA polymerase δ and polymerase ɛ. The switch from
polymerase α to polymerase δ and polymerase ɛ is mediated by
replication factor C (RFC) (26,27).
DNA polymerases and RFC are important not only for DNA replication,
but also for cell cycle control (26). RFC3 and RFC4 were reported to
promote tumor cell proliferation (28,29).
High RFC3 expression was associated with poor prognosis in a
variety of cancers (28).
Importantly, SIX1 not only increased the expression of DNA
polymerase δ3 (POLD3) and polymerase ɛ2 (POLE2), but also
upregulated the expression of RFC (RFC3, RFC4, RFC5). Therefore,
SIX1 might not only promote the initiation of DNA replication by
increasing the expression of primase and DNA polymerase α, but also
promote the further synthesis of DNA strands by upregulating the
expression of DNA polymerase δ3, polymerase ɛ2 and RFC.
SIX1 could significantly promote tumor cell
proliferation in vitro. Correspondingly, ectopic and
orthotopic xenograft models of cervical cancer further demonstrated
that SIX1 could promote tumor growth in vivo. Intriguingly,
colony formation assays in soft agar showed that SIX1 remarkably
promoted anchorage-independent cell growth, suggesting that the
promoting effect of SIX1 on tumor cell proliferation might promote
not only the development of primary tumor but also the
proliferation of disseminated tumor at metastatic sites.
In summary, this study revealed the important role
of SIX1 in modulating DNA replication. SIX1 upregulated the
expression of multiple key genes involved in the initiation of DNA
replication. These genes coordinate to accelerate G1 to S phase
progression, promote proliferation of cervical cancer cells and the
growth of cervical cancer. These results implied that SIX1 might
play a crucial role in the tumorigenesis and progression of
cervical cancer, and also suggested that targeting SIX1 might have
significant therapeutic value in cervical cancer treatment.
Acknowledgements
We thank Dr Qi-Lin Ao and Dr Shuang Guo for
reviewing the histology data. This study was supported by National
Science Foundation of China (nos. 81072135, 81372801, 81172466,
81072132 and 81372781).
References
|
1
|
Moody CA and Laimins LA: Human
papillomavirus oncoproteins: pathways to transformation. Nat Rev
Cancer. 10:550–560. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferenczy A and Franco E: Persistent human
papillomavirus infection and cervical neoplasia. Lancet Oncol.
3:11–16. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schiffman M, Castle PE, Jeronimo J,
Rodriguez AC and Wacholder S: Human papillomavirus and cervical
cancer. Lancet. 370:890–907. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wan F, Miao X, Quraishi I, Kennedy V,
Creek KE and Pirisi L: Gene expression changes during HPV-mediated
carcinogenesis: a comparison between an in vitro cell model and
cervical cancer. Int J Cancer. 123:32–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McCoy EL, Iwanaga R, Jedlicka P, et al:
Six1 expands the mouse mammary epithelial stem/progenitor cell pool
and induces mammary tumors that undergo epithelial-mesenchymal
transition. J Clin Invest. 119:2663–2677. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Christensen KL, Brennan JD, Aldridge CS
and Ford HL: Cell cycle regulation of the human Six1 homeoprotein
is mediated by APC(Cdh1). Oncogene. 26:3406–3414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Micalizzi DS, Wang CA, Farabaugh SM,
Schiemann WP and Ford HL: Homeoprotein Six1 increases TGF-beta type
I receptor and converts TGF-beta signaling from suppressive to
supportive for tumor growth. Cancer Res. 70:10371–10380. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ford HL, Kabingu EN, Bump EA, Mutter GL
and Pardee AB: Abrogation of the G2 cell cycle checkpoint
associated with overexpression of HSIX1: a possible mechanism of
breast carcinogenesis. Proc Natl Acad Sci USA. 95:12608–12613.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coletta RD, Christensen K, Reichenberger
KJ, et al: The Six1 homeoprotein stimulates tumorigenesis by
reactivation of cyclin A1. Proc Natl Acad Sci USA. 101:6478–6483.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cho YS, Kang JW, Cho M, et al: Down
modulation of IL-18 expression by human papillomavirus type 16 E6
oncogene via binding to IL-18. FEBS Lett. 501:139–145. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun C, Li N, Yang Z, et al: miR-9
Regulation of BRCA1 and ovarian cancer sensitivity to cisplatin and
PARP inhibition. J Natl Cancer Inst. 105:1750–1758. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xiong T, Zhao Y, Hu D, et al:
Administration of calcitonin promotes blastocyst implantation in
mice by up-regulating integrin beta3 expression in endometrial
epithelial cells. Hum Reprod. 27:3540–3551. 2012. View Article : Google Scholar
|
|
13
|
Shen Y, Li Y, Ye F, Wang F, Lu W and Xie
X: Identification of suitable reference genes for measurement of
gene expression in human cervical tissues. Anal Biochem.
405:224–229. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang W, Fang Y, Sima N, et al: Triggering
of death receptor apoptotic signaling by human papillomavirus 16 E2
protein in cervical cancer cell lines is mediated by interaction
with c-FLIP. Apoptosis. 16:55–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: a Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Anders S, McCarthy DJ, Chen Y, et al:
Count-based differential expression analysis of RNA sequencing data
using R and bioconductor. Nat Protoc. 8:1765–1786. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Draghici S, Khatri P, Bhavsar P, Shah A,
Krawetz SA and Tainsky MA: Onto-Tools, the toolkit of the modern
biologist: Onto-Express, Onto-Compare, Onto-Design and
Onto-Translate. Nucleic Acids Res. 31:3775–3781. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Subramanian A, Tamayo P, Mootha VK, et al:
Gene set enrichment analysis: a knowledge-based approach for
interpreting genome-wide expression profiles. Proc Natl Acad Sci
USA. 102:15545–15550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mazurek A, Luo W, Krasnitz A, Hicks J,
Powers RS and Stillman B: DDX5 regulates DNA replication and is
required for cell proliferation in a subset of breast cancer cells.
Cancer Discov. 2:812–825. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cairns RA and Hill RP: A fluorescent
orthotopic model of metastatic cervical carcinoma. Clin Exp
Metastasis. 21:275–281. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Costa A, Hood IV and Berger JM: Mechanisms
for initiating cellular DNA replication. Annu Rev Biochem.
82:25–54. 2013. View Article : Google Scholar
|
|
22
|
Soultanas P: Loading mechanisms of ring
helicases at replication origins. Mol Microbiol. 84:6–16. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Urban M, Joubert N, Purse BW, Hocek M and
Kuchta RD: Mechanisms by which human DNA primase chooses to
polymerize a nucleoside triphosphate. Biochemistry. 49:727–735.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gan N, Du Y, Zhang W and Zhou J: Increase
of Mcm3 and Mcm4 expression in cervical squamous cell carcinomas.
Eur J Gynaecol Oncol. 31:291–294. 2010.PubMed/NCBI
|
|
25
|
Cook JG, Park CH, Burke TW, et al:
Analysis of Cdc6 function in the assembly of mammalian
prereplication complexes. Proc Natl Acad Sci USA. 99:1347–1352.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Masuda Y, Suzuki M, Piao J, Gu Y,
Tsurimoto T and Kamiya K: Dynamics of human replication factors in
the elongation phase of DNA replication. Nucleic Acids Res.
35:6904–6916. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maga G, Stucki M, Spadari S and Hubscher
U: DNA polymerase switching: I. Replication factor C displaces DNA
polymerase alpha prior to PCNA loading. J Mol Biol. 295:791–801.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lockwood WW, Thu KL, Lin L, et al:
Integrative genomics identified RFC3 as an amplified candidate
oncogene in esophageal adenocarcinoma. Clin Cancer Res.
18:1936–1946. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Arai M, Kondoh N, Imazeki N, et al: The
knockdown of endogenous replication factor C4 decreases the growth
and enhances the chemosensitivity of hepatocellular carcinoma
cells. Liver Int. 29:55–62. 2009. View Article : Google Scholar : PubMed/NCBI
|